
Abstract
Introduction
Acetyl salicylic acid (ASA) and nonsteroidal anti-inflammatory drugs (NSAIDs) may have potential as adjunctive agents for sepsis.
Materials
This review considers the large body of literature that indicates a basis for sepsis therapy with ASA and suggests an agenda for future intervention studies in sepsis prevention and treatment. ASA and NSAIDs have beneficial effects in numerous experimental models of sepsis. Low doses of ASA of 100 mg/day or less trigger synthesis of lipoxins that are anti-inflammatory and aid in resolution of inflammation. Higher doses of ASA and NSAIDs act to reduce NF-κB stimulation and inhibit numerous septic pathways. While a previous randomised controlled trial of ibuprofen failed to show a reduction in mortality in sepsis, it did reduce clinical manifestations of sepsis. More recent observational studies have shown reduction in sepsis or acute lung injury leading to lower mortality in ICU patients treated with ASA.
Conclusions
Low-dose ASA appears to be beneficial in the prevention and treatment of sepsis and SIRS. If proven, this intervention would have a major, cost-effective impact on sepsis care.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acetyl salicylic acid (ASA) or aspirin has had a profound effect on human health since its discovery in 1897 [1]. This nonspecific cyclo-oxygenase (COX) inhibitor is one of the most widely used drugs in the world because of its potent vascular disease prevention [2]. ASA contributes to systemic signalling in plants in response to parasitic invasion acting to limit infection severity [3].
ASA and other non-steroidal anti-inflammatory drugs (NSAIDs) have beneficial actions on inflammatory pathways contributing to sepsis. Low doses of ASA (75–81 mg/day) trigger lipoxin synthesis [4], mediating both anti-inflammatory and inflammation-resolving effects [5]. Additionally, the NF-kappaB (NF-κB) cellular signalling pathway can be inhibited by ASA and NSAIDs [6].
The full range of therapeutic effects of ASA in sepsis is unknown. NSAIDs were shelved as agents for the therapy of sepsis because of the negative result of the single randomised controlled trial in this area [7]. ASA has activity against numerous cellular pathways and cytokine mediators of sepsis, and demonstrated benefits in animal models with treatment after establishment of sepsis or tissue injury. Human pharmacokinetics show pharmacological levels achieved with safe doses. Finally, numerous observational studies suggest benefits of ASA in sepsis.
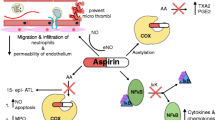
ASA triggers lipoxins, reducing and resolving inflammation (Fig. 1)
Lipoxin A4 is a natural lipid mediator that actively alters a variety of inflammatory processes including nitric oxide production [8], the inhibition of superoxide production by neutrophils and prevention of neutrophil/endothelial interactions (reviewed in [5]). ASA-triggered 15-epi-lipoxin A4 (ATL) has the same activity as lipoxin A4 [9]. ASA is the only NSAID that can directly acetylate the COX2 site in endothelial and epithelial cells to induce the formation of ATL.

Pathways and effects of aspirin-triggered lipoxin synthesis. Aspirin promotes the generation of 15R-hydroxyeicosatetraenoic acid (HETE) from arachidonic acid via the acetylation of COX-2. HETE is rapidly metabolised through the action of 5-lipoxygenase (5-LOX), leading to production of 15-epi lipoxin A4. This aspirin-triggered lipoxin (ATL) pathway mirrors classic lipoxin synthesis and function. ATL then mediates anti-inflammatory effects via reduced proinflammatory cytokines formed directly by stimulated lymphocytes as well as effects on phagocytes that contribute to additional pro-resolution effects on inflammation. Inhibition of plasma-derived growth factor (PDGF), epidermal growth factor (EGF) and leukotriene D4 (LTD4)-mediated signalling in neutrophils leads to reduced migration and promotes apoptosis while increasing phagocytosis in macrophages. The combined effects promote resolution of inflammation
Lipopolysaccharide (LPS)-stimulated polymorphonuclear leukocytes (PMNs) exhibit delayed apoptosis contributing to ongoing inflammation in sepsis [10]. PMN apoptosis was reinstated by the addition of pharmacological concentrations of ASA and was mediated through both ATL production [11, 12] and NF-κB inhibition [13], resulting in a reduction in proinflammatory cytokines [11]. ATL also reduces the secretion of TNF-alpha by T lymphocytes [14]. Importantly, such ATL-mediated actions are both anti-inflammatory and contribute to the resolution of the sepsis cascade. Recent studies also demonstrate that ASA triggers synthesis of resolvins, a novel class of lipid mediators with activities that resemble ATL [5].
