
Abstract
The subchondral bone is involved in the pathophysiology of osteoarthritis (OA), both by biochemical and mechanical pathways. Overloaded OA subchondral bone osteoblasts express a pro-angiogenic and pro-inflammatory phenotype which contributes to explain the structural changes (sclerosis and bone marrow lesion) visible in OA subchondral bone. Further, microfractures and conjonctivo-vascular structures constitute exchange routes between bone and the overlying cartilage for mediators produced by osteoblasts. This narrative review describes these physiopathological mechanisms and identifies possible therapeutic targets for pharmacological modalities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA) is considered as a global joint disease in which all articular and periarticular tissues are involved including synovial membrane, Hoffa’s infrapatellar fat pad, capsule, and ligaments. Further, several OA phenotypes can be distinguished according to the mechanical and the genetic or metabolic origin of the disease suggesting the existence of multiple clinical forms of the disease. However, the hallmark of OA remains a progressive degradation of cartilage. The progression of cartilage degradation is not linear, but interrupted by attempts at repair suggesting that chondrocytes keep potential synthetic capacities even at end stage of the disease. The mechanisms that led to cartilage failure have been largely investigated. They primarily involved an excessive production of matrix metalloproteinases and aggrecanases by the chondrocytes and the hypertrophic differentiation of chondrocytes leading to the calcification of the cartilage matrix. Under these numerous structural and metabolic alterations, a progressive loss of normal mechanical properties of the joint cartilage is observed, which gradually becomes less able to withstand mechanical loads.
Beside the cartilage, the subchondral bone plays a critical role in OA progression. The osteochondral plate is composed of a thin layer of cortical bone, so-called the subchondral plate, and the calcified layer of cartilage which is separated from the adjacent articular cartilage by the tidemark [1]. The subchondral plate is a layer of highly vascular cortical bone located between the calcified cartilage layer and the trabecular bone [2]. The osteochondral plate is an exchange area between bone and cartilage through which the bone supplies the cartilage with nutrients. The terminal blood vessels in the subchondral plate are directly in contact with the deep cartilage layer [3] and provide the cartilage with about 50 % of its glucose and oxygen requirements [2]. The subchondral bone is a bone area located below the cartilage and formed by the subchondral plate and a 6-mm layer of trabecular bone.
Imaging techniques have revealed many structural changes in the osteochondral pate and subchondral bone of OA joint. These changes are calcified cartilage thickening and tidemark duplication, subchondral bone sclerosis, cysts, bone marrow lesions, and osteophytes. The question of whether these bone changes occur prior to the cartilage degradation and initiate the osteoarthritic disease remains unresolved. In this review, we attempt to give a general view of the osteochondral plate and subchondral bone modifications in OA and their role in the physiopathology of OA.
Subchondral osteoblasts have an abnormal phenotype
Subchondral bone sclerosis is characterized by a trabecular thickening, an increase of the osteoid volume, and a decrease of calcium bind to collagen fiber. This abnormal mineralization is due to the overproduction of the homotrimeric alpha1 form of type I collagen by osteoblasts. This (α1)3 type I collagen has less affinity for the calcium than the (α1)2 α2 type I collagen [4, 5]. In 1999, Pelletier and collaborators first characterized phenotypic changes in OA subchondral osteoblasts [6–9]. They compared the synthetic activity of osteoblasts coming from the subchondral bone of OA with those of normal subjects. Compared to normal osteoblasts, OA cells showed a decreased cAMP response to PTH and an increased production of alkaline phosphatase and osteocalcin in response to vitamin D3. Further, in basal condition, OA osteoblasts produced more insulin-like growth factor-1 and urokinase than normal cells. In addition, we showed that osteoblasts coming from the thickening (called sclerotic, SC) subchondral bone located just below the main cartilage lesion had an increased alkaline phosphatase activity and produced more interleukin (IL)-6, IL-8, osteopontin, osteocalcin, transforming growth factor (TGF)-β1, and type I collagen than osteoblasts coming from the non-thickening neighboring area (called non-sclerotic, NSC) [10]. Increased Wnt signaling in the subchondral bone can also contribute to OA development. Microarray analysis also revealed that many of differentially expressed genes identified in OA bone [11] or in OA bone marrow mesenchymal stem cells [12] are related to the Wnt/β-catenin signaling pathway. Recently, sclerostin, a soluble antagonist of the Wnt signaling pathway, has been shown to be decreased in sclerotic subchondral bone [13]. Furthermore, we can mention that the phenotype of the osteoblasts coming from the osteophytes has recently been investigated. In comparison to normal subchondral osteoblasts, they produced more IL-6, IL-8, and matrix metalloproteinase (MMP)-13 [14].
