
Abstract
Summary
The efficacy and safety of denosumab were evaluated in Japanese postmenopausal women with osteoporosis. Total hip and distal 1/3 radius bone mineral densities (BMDs) were increased, and lumbar spine BMD was increased in magnitude with increasing dose. Bone turnover markers significantly decreased compared with placebo. Denosumab was well tolerated in Japanese subjects.
Introduction
The efficacy and safety of three doses of denosumab were compared with a placebo over 12 months in Japanese postmenopausal women with osteoporosis.
Methods
In this phase 2 multicenter, randomized, placebo-controlled study, 226 subjects were randomized and 212 subjects received at least 1 dose of investigational product, subcutaneously. All subjects also received daily supplements of at least 600 mg elemental calcium and 400 IU vitamin D from the beginning of screening through 12 months of treatment.
Results
Compared with placebo, denosumab (14, 60, and 100 mg) showed significant increases in percent BMD values of lumbar spine (5.25, 6.27, and 7.00) and total hip (3.90, 3.69, and 4.35) from baseline in 12 months. Distal 1/3 radius BMD was also significantly increased except at the 100-mg dose (1.82, 1.35, and 1.15). Denosumab significantly decreased the serum C-terminal crosslinking telopeptide of type 1 collagen and urinary N-terminal crosslinking telopeptide of type I collagen/urinary creatinine levels in 8 days, and bone alkaline phosphatase in 3 months. No new vertebral fracture was observed on spinal radiographs in either group. The overall incidences of adverse events were similar in the denosumab groups and the placebo group. No subject developed antibodies to denosumab. These results were similar to those obtained in the US phase 2 study.
Conclusions
Denosumab 60 mg could be an effective dose for Japanese postmenopausal women with osteoporosis as was shown in the Caucasian population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteoporosis is defined as a skeletal disorder characterized by compromised bone strength predisposed to an increased risk of fracture [1]. The prevalence of osteoporosis was estimated to be approximately 200 million people worldwide [2]. The number of patients with osteoporosis in Japan has been assessed to be about ten million [3].
Osteoporosis is largely due to increased bone resorption, resulting from increased recruitment, activation, and/or activity of osteoclasts driven by the signal through a receptor activator of nuclear factor-kB ligand (RANKL) [4]. Denosumab is a fully human monoclonal IgG2 antibody to RANKL that binds to the soluble and cell membrane-bound forms of human RANKL with high affinity and specificity. Denosumab binding to RANKL prevents RANK activation and inhibits the formation, activation, and survival of osteoclasts. Denosumab has been approved for the treatment of osteoporosis in several countries.
The denosumab US phase 2 dose–response study (US study) showed that denosumab increased bone mineral density (BMD) and decreased bone turnover markers significantly compared with placebo in postmenopausal women with low BMD at 12, 24, and 48 months [5–7]. The Fracture REduction Evaluation of Denosumab in Osteoporosis given every 6 Months (FREEDOM) trial was a randomized, double-blind, placebo-controlled, large international study that has been shown to significantly reduce fracture risks [8].
To date, however, the dose–response of denosumab on BMD and bone turnover markers has not been studied extensively in the Asian population with osteoporosis. The purpose of this study is to evaluate the dose–response of denosumab on efficacy and safety in Japanese women. This report presents the effects of 12 months of denosumab treatment on BMD, bone turnover markers, and adverse events in Japanese subjects.
Methods
Study design and subjects
This randomized, placebo-controlled, phase 2 dose–response study included four double-blind groups. Subjects were randomized to one of four treatment groups which were denosumab (14, 60, or 100 mg) and placebo. These subjects were administered with the investigational drug subcutaneously every 6 months for 12 months (total of two doses). All subjects also received daily supplements of at least 600 mg elemental calcium and 400 IU vitamin D from the beginning of screening through 12 months of treatment.