Salicylate and NSAID effects on inflammatory pathways, particularly NF-κB pathway modulation
Investigators searching for potential antitumor effects of ASA determined its ability to inhibit NF-κB activation, an anti-inflammatory pathway additional to COX inhibition. NF-κB contributes to activation of genes involved in cell cycle control (cyclin-D1) [15], inflammation (TNF-alpha, IL-6, COX) [6] and coagulation (tissue factor, TF) [16]. For example, there is a dose-dependent reduction in NF-κB gene transcription seen in LPS-stimulated human monocytes treated with salicylates and NSAIDs [17].
Cellular stimulation by various noxious stimuli, like tissue damage, infection or cytokines, releases NF-κB from binding to its inhibitory cytoplasmic protein complex, IκB, allowing NF-κB to translocate to the cell’s nucleus and transcribe genes as above. Salicylates and NSAIDs inhibit NF-κB activation by blocking ATP binding and phosphorylation of the cellular kinase IKK-β [18], preserving expression of IκB [6]. It has been shown that ASA is a less potent inhibitor of NF-κB activation than many NSAIDs and other unrelated drugs including tamoxifen and curcumin [15]. ASA’s IC50 for inhibition of TNF-alpha-induced NF-κB activation is 5.67 mM, 10-fold less potent than indomethacin and 500 times less potent than the anti-estrogen agent, tamoxifen [15].
The in vitro ASA concentration required for COX inhibition is 1,000-fold less than the dose required for NF-κB inhibition [15]. Previous authors considering the potential of ASA as an NF-κB inhibitor in critical care therapeutics have raised concerns that potentially toxic, conventional anti-inflammatory doses are required [19]. However, low doses of ASA are required to produce ATL-mediated anti-inflammatory effects [9].
Salicylates block some of the microbial mediators of sepsis
Additional benefits of ASA therapy in sepsis may arise from demonstrated inhibition of prominent microbial mediators of sepsis particularly in gram-positive infection. Salicylic acid (SA), ASA’s major metabolite, has been shown to exert its in vitro effects on S. aureus virulence through hyper-activation of the stress response regulon, sig B [20, 21]. This results in reduced expression of at least two staphylococcal structural genes crucial to pathogenesis, hla (the α-toxin gene) and fnbA (a major fibronectin-binding adhesin) [22, 23]. As ASA and NSAIDs reduce NF-κB activation these drugs may have profound effects in endotoxemia due to gram-negative sepsis. Cell-associated bacteria like rickettsia also activate NF-κB, which when blocked by ASA, attenuated vascular endothelium infection [24].
Staphylococcus aureus infective endocarditis (IE) may be a condition for which ASA has specific effects on microbial pathogenesis leading to improved patient outcomes. Recent insights into IE pathogenesis suggest how the presence of ASA in the early stages of IE may reduce the extent of valvular and perivalvular infection in IE. ASA-mediated, platelet-dependent effects include a reduction in platelet aggregation, yielding smaller sterile vegetations, the platform upon which IE is initiated [25]. FnbA is a key determinant in both the initial vegetation colonization and persistence stages in IE [22, 23]. In contrast, hla is important in the post-colonization, progression phases of this infection [26].
In experimental animal models of staphylococcal IE, improved microbiologic and embolic outcomes are seen, especially when ASA is provided to animals prior to their infectious challenge [27] or when S. aureus is pre-exposed to ASA prior to IV challenge [25]. Importantly, improved microbiologic and embolic outcomes have also been seen in animals with staphylococcal IE given ASA after the induction of experimental IE [21, 25]. Recent human cohort study data [28, 29] indicate the benefits of ASA in S. aureus IE. Concerns relating to an increased risk of major bleeding in ASA-treated patients with S. aureus IE [30] have not been proven to date.
The role of activated platelets in sepsis
Activated platelets contribute to sepsis pathogenesis with benefits inherent in ASA therapy. Early animal models of endotoxemia showed prolonged survival time in ASA pretreated animals through antiplatelet effects [31]. Organ sequestration of activated platelets plays an important role in sepsis, and pretreatment with ASA 30 min before endotoxin challenge in sheep reduced accumulation in the lungs and liver [32]. Platelet sequestration was not prevented, however, by indomethacin pretreatment in endotoxin challenged rats [33]. The thrombocytopenia commonly seen in patients with sepsis may similarly be contributed to by sequestration of platelets, as has been demonstrated in human lungs, liver and intestines [34, 35]. Sepsis induces changes in platelet aggregation although reported changes have been inconsistent, potentially as a result of the different models studied [36]. In vitro examination of Streptococcus pneumoniae-induced platelet aggregation showed that this was dependent on toll-like receptor 2 [37].
Antiplatelet agents other than ASA may also improve sepsis mediators as clopidogrel given 5 days before endotoxin challenge reduced TNF-alpha and IL-6 levels in rats [38], and prevented thrombocytopenia in a mouse polymicrobial sepsis model [39].