OA subchondral bone changes: osteopenia and sclerosis
A recent study analyzed subchondral trabecular bone structure in knee OA patients using 3-T MRI [15]. This study indicates that subchondral trabecular bone structural changes due to OA were strongly related to attrition of cartilage in the medial joint. As cartilage in the medial joint decreased, bone volume fraction, trabecular thickness, and connectivity of the subchondral bone in the medial tibia increased. This is thought to be an adaptive response to increased loading on the medial joint or a repair mechanism of microfractures. These changes occurred more frequently in the tibia than in the femur [15]. In contrast, as medial joint cartilage decreased, in the lateral compartment, bone volume fraction, trabecular thickness, trabecular number, and connectivity of subchondral bone decreased [15]. This is attributed to a shift in loading on the medial joint, associated with decreased loading on the lateral compartment and consequently the occurrence of regional osteoporosis.
Tidemark duplication and calcified cartilage layer thickening
Calcified cartilage layer (CCL) shows important modification during OA. Multiple tidemarks are visible and an increase of CCL thickness is observed. These changes increase cartilage stiffness and reduce its capacity to absorb mechanical strain. In parallel, the CCL is invaded by blood vessels—a phenomenon associated with the hypertrophic differentiation of chondrocytes [1]. Interestingly, in vitro, chondrocytes cultured in alginate beads with serum undergo a hypertrophic differentiation and produce type X collagen and alkaline phosphatase—two markers of the hypertrophic chondrocyte differentiation. In turn, hypertrophic chondrocytes synthesize high levels of vascular endothelial growth factor (VEGF) and bone sialoprotein—two factors promoting endothelial cell proliferation and migration.
Mechanical stress induces osteoblast sclerotic phenotype
We recently demonstrated that many genes expressed by sclerotic osteoblasts are mechanosensitives [16, 17]. We showed that compression upregulated IL-6, prostaglandin E2 (PGE2), and receptor activator of nuclear factor kappa-B ligand (RANKL), but downregulated osteoprotegerin (OPG) gene and protein production. These factors play a key role in the osteoblast/osteoclast crosstalk and finely regulate the osteoclastic activity [18]. IL-6 and PGE2 stimulate RANKL and inhibit OPG production by osteoblasts and increase osteoclast differentiation and activation via an effect on the RANK/RANKL/OPG system [19]. These findings show that in osteoblast-mechanosensitive genes code for bone remodeling and angiogenic/inflammatory proteins rather than for matrix proteins. This particular pattern could be associated to the formation of bone marrow lesions visible by MRI in OA trabecular bone [20, 21].
We have also observed than SC osteoblasts are less sensitive to mechanical stimuli than NSC ones. This lack of sensitivity of SC osteoblasts to mechanical strains can be explained by a decreased connexin (Cx) 43 expression. Indeed, we have shown that Cx43 expression was decreased by 30 % in SC osteoblasts compared to NSC osteoblasts. Cx43 is the major gap junction present in osteoblasts and in osteocytes. This membrane protein is very important for osteoblast metabolism and response to mechanical strain [22]. Thus, more than explaining the decrease of sensitivity to load of SC osteoblasts, a decrease of Cx43 could explain many characteristics of SC osteoblasts, as decreasing mineralization ability [23], response to PTH [6] and OPG production.
