
Abstract
Effect of selenium pretreatment (0.2 mg/kg/day, as sodium selenite), 4 h prior to mercury treatment (0.4 mg/kg/day, as mercuric chloride), administered intraperitoneally, was examined after daily exposure for 20 days’ in rats. Liver, kidney and brain tissues were assayed for malondialdehyde (MDA) level, glutathione (GSH) content and mercury concentration. Mercury induced MDA levels, which was also observed in selenium pretreated animals. Significant reduction in GSH levels was observed in mercury alone and selenium pretreated animals. Mercury accumulation was in the order of kidney, liver and brain. Selenium pretreatment resulted in further enhancement in mercury accumulation in liver and kidney.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Mercury and its compounds are released into the environment through natural geological processes, such as volcanic eruptions, dissolution and volatilization from rocks, soils and sediments. Mercury is also released into the environment due to anthropogenic activities such as combustion of fossil fuels, incineration of waste, mining and industrial discharge (Goering et al. 2002). Several mercury compounds have been used as preservatives in drugs and cosmetics. Mercuric chloride is one of the first mercurial compounds to be used as antiseptic and disinfectant. Exposure to mercury may primarily occur by ingestion and inhalation through the food chain. Mercurial compounds, organic or inorganic forms exhibit a variety of toxicological effects including neurotoxicity, nephrotoxicity and gastrointestinal toxicity with ulceration and hemorrhage (Stohs and Bagchi 1995). Mercury is known to induce reactive oxygen species (ROS) in animals. These ROS may induce peroxidation injury in the membrane of lipids and protein as well as DNA fragmentation, which can result in disruption of nerve cell function and integrity (Goering et al. 2002). Inorganic mercury in the Hg2+ form has a great affinity for SH groups of endogenous biomolecules, reaches in the cells and tissues attached to thiol-containing proteins and low-molecular-weight thiols such as cysteine and glutathione (GSH) (Perottoni et al. 2004). Thus, it was suggested that in addition to depletion of intracellular thiol pools, the oxidant pathway might be a primary mechanism of induction of the response for mercury to induce oxygen free radicals or promote formation of lipid peroxides. Mercuric compounds can also alter protein conformation by covalently binding to sulfhydryl groups, or creating protein adducts through modification of side chains leading to changes in protein shape and activity. Such changes are known to be the result of the generation of free radicals by metals. It is also reported that acute treatment with mercury induces dramatic increase in reactive oxygen species accumulating in rat brain cell cultures leading to lipid peroxidation, protein degradation, and finally to cell death. Chronic exposure of mercury at low concentration is known to induce reactive oxygen species and inhibit the enzymes (antioxidant defense mechanism) that neutralize reactive oxygen species, but during chronic exposure reactive oxygen species seems to be neutralized by antioxidant defense mechanisms of the cell (Sener et al. 2003). Toxic response of mercury exposure may result in enhanced lipid peroxidation due to the formation of reactive oxygen species and in the depletion of glutathione content because mercury binds with SH group and produces its toxicity (Rocha et al. 2001).
Selenium is an essential trace element for animals and humans. It is well established that selenium deficiency causes health implications in humans and animals. Selenium is also toxic and can cause poisoning (selenosis) in humans and animals (Tinggi 2003). Selenium is known to have the ability to reduce the toxicity of several xenobiotics including heavy metals. Selenium (selenite after reduction to selenide) is known to possess an affinity constant for mercury higher than that of sulfhydryl compounds whereby the protection offered by sodium selenite in mercury intoxication has been attributed to the formation of Hg–Se–S complex which is stated to be non-toxic. Despite the essentiality of selenium and its protective effect against mercury toxicity, selenium is not currently used in the treatment of human mercury intoxication, possibly because selenium toxicity may be manifested after ingestion of element levels only slightly higher than those nutritionally required (Perottoni et al. 2004). There has been increased interest in the action of selenium as an antioxidant, while its role in prooxidant effects is also being probed. Studies were carried out on the toxic manifestations related to oxidative damage due to mercury and role of selenium in mercury intoxication. The present work was undertaken to examine the effect of mercury and the effect of selenium pretreatment in mercury intoxication (repeated intraperitoneal administration at low dose) in terms of thiobarbituric acid reactive substances (TBARS) generation, glutathione (GSH) content and mercury concentration in liver, kidney and brain of rats and also to evaluate the role of sodium selenite when administered prior to mercury injection (4 h).