Isolated organ and whole animal models showing the benefit of ASA and NSAIDs on organ specific and general effects of sepsis
Bacterial virulence determinants contribute to depression of cardiac function in septic shock. Staphylococcal α-toxin is known to reduce myocardial function, with this effect being prevented by indomethacin (100 μmol/l) or ASA (500 μmol/l) [40]. When isolated rat hearts were exposed to LPS, increases in myocyte-derived TNF-alpha induced a reduction in cardiac contractility but not coronary perfusion. Indomethacin was able to partially reverse this TNF-alpha-related impairment in myocardial function [41].
Alveolar macrophages (AM) become activated during experimental septic shock with an increase in TNF-alpha production via the NF-κB pathway. Macrophage inhibitory protein-2 (MIP) production is also increased in activated AM, leading to increased PMN migration into the pulmonary interstitium. NF-κB inhibition reduced both TNF-alpha and MIP production by LPS-stimulated rat AMs [42, 43]. Staphylococcal α-toxin also produces ventilation perfusion mismatch in perfused rabbit lungs [44]. This exotoxin-mediated process was inhibited by ASA. Importantly the vasculature changes resulting in pulmonary hypertension could also be potentiated by priming with endotoxin [45]. Activation of AM by LPS stimulation led to pulmonary vasoconstriction in a perfused rabbit lung model [46]. Here, rabbit lungs primed with LPS then exposed to arachidonic acid showed up to threefold increases in pulmonary artery pressure. This was completely reversed by pre-incubation with 1 μmol ASA. Resolution of LPS-induced acute lung injury (ALI) in mice was improved by treatment with ATL administered at the height of the inflammatory response [47]. ATL has been shown to be effective in treating as well as preventing ALI (carrageenan and LPS-induced) in mouse models [12, 47]. COX-2 expression has been shown to mediate recovery of ALI in mice via reduced leukocyte recruitment and resolution of epithelial integrity. Selective COX-2 inhibition blocked these effects, while non-selective COX inhibition via ASA did not [48].
Many animal models of sepsis have shown beneficial effects of ASA or NSAIDs, particularly with ibuprofen [49–52]. Rat-endotoxemia models showed that pretreatment with ASA [51] substantially reduced mortality. In an ovine endotoxic shock model, ibuprofen given before and after endotoxin infusion reduced early stage hypovolemia and hypoxia without effecting late changes [53]. Another canine endotoxic shock model mirrored the findings of isolated rat hearts described earlier as ibuprofen was shown to protect against depression of the cardiac index [52]. Finally, in a rabbit group B streptococcal shock model, ibuprofen significantly improved short-term survival [54].
Not all animal model data on NSAIDs in experimental sepsis indicate a beneficial role. COX-2 inhibition increased mortality in a rodent CLP model, whereas it improved survival in an endotoxemia model [55]. The murine CLP model has been shown to have a different cytokine profile from endotoxemia models with more prolonged elevation of TNF-alpha, IL-6 and MIP-2 [56]. These differences may account for conflicting results with NSAID intervention in the different models described above.
Pharmacokinetic data relating to low-dose ASA
The in vitro concentrations required to produce NF-κB inhibition indicate that 100 mg ASA/day cannot mediate sepsis outcomes via this pathway. However, multiple clinical trials in healthy hosts have shown that 81 mg of ASA per day is sufficient to increase ATL [57, 58]. The ASA concentration required in vitro to achieve 50 % inhibition of NFκB is 5.67 mM [15]. The steady-state ASA blood concentration in healthy human volunteers following 7 days of 160 mg ASA daily is only 0.31 mM [59]. Maximal concentrations after 7 days of the same dose were 2.99 mM [59]. Maximal ASA concentrations seen after a single dose of 325 mg enteric-coated ASA were 3.99–7.92 mM [60]. First order kinetics apply to ASA up to doses of 400 mg [61]. The pharmacokinetics and pharmacodynamics of ASA in critically ill patients has not been defined as yet, but a prospective trial is underway. (http://www.anzctr.org.au/trial_view.aspx?id=343088). As ASA absorption is rapidly achieved from the stomach, reduced splanchnic blood supply consequent on sepsis and hypotension will impact relatively minimally [62].
Studies of ASA and NSAIDs in inflammation in humans
A recent human endotoxin challenge study showed that high-dose ASA (425 mg bd) inhibited endotoxin-induced changes in platelet plug formation [63]. Other human-endotoxin challenge studies failed to show benefits of ASA on different aspects of the sepsis cascade. LPS-induced coagulation was not inhibited by ASA with no reduction in thrombin formation or TF production [63, 64]. Expression of endothelial cell adhesion molecules such as e-selectin and von Willebrand factor antigen that recruit inflammatory cells were not reduced by pre-dosing with 1,000 mg ASA [65]. Mean serum ASA levels in the experimental subjects were ~0.2 mM at the time of endotoxin challenge [64, 65], which is substantially lower than 5.67 mM ASA concentration shown to block NF-κB activation in vitro [15], but clearly in the range required for ATL activation. Interestingly, a placebo-controlled human endotoxemia model study of ibuprofen use showed TNF-alpha and IL-8 responses were significantly higher in ibuprofen-treated subjects [66].