Subjects that were enrolled in this trial were ambulatory Japanese postmenopausal women that were no older than 80 years old. All the subjects had osteoporosis, a BMD T-score of −2.5 to −4.0 at the L1–L4 spine or −2.5 to −3.5 at either the femoral neck or total hip. The eligible criteria of BMD T-score for Japanese subjects were obtained previously in the study including 12,419 Japanese females at the ages from 20 to 85 years or more [9]. The absolute values of BMD in Japanese women are apparently smaller than those of Caucasian, and WHO T-score was obtained from Caucasian women. Thus, we applied the Japanese women’s T-score in this study. The validity of the T-score in relation to fracture occurrence was also confirmed in Japanese [10]. Only Hologic dual-energy X-ray absorptiometry (DXA) instruments were used to measure BMD in the study. Subjects were excluded if they had received any oral bisphosphonate treatment, parathyroid hormone (PTH), or PTH derivatives within the last 12 months before initial administration of the investigational product, systemic hormone replacement therapy, selective estrogen receptor modulators, calcitonin, calcitriol, maxacalcitol, falecalcitriol, or alfacalcidol within the last 3 months. Patients were also excluded if they had hypocalcemia, more than two moderate vertebral fractures (grade 2), or any severe vertebral fracture (grade 3) on spinal X-rays as determined by the Genant criteria [11]. Institutional review boards at each study site approved the study protocol, and all subjects provided written informed consent.
Study procedure
BMD values of the lumbar spine (L1–L4) and hip of all subjects were obtained by DXA at baseline, 1, 3, 6, and 12 months of treatment. The BMD of the forearm was measured at baseline, 6, and 12 months. Quality control and analysis of DXA scans were performed by Synarc Inc. (Oregon, USA).
To diagnose vertebral fractures, anterior–posterior and lateral X-ray films of the thoracic and lumbar spine were taken at baseline and 12 months. If a subject presented with acute back pain at a time point other than a scheduled visit for the X-ray and then occurrence of a vertebral fracture was suspected, the investigator should obtain a lateral spinal X-ray and submit it to Synarc Inc. for evaluation. All X-ray films showing evidence of a vertebral fracture were adjudicated by radiologists in Synarc Inc., who were blinded to the treatment group assignment. The levels of serum C-terminal crosslinking telopeptide of type 1 collagen (CTX-I; Serum Crosslaps ELISA kit, Nordic Biosciences, Herlev, Denmark) and urinary N-terminal crosslinking telopeptide of type I collagen (NTX-I; Osteomark® NTx Urine, Wampole Laboratories, NJ, USA) were measured at baseline, 8 days (day 1 is the first day the investigational product is administrated), and monthly through 12 months with an additional measurement 7 days after the second subcutaneous injection. NTX-I was adjusted for urinary creatinine (Cr) excretion. Bone-specific alkaline phosphatase (bone ALP; Tandem®-Ostase®, Beckman Coulter, CA, USA) was assessed at baseline, 1, 3, 6, 9, and 12 months. Intact PTH (iPTH; Elecsys PTH, Roche Diagnostics K.K., Tokyo, Japan) was measured at baseline, 1, 3, 6, 7, 9, and 12 months.
The subject must be fasting for at least 10 h prior to serum and urine collection. The urine sample should be collected in the morning from the second urine void of the day.
Safety assessment
All subjects were questioned about adverse events at each treatment visit (at least once a month). Adverse events were reported without any consideration of the relationship between the adverse event and the study drug. Investigators were blinded to the treatment group assignment and classified each adverse event based on seriousness (yes or no), severity (mild, moderate, or severe), and the relationship to the study drug.
Hematologic and chemical tests were examined at baseline, 8 days, 1, 2, 3, 6, 7, 9, and 12 months, and 7 days after the second administration. Hematologic and chemical tests were analyzed at the central laboratory. Serum albumin-adjusted calcium (in milligrams per deciliter) was calculated as follows: actual serum calcium (in milligrams per deciliter) − 0.8 × [serum albumin (in grams/deciliter) − 4] [only when albumin <4.0 (in grams/deciliter)].
The anti-denosumab antibodies were assessed at baseline, 6, and 12 months. A validated electrochemiluminescent immunoassay (PPD Inc., VA, USA and Amgen Inc., CA, USA) was used to detect denosumab-binding antibodies; samples with binding antibodies were later screened for denosumab-neutralizing antibodies by a cell-based assay (Amgen Inc., CA, USA).
Electrocardiograms (ECG) were conducted at baseline, 1, 6, and 12 months. All ECGs were obtained before administration of placebo or denosumab. QTc interval was evaluated using both Bazett’s and Fridericia’s correction formulae. ECGs were read manually by investigators.