Materials and Methods
1,1,3,3-Tetraethoxypropane (TEP), 2-thiobarbituric acid (TBA), 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB), glutathione (GSH), Folin Ciocalteu’s phenol reagent, butanol, tris (hydroxymethyl) aminomethane, bovine serum albumin (BSA), sodiumdodecylsulphate were obtained from Sigma Chemical Co. Sodium hydroxide, copper sulphate, sodium potassium tartrate and hydroxylammonium chloride were procured from Merck India. Potassium chloride, trichloroacetic acid, ethylenediamine tetraacetic acid, acetic acid, mercuric chloride, sodium selenite and nitric acid were obtained from Qualigens, India. Sodium carbonate from S D Fine-Chemicals Limited and perchloric acid from Ranbaxy, India were used.
Twenty-four adult male albino rats (200 ± 10 g) from ITRC, Lucknow breeding colony maintained under controlled temperature (22–25°C) were used for the study and maintained under 12 h alternate light and dark conditions, with free access to water and pellet food (Lipton India Ltd, Mumbai, India). Mercury (0.4 mg/kg/day) as mercuric chloride and/or selenium (0.2 mg/kg/day) as sodium selenite, dissolved in physiological saline were administered intraperitoneally daily for 20 days. Animals were separated into four groups (six animals each). Animals were given two injections of physiological saline at a gap of 4 h (group 1, control), physiological saline followed by mercury (group 2, mercury), selenium followed by physiological saline injection 4 h later (group 3, selenium) and selenium followed by mercury injection 4 h later (group 4, selenium + mercury). Rats were sacrificed under anesthesia, 48 h after the mercury injection. Liver, kidney and brain were quickly removed, trimmed of extraneous tissue, washed with ice-cold 0.15 M KCl solution, placed on ice chilled dish and weighed. Tissues were homogenized with ice-cold 0.15 M KCl solution (10% w/v).
LPO assay was carried out according to the method of Ohkawa et al. (1979). Levels of MDA (TBARS), an end product of polyunsaturated fatty acid peroxidation (lipid peroxidation, LPO) were measured on the basis of the reaction with thiobarbituric acid (TBA) to form a pink coloured complex. MDA values were determined with the absorbance coefficient of the MDA-TBA complex at 532 nm on GBC Cintra 20 Spectrophotometer using 1,1,3,3-tetraethoxypropane as standard.
Glutathione level was determined, by the method of Ellman (1959) using 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) for colour development and reading taken at 412 nm on GBC Cintra 20 Spectrophotometer after 15 min using reduced glutathione as standard for calibration.
Protein was assayed by the method of Lowry et al. (1951) using bovine serum albumin (BSA) as standard and O.D. recorded at 690 nm on GBC Cintra 20 Spectrophotometer.
Tissue homogenates were used for analysis of mercury concentration. They were digested according to the method of Skare (1972). Briefly, 1 ml tissue homogenates were digested with 7 ml volume of nitric acid and perchloric acid (6:1) mixture heated on water bath at 85°C for 18–20 h. When the digestion was complete, clear homogeneous digest was obtained, allowed to cool at room temperature and 2 ml volume of 20% hydroxylammonium chloride solution was added. Finally solution was raised up to 20 ml volume with the addition of distilled water and analyzed on Atomic Absorption Spectrophotometer (GBC-Avanta Sigma) equipped with vapor generation assembly.
Statistical significance of mean value of different biochemical parameters (LPO, GSH) and concentration of mercury in different tissues (liver, kidney and brain) for different treatments (control, mercury, selenium and selenium pre treatments) was tested using one-way analysis of variance (ANOVA). The analysis was repeated for each parameter and for each tissue separately. Prior to this analysis, the homogeneity of variance between treatment groups was ascertained and post hoc analysis was carried out to compare the mean values between the pair of treatments using Tukey test (Zar 1984). A probability of less than or equal to 0.05 was considered to be significant.