Surprisingly, after a 4-h mechanical stimulation, we observed that differences between NSC and SC osteoblasts are highly reduced or even abolished. These findings also suggest that the phenotypic difference observed between sclerotic and not sclerotic Ob in vitro could result from altered mechanical strain occurring in situ prior to bone sampling. Using a computational model, a recent study shows that bone adaptation in OA can be mechano-regulated. Structural changes occur independently from cartilage degeneration. In conditions associated with OA, like high joint load or malalignment, this group observed a bone remodeling leading to a bone structure close to subchondral bone sclerosis observed in OA patients [24]. This finding supports the concept that subchondral bone and the overlying cartilage form a mechanical unit responsible for the absorption of mechanical strains. As the bone sclerosis progresses in the subchondral bone plate, this could trigger cartilage degradation which leads to calcified cartilage thickening which in turn contributes to further promote bone sclerosis.
Obesity and lipid involvement in subchondral bone changes
It is now clearly established that obesity is associated with the prevalence of symptomatic knee osteoarthritis. More surprising was the relationship between obesity and hand OA. In that case, joint overloading can be excluded as the cause of OA. This finding has suggested the hypothesis that beside the mechanical factors, systemic mediators produced by fat tissue could be involved in OA pathogenesis. Recently, the role of adipokines in cartilage degradation was investigated. Contradictory effects were observed according the adipokine tested, but leptin and vistatin were systematically deleterious for cartilage [25]. Leptin and several other adipokines are produced by adipocytes, but also by the osteoblasts themselves, and play an important role in bone metabolism. In leptin knockout mice, leptin administration increases bone density and mineralization and cortical bone formation [26]. Mutabaruka et al. have recently shown that OA subchondral osteoblasts overexpress leptin compared to normal cells [27]. Furthermore, they found that leptin neutralization in OA subchondral osteoblasts induced a normalization of their phenotype as suggested by the decrease of alkaline phosphatase, osteocalcin, type I collagen, and TGF-β1 production.
Beside the leptin, Davies et al. have shown that high serum levels of total cholesterol and triglycerides were associated with the incidence of BMLs over 2 years [28]. This finding supports the recent physiopathological concepts linking metabolic syndrome and knee OA.
Subchondral bone/cartilage crosstalk—an important vascular subchondral network
Numerous connections between bone and cartilage have been identified in OA joints including perforation in the osteochondral plate and the presence of blood vessel in some of them, microcracks through calcified cartilage layer to subchondral bone and blood vessels invading calcified cartilage [29]. Via these channels, humoral messengers can be exchanged between subchondral bone and cartilage. Westacott et al. have shown that OA osteoblasts in monolayer increased glycosaminoglycan release from cartilage explants, whereas normal cells had no effects [30]. To look at the influence of soluble mediators produced by osteoblasts on chondrocytes, we developed an original co-culture model, in which OA subchondral osteoblasts in monolayer were cultured with OA chondrocytes in alginate beads. Using this co-culture model, we have shown that OA subchondral osteoblasts from the sclerotic zone modulated chondrocyte metabolism. The presence of SC subchondral osteoblasts induced a decrease of aggrecan, type II collagen, SOX-9, and parathyroid hormone-related peptide mRNA levels in chondrocytes, but an increase of those of osteoblast-specific factor 1, MMP-3, and MMP-13 [31, 32].