Statistical analyses
The primary efficacy endpoint of this study was the percent change of lumbar spine BMD from baseline to 12 months. The primary analysis used t test using last observation carried forward imputation to compare each denosumab group with the placebo group. Each denosumab group versus the placebo group was tested in a step-up fashion using Hochberg’s procedure to adjust for multiplicity. As a sensitivity analysis, an analysis of covariance model with the treatment groups as the main effect and baseline lumbar spine BMD as the single covariate was also applied. Bone turnover markers were analyzed using the nonparametric methodology. Rank tests were used to compare each denosumab group with the placebo group.
The primary endpoint analyses were performed using the full analysis set (the primary efficacy subset). This set included all randomized subjects who received at least one dose of the investigational product and had a baseline and at least one post-baseline measure of lumbar spine of DXA.
Results
The disposition of study subjects is summarized in Fig. 1. Of the 226 women randomly assigned to the treatment groups (169 denosumab, 57 placebo), 212 subjects received at least one dose of the study drug and 195 subjects completed 12 months of study. Fourteen subjects withdrew from the study before administration of the investigational product. Patient demographics are presented in Table 1. Baseline demographics were generally balanced among the groups.

Study recruitment and randomization
BMD
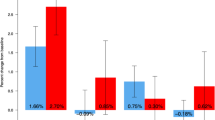
Denosumab treatments showed significant mean percent increases from baseline to 12 months in BMD at lumbar spine: 5.71% for 14 mg, 6.73% for 60 mg, and 7.45% for the 100-mg dose group, respectively. All treatment groups showed a statistical difference in magnitude with increasing dose compared with 0.46% in the placebo group (Fig. 2a). Mean percent increases from baseline to 12 months in BMD of the total hip were 3.29% for 14 mg, 3.09% for 60 mg, and 3.74% for the 100-mg dose group, respectively. All treatment groups showed a statistical difference compared with −0.61% in the placebo group (Fig. 2b). At the distal 1/3 radius, the mean percent increases from baseline to 12 months in BMD were 1.28% for 14 mg, 0.81% for 60 mg, and 0.61% for the 100-mg dose group, respectively. All treatment groups showed a statistical difference compared with −0.54% in the placebo group (p < 0.05), except at 100-mg dose group (p = 0.0536) (Fig. 2c).

Mean percent change from baseline to specific time points in bone mineral density (BMD) at lumbar spine (L1–L4) (a), total hip (b), and distal 1/3 radius (c). Values are means and error bars show standard deviation. *p value < 0.05 vs. placebo based on t test; **p value < 0.0001 vs. placebo based on t test.
Bone turnover markers
The effects of denosumab compared with placebo on the bone turnover markers (CTX-I, NTX-I/Cr, and bone ALP) are presented in Fig. 3a, b, and c. Denosumab treatment significantly reduced CTX-I and NTX-I/Cr concentrations from baseline by 8 days relative to placebo. All doses of denosumab maintained the decrease of CTX-I levels for 6 months, whereas values for the 14-mg dose showed signs of diminishing reduction following the 4-month time point, but were still reduced compared to placebo at all time points. Bone ALP levels significantly decreased by 3 months relative to placebo, reaching 45% to 65% of the baseline at 6, 9, and 12 months in all denosumab dose groups.


Median percent change from baseline to specific time points in serum CTX-I (a), urine NTX-I/Cr (b), bone ALP (c), and iPTH (d). Values are medians and error bars show interquartile. Mean change from baseline to specific time points in serum albumin-adjusted calcium (e). Values are means and error bars show standard deviation.
iPTH and calcium levels
Increases from baseline in iPTH concentrations were observed in the denosumab groups at 1 month, and then returned toward baseline in 6 months (Fig. 3d). The mean albumin-adjusted serum levels of calcium in denosumab-treated subjects demonstrated early and small decreases from baseline, as compared with the placebo (Fig. 3e).
Vertebral fracture
None of the subjects had a new or worsening (defined by an increase of at least one grade from baseline on the Genant criteria) vertebral fracture during the study.
Safety
The most common adverse events and the system organ classes in which adverse events were frequently reported showed no difference between the groups (Table 2). The number of subjects reporting serious adverse events did not differ (Table 3). There was no case of osteonecrosis of the jaw.