Two randomised controlled studies have shown that low doses of ASA trigger ATL. These studies have both been in healthy volunteers [57, 58]. The benefits of low-dose ASA (75 mg) were recently illustrated using a human skin blister model of inflammation in healthy subjects [9]. In this model, low-dose ASA’s anti-inflammatory effect was due to reduced neutrophil migration mediated by ATL synthesis and nitric oxide secretion. Here, ATL was increased in blister fluid 24 h after ASA dosing [9]. Other septic cascade pathways affected by ATL have not been adequately explored.
Clinical experience of NSAIDs and ASA in sepsis (Table 1)
A large-scale randomised controlled sepsis trial of ibuprofen was performed involving septic patients treated with 48 h of intravenous ibuprofen [7]. In the study, ibuprofen treatment led to substantial reductions in sepsis-induced prostacyclin and thromboxane excretion. Reductions in temperature, lactic acid levels and oxygen consumption were also shown in the ibuprofen-treated group. There was no significant improvement in the incidence or duration of septic shock, but a non-significant, 3 % absolute reduction in mortality was found in ibuprofen-treated patients [7]. In a post hoc subgroup analysis of the small group of patients who entered the study with hypothermic septic shock and who had the highest mortality, ibuprofen was associated with a survival benefit (36 % absolute risk reduction) [67]. Ibuprofen-treated patients in the sub-analysis were significantly younger than placebo-treated controls as there had been no randomisation stratification for temperature [67].
The failure of the ibuprofen study in sepsis [7] predominantly relates to its small sample size based on exaggerated estimates of reduction in mortality as it was powered to show an unrealistic 35 % reduction in mortality. Subsequent trials showing small mortality improvements in sepsis have required far greater sample sizes [68]. The study groups were well matched for disease severity [7]. Adequate therapy for proven blood stream infection was provided in both groups, but there is no quantitation of the suitability of treatment for non-blood stream infection, which may have introduced a bias between the study groups. The 2-day ibuprofen regimen may also have been insufficient. As is a perennial issue in sepsis trials, the inability to identify patients early in their disease may have limited the effect of ibuprofen as the effects of the established sepsis cascade may have been too great to respond to COX inhibition. Lastly, with subsequent knowledge of ATL’s inflammation-resolving effects, it may be that ASA is a superior agent to ibuprofen for management of sepsis.
A number of recent, observational studies have shown potential benefits of ASA or anti-platelet drugs in patients with sepsis. Septic ICU patients with no increased bleeding risk were observed in a single-centre study to have lesser mortality if they were treated with antiplatelet agents, most commonly ASA [69]. A smaller series of patients with community-acquired pneumonia was studied by the same investigators, who showed reduced length of hospital stay in those treated with antiplatelet agents [70]. Fears that ASA or NSAIDs may predispose to severe sepsis do not seem to be borne out [71]. A large cohort study of ICU patients has shown an association between administration of ASA to patients within 24 h of the onset of SIRS or sepsis and reduced mortality. These patients had been treated with ASA prior to hospitalisation [72]. These studies [69, 70, 72] showed that 25–37 % of patients in the ICUs examined were administered ASA or antiplatelet agents.
Recent studies have concentrated on the possibility that ASA may prevent ALI in patients at high risk for this manifestation of sepsis or trauma. A population-based study involving a tightly defined group showed reduced ALI and ARDS in patients admitted to the medical ICU who had been receiving ASA [73]. A substantially larger study of a more heterogeneous ICU population from 22 US and Turkish hospitals failed to confirm this beneficial association, although a trend to reduced ALI remained in ASA-treated patients [74]. The apparent beneficial effects of statins in preventing sepsis and ALI in another ICU cohort were both potentiated by concomitant ASA use [75]. Studies to date, including the ibuprofen trial [7], provide evidence of clinical equipoise for the effect of ASA in critically ill patients.
There may be deleterious effects of salicylates or NSAIDs in sepsis. Renal impairment is a common and serious side effect of NSAID use [76]. Increases in bleeding due to salicylates and ASA are also of major concern although they are not shown to increase following low doses of ASA. For instance, there was no overall increase in gastrointestinal bleeding risk in large-scale primary prevention studies involving ASA in participants taking ≤70 mg/day [77]. It is difficult to be precise about the risk of bleeding in ASA-treated critically ill patients because of a paucity of data.