Concluding remarks and therapeutic perspectives
The osteochondral junction is involved in the pathophysiology of OA at many levels and constitutes an attractive therapeutic target (Fig. 1). We consider that the optimal amount and structure of bone are found in normal bone. Both bone resorption and bone sclerosis area have been found in the subchondral bone of OA patients. Therefore, the optimal treatment would be the treatment that induces a normalization of the subchondral bone metabolism, density, and structure. Recently, clinical trials have been initiated to investigate the symptomatic and structural effects of biphosphonate, strontium ranelate, and calcitonin. This approach was supported by in vitro studies reporting the beneficial effects of these drugs on cartilage. Using a mouse model with an increased bone turnover, Kardi et al. have shown that pamidronate decreased the number of osteoclasts and the OA score [33]. Zoledronate reduced subchondral bone loss in dogs [34], displayed chondroprotective effects in rabbit [35], and, in rats with advanced joint degeneration, partially restored BMD and reduced pain [36]. Recently, zoledronate treatment in people with BMLs was associated with reduced BMLs and improved knee pain [37]. In vitro, strontium ranelate inhibits MMP-2 and MMP-9 expression by subchondral osteoblasts [38]. In a 3-year placebo-controlled trial including patients with knee OA, strontium ranelate slows down joint space narrowing as evaluated on standard X-ray and improves the algo-functional status of these patients [39]. Further in vitro and in vivo studies investigating their actions on subchondral bone are needed before concluding on the relevance of this therapeutic approach.

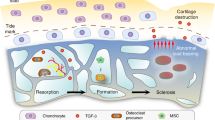
Hypothetical crosstalk between cartilage and subchondral bone in OA. Subchondral OA osteoblasts expressed more biochemical factors like IL-6 and adipokines, which could reach articular cartilage and alter the chondrocyte metabolism, by increasing MMPs or angiogenic factors like VEGF. In parallel, subchondral OA osteoblasts produced more VEGF, which could promote cartilage angiogenesis and induce chondrocyte hypertrophy and cartilage mineralization. Furthermore, OA chondrocytes produced higher levels of RANKL, promoting osteoclast activation and increasing subchondral bone remodeling. AP alkaline phosphatase, coll I type I collagen, IGF-1 insulin-like growth factor-1, IL-6 interleukin-6, MMP matrix metalloproteinase, RANKL receptor activator of nuclear factor kappa-B ligand, VEGF vascular endothelial growth factor
References
Pesesse L, Sanchez C, Henrotin Y (2011) Osteochondral plate angiogenesis: a new treatment target in osteoarthritis. Joint Bone Spine 78:144–149
Imhof H, Sulzbacher I, Grampp S, Czerny C, Youssefzadeh S, Kainberger F (2000) Subchondral bone and cartilage disease: a rediscovered functional unit. Invest Radiol 35:581–588
Duncan H, Jundt J, Riddle JM, Pitchford W, Christopherson T (1987) The tibial subchondral plate. A scanning electron microscopic study. J Bone Joint Surg Am 69:1212–1220
Mansell JP, Bailey AJ (1998) Abnormal cancellous bone collagen metabolism in osteoarthritis. J Clin Invest 101:1596–1603
Bailey AJ, Sims TJ, Knott L (2002) Phenotypic expression of osteoblast collagen in osteoarthritic bone: production of type I homotrimer. Int J Biochem Cell Biol 34:176–182
Hilal G, Massicotte F, Martel-Pelletier J, Fernandes JC, Pelletier JP, Lajeunesse D (2001) Endogenous prostaglandin E2 and insulin-like growth factor 1 can modulate the levels of parathyroid hormone receptor in human osteoarthritic osteoblasts. J Bone Miner Res 16:713–721