No clinically significant trends were noted in serum chemistry or hematology safety parameters, other than decreases in serum albumin-adjusted calcium, phosphorus, and total alkaline phosphatase. One case of treatment-related mild hypocalcemia was reported in the 14-mg group. Denosumab administration was not associated with any significant ECG abnormalities. No subject developed antibodies to denosumab.
Discussion
To date, dose–response study results of denosumab for osteoporosis are available only in the US study, which investigated mainly Caucasians. This study clearly demonstrates that BMD increases and changes in bone turnover markers in Japanese osteoporosis patients in equivalence with those in Caucasians. No difference was observed in the data of adverse events. The optimal dose, considered to be 60 mg per 6 months, is consistent with that of Caucasians.
Mean percent changes from baseline in BMD at lumbar spine and total hip were significantly greater in the denosumab groups than in the placebo group at 1 month and thereafter. The BMD response of lumbar spine at 12 months was observed regardless of the patient’s baseline demographics; subjects’ years since menopause, baseline BMD T-scores of lumbar spine, and total hip, previous osteoporosis medication use, baseline bone marker levels, smoking status, baseline weight/BMI, or the presence of prevalent fractures (data not shown). Therefore, BMD increases by denosumab treatment could be consistently expected in Japanese patients with osteoporosis. Denosumab also increased distal 1/3 radius BMD. These results indicate that denosumab can increase bone density at trabecular bone and cortical bone as well as those observed previously in Caucasians [5–7, 12]. BMD increases in all three denosumab groups were observed in all measured sites. However, mean percent increase in magnitude with increasing dose by denosumab was more marked in lumbar spine than in total hip and 1/3 distal radius. Thus, lumbar spine, rich in trabecular bone, could be more sensitive in detecting the response of BMD increases to denosumab. The percent increases of lumbar spine BMD in this study seemed to be slightly higher than the study of Caucasians [5]. Smaller absolute BMD values in Japanese may have contributed to this small difference.
Bone resorption markers, CTX-I and NTX-I, rapidly decreased after the start of denosumab administration and the effects were sustained during the dosing interval of 6 months in 60- and 100-mg doses. The dose of 14 mg, while rapidly reduced bone resorption markers to similar levels of those in 60- and 100-mg doses, but the reduction was less pronounced towards the end of the treatment cycle. These findings clearly indicate that a 14-mg dose in 6 months is not enough to maintain the pronounced reduction of bone resorption. The bone ALP level was significantly decreased in denosumab groups compared to the placebo groups at 3 months. The delayed decrease in bone ALP concentration suggests that denosumab acts primarily to inhibit bone resorption, and then coupling between bone resorption and formation secondarily reduces bone formation. This time difference was apparently similar to that observed in Caucasians [5–7]. The maintenance of bone ALP suppression did not seem to be enough in the 14-mg dosing. The response of iPTH did not seem to greatly differ from that in Caucasians.
Although a maximized effect on lumbar BMD was not apparent, the data sets of bone turnover markers clearly indicate that systemic inhibition of bone resorption by denosumab seems to be maximized at 60 mg in every 6-month dosing regimen in patients with osteoporosis. The degree of bone resorption inhibition did not differ in 3 months after dosing of denosumab. Thus, the dose of denosumab does not seem to influence the extent of inhibition of bone resorption, but affects the duration of inhibition of bone resorption. Therefore, the maximized effective dose of denosumab, when used in a 6-month interval, is apparently 60 mg in Japanese women with osteoporosis as determined in Caucasians.
The 12-month treatment of denosumab 14, 60, and 100 mg was well tolerated among Japanese postmenopausal women in this study. The overall incidences of adverse events, treatment-related adverse events, withdrawals due to adverse events, and serious adverse events were similar in the denosumab groups and the placebo group, with no dose-related adverse events being observed. The decreases in serum calcium, phosphorus, and total alkaline phosphatase were mild and consistent with known effects on bone metabolism by denosumab.
In the FREEDOM trial, the incidences of eczema, flatulence, and hospitalization for cellulitis were only reported slightly but showed significantly higher results in the denosumab group than in the placebo group, for the duration of 3 years in 7,868 subjects. There was no significant difference in the overall incidence of cellulitis between the two groups [8]. In this study, cellulitis and flatulence were not found and the incidence of eczema in the denosumab groups did not significantly differ to that of the placebo group for the period of 1 year in 212 subjects. Thus, although the number of cases in this study is obviously small, these adverse events do not seem to raise major concern for exploring further clinical study in Japanese subjects.