Potential treatment strategies for prevention or treatment of sepsis
The substantial body of literature reviewed from cellular, animal models and the trends from human studies suggest that ASA and NSAIDs may have beneficial roles in sepsis and that further study is warranted. Sepsis prevention and reduction of infectious disease mortality may be shown in planned analysis of current ASA primary prevention studies [78]. Targeted prevention could also be considered in high-risk populations such as hospital inpatients with the aim of reducing the frequency and severity of nosocomial sepsis. Finally, ASA may be shown in future interventional trials to be beneficial in treating established sepsis. The demonstrated impact of low-dose ASA on human models of inflammation [9] suggests that this agent rather than other NSAIDs is probably the best agent to consider for sepsis interventions. ASA alone stimulates ATLs with their anti-inflammatory and pro-resolution effects on sepsis [5], avoiding the greater toxicity of NSAIDs in the critically ill and potential deleterious, pro-inflammatory effects of selective COX-2 inhibition [48]. Properly targeted treatment with low-dose ASA could hold promise as a relatively safe, extremely cheap agent to use in sepsis even if it is shown to have only modest overall clinical benefit.
References
Sneader W (2000) The discovery of aspirin: a reappraisal. BMJ 321:1591–1594
Goodman SG, Menon V, Cannon CP, Steg G, Ohman EM, Harrington RA (2008) Acute ST-segment elevation myocardial infarction: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th edition). Chest 133:708S–775S
Yalpani N, Raskin I (1993) Salicylic acid: a systemic signal in induced plant disease resistance. Trends Microbiol 1:88–92
Claria J, Serhan CN (1995) Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci USA 92:9475–9479
Spite M, Serhan CN (2010) Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res 107:1170–1184
Kopp E, Ghosh S (1994) Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 265:956–959
Bernard GR, Wheeler AP, Russell JA, Schein R, Summer WR, Steinberg KP, Fulkerson WJ, Wright PE, Christman BW, Dupont WD, Higgins SB, Swindell BB (1997) The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N Engl J Med 336:912–918
Paul-Clark MJ, Van Cao T, Moradi-Bidhendi N, Cooper D, Gilroy DW (2004) 15-epi-lipoxin A4-mediated induction of nitric oxide explains how aspirin inhibits acute inflammation. J Exp Med 200:69–78
Morris T, Stables M, Hobbs A, de Souza P, Colville-Nash P, Warner T, Newson J, Bellingan G, Gilroy DW (2009) Effects of low-dose aspirin on acute inflammatory responses in humans. J Immunol 183:2089–2096
Watson RW, Rotstein OD, Nathens AB, Parodo J, Marshall JC (1997) Neutrophil apoptosis is modulated by endothelial transmigration and adhesion molecule engagement. J Immunol 158:945–953
El Kebir D, Jozsef L, Khreiss T, Pan W, Petasis NA, Serhan CN, Filep JG (2007) Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid A in human neutrophils: a novel mechanism for resolution of inflammation. J Immunol 179:616–622
El Kebir D, Jozsef L, Pan W, Wang L, Petasis NA, Serhan CN, Filep JG (2009) 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med 180:311–319
Negrotto S, Malaver E, Alvarez ME, Pacienza N, D’Atri LP, Pozner RG, Gomez RM, Schattner M (2006) Aspirin and salicylate suppress polymorphonuclear apoptosis delay mediated by proinflammatory stimuli. J Pharmacol Exp Ther 319:972–979
Claria J, Planaguma A (2005) Liver: the formation and actions of aspirin-triggered lipoxins. Prostaglandins Leukot Essent Fatty Acids 73:277–282
Takada Y, Bhardwaj A, Potdar P, Aggarwal BB (2004) Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-kappaB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene 23:9247–9258
Oeth P, Mackman N (1995) Salicylates inhibit lipopolysaccharide-induced transcriptional activation of the tissue factor gene in human monocytic cells. Blood 86:4144–4152
Weber C, Erl W, Pietsch A, Weber PC (1995) Aspirin inhibits nuclear factor-kappa B mobilization and monocyte adhesion in stimulated human endothelial cells. Circulation 91:1914–1917
Yin MJ, Yamamoto Y, Gaynor RB (1998) The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 396:77–80
Zingarelli B, Sheehan M, Wong HR (2003) Nuclear factor-kappaB as a therapeutic target in critical care medicine. Crit Care Med 31:S105–S111