Hilal G, Martel-Pelletier J, Pelletier JP, Duval N, Lajeunesse D (1999) Abnormal regulation of urokinase plasminogen activator by insulin-like growth factor 1 in human osteoarthritic subchondral osteoblasts. Arthritis Rheum 42:2112–2122
Massicotte F, Fernandes JC, Martel-Pelletier J, Pelletier JP, Lajeunesse D (2006) Modulation of insulin-like growth factor 1 levels in human osteoarthritic subchondral bone osteoblasts. Bone 38:333–341
Hilal G, Martel-Pelletier J, Pelletier JP, Ranger P, Lajeunesse D (1998) Osteoblast-like cells from human subchondral osteoarthritic bone demonstrate an altered phenotype in vitro: possible role in subchondral bone sclerosis. Arthritis Rheum 41:891–899
Sanchez C, Deberg MA, Bellahcene A, Castronovo V, Msika P, Delcour JP, Crielaard JM, Henrotin YE (2008) Phenotypic characterization of osteoblasts from the sclerotic zones of osteoarthritic subchondral bone. Arthritis Rheum 58:442–455
Hopwood B, Tsykin A, Findlay DM, Fazzalari NL (2007) Microarray gene expression profiling of osteoarthritic bone suggests altered bone remodelling, Wnt and transforming growth factor-beta/bone morphogenic protein signalling. Arthritis Res Ther 9:R100
Lamas JR, Rodriguez-Rodriguez L, Vigo AG et al (2010) Large-scale gene expression in bone marrow mesenchymal stem cells: a putative role for COL10A1 in osteoarthritis. Ann Rheum Dis 69:1880–1885
Chan BY, Fuller ES, Russell AK et al (2011) Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthr Cartil 19:874–885
Sakao K, Takahashi KA, Arai Y et al (2009) Osteoblasts derived from osteophytes produce interleukin-6, interleukin-8, and matrix metalloproteinase-13 in osteoarthritis. J Bone Miner Metab 27:412–423
Chiba K, Uetani M, Kido Y, Ito M, Okazaki N, Taguchi K, Shindo H (2011) Osteoporotic changes of subchondral trabecular bone in osteoarthritis of the knee: a 3-T MRI study. Osteoporos Int 23(2):589–597
Sanchez C, Gabay O, Salvat C, Henrotin YE, Berenbaum F (2009) Mechanical loading highly increases IL-6 production and decreases OPG expression by osteoblasts. Osteoarthr Cartil 17:473–481
Sanchez C, Pesesse L, Gabay O, Delcour JP, Msika P, Baudouin C, Henrotin Y (2012) Regulation of subchondral bone osteoblast metabolism by cyclic compression. Arthritis Rheum 64(4):1193–203
Liu XH, Kirschenbaum A, Yao S, Levine AC (2006) Interactive effect of interleukin-6 and prostaglandin E2 on osteoclastogenesis via the OPG/RANKL/RANK system. Ann N Y Acad Sci 1068:225–233
Liu XH, Kirschenbaum A, Yao S, Levine AC (2005) Cross-talk between the interleukin-6 and prostaglandin E(2) signaling systems results in enhancement of osteoclastogenesis through effects on the osteoprotegerin/receptor activator of nuclear factor-{kappa}B (RANK) ligand/RANK system. Endocrinology 146:1991–1998
Bennell KL, Creaby MW, Wrigley TV, Bowles KA, Hinman RS, Cicuttini F, Hunter DJ (2010) Bone marrow lesions are related to dynamic knee loading in medial knee osteoarthritis. Ann Rheum Dis 69:1151–1154
Hunter DJ, Zhang Y, Niu J, Goggins J, Amin S, LaValley MP, Guermazi A, Genant H, Gale D, Felson DT (2006) Increase in bone marrow lesions associated with cartilage loss: a longitudinal magnetic resonance imaging study of knee osteoarthritis. Arthritis Rheum 54:1529–1535
Grimston SK, Screen J, Haskell JH, Chung DJ, Brodt MD, Silva MJ, Civitelli R (2006) Role of connexin43 in osteoblast response to physical load. Ann N Y Acad Sci 1068:214–224
Grynpas MD, Alpert B, Katz I, Lieberman I, Pritzker KP (1991) Subchondral bone in osteoarthritis. Calcif Tissue Int 49:20–26
Cox LG, van Rietbergen B, van Donkelaar CC, Ito K (2011) Bone structural changes in osteoarthritis as a result of mechanoregulated bone adaptation: a modeling approach. Osteoarthr Cartil 19:676–682