Conclusion
Denosumab effectively increased BMD at lumbar spine, total hip, and distal 1/3 radius in Japanese postmenopausal women with osteoporosis and rapidly decreased bone turnover markers compared with placebo. Denosumab is well tolerated. These results were similar to the US phase 2 study. The maximized effective dose of denosumab, when used in a 6-month interval, is apparently 60 mg in Japanese women with osteoporosis. This result may warrant a further study to examine the efficacy on fracture risk reduction in Japanese subjects with postmenopausal osteoporosis.
References
NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy (2001) Osteoporosis prevention, diagnosis, and therapy. JAMA 285:785–795
Reginster JY, Burlet N (2006) Osteoporosis: a still increasing prevalence. Bone 38(2 suppl 1):S4–S9
Sone T, Fukunaga M (2004) Prevalence of osteoporosis in Japan and the international comparison. Nippon Rinsho 62(Suppl 2):197–200
Rodan GA, Martin TJ (2000) Therapeutic approaches to bone diseases. Science 289:1508–1514
McClung MR, Lewiecki EM, Cohen SB, Bolognese MA, Woodson GC, Moffett AH, Peacock M, Miller PD, Lederman SN, Chesnut CH, Lain D, Kivitz AJ, Holloway DL, Zhang C, Peterson MC, Bekker PJ, AMG 162 Bone Loss Study Group (2006) Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 354:821–831
Lewiecki EM, Miller PD, McClung MR, Cohen SB, Bolognese MA, Liu Y, Wang A, Siddhanti S, Fitzpatrick LA, AMG 162 Bone Loss Study Group (2007) Two-year treatment with denosumab (AMG 162) in a randomized phase 2 study of postmenopausal women with low BMD. J Bone Miner Res 22:1832–1841
Miller PD, Bolognese MA, Lewiecki EM, McClung MR, Ding B, Austin M, Liu Y, San Martin J, AMG 162 Bone Loss Study Group (2008) Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone 43:222–229
Cummings SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, Delmas P, Zoog HB, Austin M, Wang A, Kutilek S, Adami S, Zanchetta J, Libanati C, Siddhanti S, Christiansen C, FREEDOM Trial (2009) Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 361:756–765
Orimo H, Sugioka Y, Fukunaga M, Muto Y, Hotokebuchi T, Gorai I, Nakamura T, Kushida K, Tanaka H, Ikai T, Oh-hashi Y, The Committee of the Japanese Society for Bone and Mineral Research for Development of Diagnostic Criteria of Osteoporosis (1998) Diagnostic criteria of primary osteoporosis. J Bone Miner Metab 16:139–150
Orimo H, Hayashi Y, Fukunaga M, Sone T, Fujiwara S, Shiraki M, Kushida K, Miyamoto S, Soen S, Nishimura J, Oh-hashi Y, Hosoi T, Gorai I, Tanaka H, Igai T, Kishimoto H, The Osteoporosis Diagnostic Criteria Review Committee: Japanese Society for Bone and Mineral Research (2001) Diagnostic criteria for primary osteoporosis: year 2000 revision. J Bone Miner Metab 19:331–337
Genant HK, Wu CY, Van Kuijk C, Nevitt MC (1993) Vertebral fracture assessment using a semiquantitative technique. J Bone Miner Res 8:1137–1148
Seeman E, Delmas PD, Hanley DA, Sellmeyer D, Cheung AM, Shane E, Kearns A, Thomas T, Boyd SK, Boutroy S, Bogado C, Majumdar S, Fan M, Libanati C, Zanchetta J (2010) Microarchitectural deterioration of cortical and trabecular bone: differing effects of denosumab and alendronate. J Bone Miner Res 25(8):1886–1894
Acknowledgment
This study was supported by funding from Amgen Inc.
Conflicts of interest
Drs. Nakamura, Matsumoto, Sugimoto, and Shiraki have received consultant/honorarium fees from Amgen Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nakamura, T., Matsumoto, T., Sugimoto, T. et al. Dose–response study of denosumab on bone mineral density and bone turnover markers in Japanese postmenopausal women with osteoporosis. Osteoporos Int 23, 1131–1140 (2012). https://doi.org/10.1007/s00198-011-1786-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-011-1786-8