Palma M, Bayer A, Kupferwasser LI, Joska T, Yeaman MR, Cheung A (2006) Salicylic acid activates sigma factor B by rsbU-dependent and -independent mechanisms. J Bacteriol 188:5896–5903
Kupferwasser LI, Yeaman MR, Nast CC, Kupferwasser D, Xiong YQ, Palma M, Cheung AL, Bayer AS (2003) Salicylic acid attenuates virulence in endovascular infections by targeting global regulatory pathways in Staphylococcus aureus. J Clin Invest 112:222–233
Que YA, Haefliger JA, Piroth L, Francois P, Widmer E, Entenza JM, Sinha B, Herrmann M, Francioli P, Vaudaux P, Moreillon P (2005) Fibrinogen and fibronectin binding cooperate for valve infection and invasion in Staphylococcus aureus experimental endocarditis. J Exp Med 201:1627–1635
Xiong YQ, Willard J, Yeaman MR, Cheung AL, Bayer AS (2006) Regulation of Staphylococcus aureus alpha-toxin gene (hla) expression by agr, sarA, and sae in vitro and in experimental infective endocarditis. J Infect Dis 194:1267–1275
Clifton DR, Rydkina E, Freeman RS, Sahni SK (2005) NF-kappaB activation during Rickettsia rickettsii infection of endothelial cells involves the activation of catalytic IkappaB kinases IKKalpha and IKKbeta and phosphorylation-proteolysis of the inhibitor protein IkappaBalpha. Infect Immun 73:155–165
Kupferwasser LI, Yeaman MR, Shapiro SM, Nast CC, Sullam PM, Filler SG, Bayer AS (1999) Acetylsalicylic acid reduces vegetation bacterial density, hematogenous bacterial dissemination, and frequency of embolic events in experimental Staphylococcus aureus endocarditis through antiplatelet and antibacterial effects. Circulation 99:2791–2797
Bayer AS, Ramos MD, Menzies BE, Yeaman MR, Shen AJ, Cheung AL (1997) Hyperproduction of alpha-toxin by Staphylococcus aureus results in paradoxically reduced virulence in experimental endocarditis: a host defense role for platelet microbicidal proteins. Infect Immun 65:4652–4660
Nicolau DP, Marangos MN, Nightingale CH, Quintiliani R (1995) Influence of aspirin on development and treatment of experimental Staphylococcus aureus endocarditis. Antimicrob Agents Chemother 39:1748–1751
Anavekar NS, Tleyjeh IM, Mirzoyev Z, Steckelberg JM, Haddad C, Khandaker MH, Wilson WR, Chandrasekaran K, Baddour LM (2007) Impact of prior antiplatelet therapy on risk of embolism in infective endocarditis. Clin Infect Dis 44:1180–1186
Eisen DP, Corey GR, McBryde ES, Fowler VG Jr, Miro JM, Cabell CH, Street AC, Paiva MG, Ionac A, Tan RS, Tribouilloy C, Pachirat O, Jones SB, Chipigina N, Naber C, Pan A, Ravasio V, Gattringer R, Chu VH, Bayer AS (2009) Reduced valve replacement surgery and complication rate in Staphylococcus aureus endocarditis patients receiving acetyl-salicylic acid. J Infect 58:332–338
Chan KL, Dumesnil JG, Cujec B, Sanfilippo AJ, Jue J, Turek MA, Robinson TI, Moher D (2003) A randomized trial of aspirin on the risk of embolic events in patients with infective endocarditis. J Am Coll Cardiol 42:775–780
Almqvist PM, Kuenzig M, Schwartz SI (1984) Treatment of experimental canine endotoxin shock with ibuprofen, a cyclooxygenase inhibitor. Circ Shock 13:227–232
Sigurdsson GH, Youssef HA, Owunnwanne A (1994) Effects of two different inhibitors of the arachidonic acid metabolism on platelet sequestration in endotoxic shock. Res Exp Med (Berl) 194:287–295
Itoh H, Cicala C, Douglas GJ, Page CP (1996) Platelet accumulation induced by bacterial endotoxin in rats. Thromb Res 83:405–419
Sigurdsson GH, Christenson JT, el-Rakshy MB, Sadek S (1992) Intestinal platelet trapping after traumatic and septic shock. An early sign of sepsis and multiorgan failure in critically ill patients? Crit Care Med 20:458–467
Singer G, Urakami H, Specian RD, Stokes KY, Granger DN (2006) Platelet recruitment in the murine hepatic microvasculature during experimental sepsis: role of neutrophils. Microcirculation 13:89–97
Vincent JL, Yagushi A, Pradier O (2002) Platelet function in sepsis. Crit Care Med 30:S313–S317
Keane C, Tilley D, Cunningham A, Smolenski A, Kadioglu A, Cox D, Jenkinson HF, Kerrigan SW (2010) Invasive Streptococcus pneumoniae trigger platelet activation via Toll-like receptor 2. J Thromb Haemost 8:2757–2765
Hagiwara S, Iwasaka H, Hasegawa A, Oyama M, Imatomi R, Uchida T, Noguchi T (2011) Adenosine diphosphate receptor antagonist clopidogrel sulfate attenuates LPS-induced systemic inflammation in a rat model. Shock 35:289–292