Gosset M, Berenbaum F, Salvat C, Sautet A, Pigenet A, Tahiri K, Jacques C (2008) Crucial role of visfatin/pre-B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: possible influence on osteoarthritis. Arthritis Rheum 58:1399–1409
Griffin TM, Huebner JL, Kraus VB, Guilak F (2009) Extreme obesity due to impaired leptin signaling in mice does not cause knee osteoarthritis. Arthritis Rheum 60:2935–2944
Mutabaruka MS, Aoulad Aissa M, Delalandre A, Lavigne M, Lajeunesse D (2010) Local leptin production in osteoarthritis subchondral osteoblasts may be responsible for their abnormal phenotypic expression. Arthritis Res Ther 12:R20
Davies-Tuck ML, Hanna F, Davis SR, Bell RJ, Davison SL, Wluka AE, Adams J, Cicuttini FM (2009) Total cholesterol and triglycerides are associated with the development of new bone marrow lesions in asymptomatic middle-aged women—a prospective cohort study. Arthritis Res Ther 11:R181
Walsh DA, McWilliams DF, Turley MJ, Dixon MR, Franses RE, Mapp PI, Wilson D (2010) Angiogenesis and nerve growth factor at the osteochondral junction in rheumatoid arthritis and osteoarthritis. Rheumatology (Oxford) 49:1852–1861
Westacott CI, Webb GR, Warnock MG, Sims JV, Elson CJ (1997) Alteration of cartilage metabolism by cells from osteoarthritic bone. Arthritis Rheum 40:1282–1291
Sanchez C, Deberg MA, Piccardi N, Msika P, Reginster JY, Henrotin Y (2005) Osteoblasts from the sclerotic subchondral bone downregulate aggrecan but upregulate metalloproteinases expression by chondrocytes. This effect is mimicked by Interleukin-6, -1β and oncostatin M pre-treated non sclerotic osteoblasts. Osteoarthr Cartil 13:979–987
Sanchez C, Deberg MA, Piccardi N, Msika P, Reginster JY, Henrotin Y (2005) Subchondral bone osteoblasts induce phenotypic changes in human osteoarthritic chondrocytes. Osteoarthr Cartil 13:988–997
Kadri A, Funck-Brentano T, Lin H, Ea HK, Hannouche D, Marty C, Liote F, Geoffroy V, Cohen-Solal ME (2010) Inhibition of bone resorption blunts osteoarthritis in mice with high bone remodelling. Ann Rheum Dis 69:1533–1538
Agnello KA, Trumble TN, Chambers JN, Seewald W, Budsberg SC (2005) Effects of zoledronate on markers of bone metabolism and subchondral bone mineral density in dogs with experimentally induced cruciate-deficient osteoarthritis. Am J Vet Res 66:1487–1495
Podworny NV, Kandel RA, Renlund RC, Grynpas MD (1999) Partial chondroprotective effect of zoledronate in a rabbit model of inflammatory arthritis. J Rheumatol 26:1972–1982
Strassle BW, Mark L, Leventhal L, Piesla MJ, Jian Li X, Kennedy JD, Glasson SS, Whiteside GT (2010) Inhibition of osteoclasts prevents cartilage loss and pain in a rat model of degenerative joint disease. Osteoarthr Cartil 18:1319–1328
Laslett LL, Dore DA, Quinn SJ, Boon P, Ryan E, Winzenberg TM, Jones G (2012) Zoledronic acid reduces knee pain and bone marrow lesions over 1 year: a randomised controlled trial. Ann Rheum Dis 71:1322–1328
Tat SK, Pelletier JP, Mineau F, Caron J, Martel-Pelletier J (2011) Strontium ranelate inhibits key factors affecting bone remodeling in human osteoarthritic subchondral bone osteoblasts. Bone 49:559–567
Reginster JY, Charpurlat R, Christiansen C et al (2012) Structure modifying effect of strontium ranelate on knee ostoearthritis. Osteoporos Int 23:58–59
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Henrotin, Y., Pesesse, L. & Sanchez, C. Subchondral bone and osteoarthritis: biological and cellular aspects. Osteoporos Int 23 (Suppl 8), 847–851 (2012). https://doi.org/10.1007/s00198-012-2162-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-012-2162-z