Results and Discussion
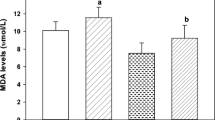
The hepatic, renal and cerebral MDA levels of mercury and/or selenium treated rats are shown in Fig. 1. Our results indicate an induction of malondialdehyde level in all the tissues examined in mercury treated animals. Selenium pretreatment also showed significant enhancement in MDA levels in all the tissues examined when compared with control group. However, significant enhancement was also observed only in liver tissue when treated with selenium alone. Magos (1991) observed an increase in lipid peroxidation in mercury intoxication when selenite has been administered before mercury treatment. However, the mechanism of the protection afforded by the preadministration of sodium selenite still needs to be unravelled.

MDA levels in rat tissues after ip administration of mercuric chloride and/or sodium selenite. MDA levels are expressed as nmoles MDA/mg protein. The results are presented as mean ± SE (n = 6). Difference between groups were considered to be significant when p < 0.05. * Values significantly different from control group
Levels of GSH in liver, kidney and brain are shown in Fig. 2. Significant reduction was observed in mercury-exposed animals in all the tissues examined however the same was observed only in kidney tissue in selenium treated animals. Administration of the two metals also resulted in significant reduction in GSH levels when compared with control group. Kidney tissue of selenium pretreated animals showed further depletion in GSH level when compared to mercury-exposed animals.

GSH Contents in rat tissues after ip administration of mercuric chloride and/or sodium selenite. GSH Contents are expressed as μg GSH/mg protein. The results are presented as mean ± SE (n = 6). Difference between groups were considered to be significant when p < 0.05. * Values significantly different from control group. # Values significantly different from mercury treated group
Concentration of mercury in all the tissues examined is shown in Fig. 3. The results indicate that the accumulation of mercury was highest in the kidney of the mercury treated rats followed by liver and brain. Selenium pretreatment resulted in enhanced mercury accumulation both in liver and kidney when compared with mercury treated animals as also supported in the study by Yamamoto (1985). However, this pattern was not observed in brain.

Mercury concentration in rat tissues after ip administration of mercuric chloride and/or sodium selenite. Mercury concentration is expressed as μg Hg/g tissue. The results are presented as mean ± SE (n = 6). Difference between groups were considered to be significant when p < 0.05. * Values significantly different from control group. # Values significantly different from mercury treated group
Mercury is a widespread environmental and industrial pollutant, which induces severe alterations in the tissues of both animals as well as humans. Various mechanisms, including lipid peroxidation have been proposed for the toxicity of mercuric chloride (HgCl2), and it has been demonstrated that lipid peroxidation occurs in the kidney, liver and other tissues of the rats and mice following parenteral administration of HgCl2. Mercuric chloride exposure causes an imbalance in the protective mechanisms, i.e. decrease in antioxidant enzymatic activity and GSH level leading to an increase in reactive oxygen species production which may result in the generation of lipid peroxides (Sener et al. 2003).
Toxic effects of mercuric chloride (chronic administration at low dose) in the vital organs, i.e. liver, kidney and brain of rats were observed in the form of enhanced LPO level, depletion in GSH level and also the accumulation of mercury in the tissues. Significant reduction in GSH level in tissues is also reported by Cantoni et al. (1984). The primary target organ for mercuric salt is kidney because of the high bonding affinity between mercury and sulphur. Mercury binds to metallothioneins and small molecular weight thiols, high content of such compounds is known to be present in kidney (Perottoni et al. 2004). This fact is demonstrated by the high mercury accumulation in kidney among the tissues examined as also reported by the results of Khan et al. (2001). Low accumulation of mercury in the brain may be due to the fact that inorganic mercury may be difficult to penetrate the blood brain barrier. However, an increase in LPO and decrease in GSH levels showed the generation of ROS in brain due to toxicity of mercury. It is well known that brain is more sensitive tissue to oxidative damage because of its high concentration of unsaturated lipids and its high rate of oxidative metabolism (Goering et al. 2002). This is also demonstrated in our results by the highest MDA level in brain among the tissues examined in mercury treated animals as well as pretreatment of selenium in mercury exposed animals.