Seidel M, Winning J, Claus RA, Bauer M, Losche W (2009) Beneficial effect of clopidogrel in a mouse model of polymicrobial sepsis. J Thromb Haemost 7:1030–1032
Sibelius U, Grandel U, Buerke M, Mueller D, Kiss L, Kraemer HJ, Braun-Dullaeus R, Haberbosch W, Seeger W, Grimminger F (2000) Staphylococcal alpha-toxin provokes coronary vasoconstriction and loss in myocardial contractility in perfused rat hearts: role of thromboxane generation. Circulation 101:78–85
Grandel U, Fink L, Blum A, Heep M, Buerke M, Kraemer HJ, Mayer K, Bohle RM, Seeger W, Grimminger F, Sibelius U (2000) Endotoxin-induced myocardial tumor necrosis factor-alpha synthesis depresses contractility of isolated rat hearts: evidence for a role of sphingosine and cyclooxygenase-2-derived thromboxane production. Circulation 102:2758–2764
Jarrar D, Kuebler JF, Rue LW 3rd, Matalon S, Wang P, Bland KI, Chaudry IH (2002) Alveolar macrophage activation after trauma-hemorrhage and sepsis is dependent on NF-kappaB and MAPK/ERK mechanisms. Am J Physiol Lung Cell Mol Physiol 283:L799–L805
Chen ZT, Li SL, Cai EQ, Wu WL, Jin JS, Zhu B (2003) LPS induces pulmonary intravascular macrophages producing inflammatory mediators via activating NF-kappaB. J Cell Biochem 89:1206–1214
Walmrath D, Scharmann M, Konig R, Pilch J, Grimminger F, Seeger W (1993) Staphylococcal alpha-toxin induced ventilation-perfusion mismatch in isolated blood-free perfused rabbit lungs. J Appl Physiol 74:1972–1980
Walmrath D, Griebner M, Kolb B, Grimminger F, Galanos C, Schade U, Seeger W (1993) Endotoxin primes perfused rabbit lungs for enhanced vasoconstrictor response to staphylococcal alpha-toxin. Am Rev Respir Dis 148:1179–1186
Steudel W, Kramer HJ, Degner D, Rosseau S, Schutte H, Walmrath D, Seeger W (1997) Endotoxin priming of thromboxane-related vasoconstrictor responses in perfused rabbit lungs. J Appl Physiol 83:18–24
Jin SW, Zhang L, Lian QQ, Liu D, Wu P, Yao SL, Ye DY (2007) Posttreatment with aspirin-triggered lipoxin A4 analog attenuates lipopolysaccharide-induced acute lung injury in mice: the role of heme oxygenase-1. Anesth Analg 104:369–377
Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD (2005) Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol 174:5033–5039
Northover BJ, Subramanian G (1962) Analgesic-antipyretic drugs as antagonists of endotoxin shock in dogs. J Pathol Bacteriol 83:463–468
Hinshaw LB, Solomon LA, Erdos EG, Reins DA, Gunter BJ (1967) Effects of acetylsalicylic acid on the canine response to endotoxin. J Pharmacol Exp Ther 157:665–671
Halushka PV, Wise WC, Cook JA (1981) Protective effects of aspirin in endotoxic shock. J Pharmacol Exp Ther 218:464–469
Jacobs ER, Soulsby ME, Bone RC, Wilson FJ Jr, Hiller FC (1982) Ibuprofen in canine endotoxin shock. J Clin Invest 70:536–541
Adams T Jr, Traber DL (1982) The effects of a prostaglandin synthetase inhibitor, ibuprofen, on the cardiopulmonary response to endotoxin in sheep. Circ Shock 9:481–489
Peevy KJ, Ronnlund RD, Chartrand SA, Boerth RC, Longenecker GL (1988) Ibuprofen in experimental group B streptococcal shock. Circ Shock 24:35–41
Reddy RC, Chen GH, Tateda K, Tsai WC, Phare SM, Mancuso P, Peters-Golden M, Standiford TJ (2001) Selective inhibition of COX-2 improves early survival in murine endotoxemia but not in bacterial peritonitis. Am J Physiol Lung Cell Mol Physiol 281:L537–L543
Remick DG, Newcomb DE, Bolgos GL, Call DR (2000) Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock 13:110–116
Chiang N, Bermudez EA, Ridker PM, Hurwitz S, Serhan CN (2004) Aspirin triggers antiinflammatory 15-epi-lipoxin A4 and inhibits thromboxane in a randomized human trial. Proc Natl Acad Sci USA 101:15178–15183
Hennekens CH, Schneider WR, Pokov A, Hetzel S, Demets D, Serebruany V, Schroder H (2010) A randomized trial of aspirin at clinically relevant doses and nitric oxide formation in humans. J Cardiovasc Pharmacol Ther 15:344–348
Cerletti C, Dell’Elba G, Manarini S, Pecce R, Di Castelnuovo A, Scorpiglione N, Feliziani V, de Gaetano G (2003) Pharmacokinetic and pharmacodynamic differences between two low dosages of aspirin may affect therapeutic outcomes. Clin Pharmacokinet 42:1059–1070
Cerletti C, Marchi S, Lauri D, Domanin M, Lorenzi G, Urso R, Dejana E, Latini R, de Gaetano G (1987) Pharmacokinetics of enteric-coated aspirin and inhibition of platelet thromboxane A2 and vascular prostacyclin generation in humans. Clin Pharmacol Ther 42:175–180
Needs CJ, Brooks PM (1985) Clinical pharmacokinetics of the salicylates. Clin Pharmacokinet 10:164–177
Muir N, Nichols JD, Stillings MR, Sykes J (1997) Comparative bioavailability of aspirin and paracetamol following single dose administration of soluble and plain tablets. Curr Med Res Opin 13:491–500
Derhaschnig U, Schweeger-Exeli I, Marsik C, Cardona F, Minuz P, Jilma B (2010) Effects of aspirin and NO-aspirin (NCX 4016) on platelet function and coagulation in human endotoxemia. Platelets 21:320–328
Pernerstorfer T, Stohlawetz P, Hollenstein U, Dzirlo L, Eichler HG, Kapiotis S, Jilma B, Speiser W (1999) Endotoxin-induced activation of the coagulation cascade in humans: effect of acetylsalicylic acid and acetaminophen. Arterioscler Thromb Vasc Biol 19:2517–2523
Jilma B, Blann A, Pernerstorfer T, Stohlawetz P, Eichler HG, Vondrovec B, Amiral J, Richter V, Wagner OF (1999) Regulation of adhesion molecules during human endotoxemia. No acute effects of aspirin. Am J Respir Crit Care Med 159:857–863
Martich GD, Danner RL, Ceska M, Suffredini AF (1991) Detection of interleukin 8 and tumor necrosis factor in normal humans after intravenous endotoxin: the effect of antiinflammatory agents. J Exp Med 173:1021–1024
Arons MM, Wheeler AP, Bernard GR, Christman BW, Russell JA, Schein R, Summer WR, Steinberg KP, Fulkerson W, Wright P, Dupont WD, Swindell BB (1999) Effects of ibuprofen on the physiology and survival of hypothermic sepsis. Ibuprofen in Sepsis Study Group. Crit Care Med 27:699–707
Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ Jr (2001) Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 344:699–709
Winning J, Neumann J, Kohl M, Claus RA, Reinhart K, Bauer M, Losche W (2010) Antiplatelet drugs and outcome in mixed admissions to an intensive care unit. Crit Care Med 38:32–37
Winning J, Reichel J, Eisenhut Y, Hamacher J, Kohl M, Deigner HP, Claus RA, Bauer M, Losche W (2009) Anti-platelet drugs and outcome in severe infection: clinical impact and underlying mechanisms. Platelets 20:50–57
Legras A, Giraudeau B, Jonville-Bera AP, Camus C, Francois B, Runge I, Kouatchet A, Veinstein A, Tayoro J, Villers D, Autret-Leca E (2009) A multicentre case-control study of nonsteroidal anti-inflammatory drugs as a risk factor for severe sepsis and septic shock. Crit Care 13:R43
Eisen DP, Reid D, McBryde ES (2012) Acetyl salicylic acid usage and mortality in critically-ill patients with the systemic inflammatory response syndrome and sepsis. Crit Care Med. doi:10.1097/CCM.0b013e318246b9df
Erlich JM, Talmor DS, Cartin-Ceba R, Gajic O, Kor DJ (2011) Prehospitalization antiplatelet therapy is associated with a reduced incidence of acute lung injury: a population-based cohort study. Chest 139:289–295
Kor DJ, Erlich J, Gong MN, Malinchoc M, Carter RE, Gajic O, Talmor DS (2011) Association of prehospitalization aspirin therapy and acute lung injury: results of a multicenter international observational study of at-risk patients. Crit Care Med 39:2393–2400
O’Neal HR Jr, Koyama T, Koehler EA, Siew E, Curtis BR, Fremont RD, May AK, Bernard GR, Ware LB (2011) Prehospital statin and aspirin use and the prevalence of severe sepsis and acute lung injury/acute respiratory distress syndrome. Crit Care Med 39:1343–1350
Clive DM, Stoff JS (1984) Renal syndromes associated with nonsteroidal antiinflammatory drugs. N Engl J Med 310:563–572
Huang ES, Strate LL, Ho WW, Lee SS, Chan AT (2011) Long-term use of aspirin and the risk of gastrointestinal bleeding. Am J Med 124:426–433
Nelson MR, Reid CM, Ames DA, Beilin LJ, Donnan GA, Gibbs P, Johnston CI, Krum H, Storey E, Tonkin A, Wolfe R, Woods R, McNeil JJ (2008) Feasibility of conducting a primary prevention trial of low-dose aspirin for major adverse cardiovascular events in older people in Australia: results from the ASPirin in Reducing Events in the Elderly (ASPREE) pilot study. Med J Aust 189:105–109
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eisen, D.P. Manifold beneficial effects of acetyl salicylic acid and nonsteroidal anti-inflammatory drugs on sepsis. Intensive Care Med 38, 1249–1257 (2012). https://doi.org/10.1007/s00134-012-2570-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-012-2570-8