Pretreatment of selenium in mercury exposed animals caused further enhancement in mercury concentration in liver and kidney tissues which resulted in the reduction of GSH level and enhanced LPO level in all the tissues examined when compared to control group. The results of Cuvin-Aralar and Furness (1991) have shown that in the presence of selenium, the accumulation of mercury in the target organ kidney is reduced but body retention of mercury on the whole is increased, especially in the liver. Our results are in agreement with their finding to the extent that enhancement of mercury concentration in liver (167% compared to mercury treated animals) was observed by selenium pretreatment. In kidney however, only 46% increase in mercury accumulation was observed in selenium pretreated mercury exposed animals compared to mercury alone treatment in animals, which may also be the cause of depleted GSH level in kidney.
Antioxidant role of any compound is demonstrated as a result of recovery of the alteration or restoration of the levels of parameters examined towards the control level. In the present study no restoration of LPO and GSH level in any of the tissue among the mercury treated animals was observed by the selenium pre treatment, rather further enhancement of mercury accumulation in liver and kidney tissues was observed. Thus the pretreatment of selenium in our experiment at the dose of 0.2 mg/kg/day did not show the antioxidant activity in mercuric chloride intoxication, when the two metals were administered chronically (once daily for 20 days) in terms of further mercury accumulation in liver and kidney tissues. Further studies are required to understand the role of selenium in mercury intoxication.
References
Cantoni O, Christie NT, Swann A, Drath DB, Costa M (1984) Mechanism of HgCl2 cytotoxicity in cultured mammalian cells. Mol Pharmacol 26:360–368
Cuvin-Aralar MLA, Furness RW (1991) Mercury and selenium interaction: a review. Ecotoxicol Environ Safety 21:348–364
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophy 82:70–77
Goering PL, Morgan DL, Ali SF (2002) Effect of mercury vapor inhalation on reactive oxygen species and antioxidant enzymes in rat brain and kidney are minimal. J Appl Toxicol 22:167–172
Khan AT, Atkinson A, Graham TC, Shireen KF (2001) Uptake and distribution of mercury in rats after repeated administration of mercuric chloride. J Environ Sci Health A Tox Hazard Subst Environ Eng. 36:2039–2045
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with folin phenol reagent. J Biol Chem 193:265–275
Magos L (1991) Overview on the protection given by selenium against mercurials. In: Suzuki K, Imura N, Clarkson TW (eds) Advances in mercury toxicology, Plenum, New York, 289–298
Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95:351–358
Perottoni J, Rodrigues OED, Paixao MW, Zeni G, Lobato LP, Braga AL, Rocha JBT, Emanuelli T (2004) Renal and hepatic ALA-D activity and selected oxidative stress parameters of rats exposed to inorganic mercury and organoselenium compounds. Food Chem Toxicol 42:17–28
Rocha JBT, Rocha LK, Emanuelli T, Pereira ME (2001) Effect of mercuric chloride and lead acetate treatment during the second stage of rapid post-natal brain growth on the behavioral response to chlorpromazine and on ALA-D activity in weaning rats. Toxicol Lett 125:143–150
Sener G, Sehirli AO, Ayanoglu-Dulger G (2003) Melatonin protects against mercury(II)-induced oxidative tissue damage in rats. Pharmacol Toxicol 93:290–296
Skare I (1972) Microdetermination of mercury in biological samples. Part III automated determination of mercury in urine, fish and blood samples. Analyst 97:148–155
Stohs SJ, Bagchi D (1995) Oxidative mechanisms in the toxicity of metal ions. Free Radical Biol Med 18:321–336
Tinggi U (2003) Essentiality and toxicity of selenium and its status in Australia: a review. Toxicol Lett 137:103–110
Yamamoto I (1985) Effect of various amounts of selenium on the metabolism of mercuric chloride in mice. Biochem Pharmacol 34:2713–2720
Zar JH (1984) Biostatistical analysis, 2nd edn. Prentice-Hall, Englewood cliffs, N.J.07632, pp 195–198
Acknowledgments
Thanks are due to Director, ITRC for his encouragement in the work. Technical assistance rendered by Mr. Ramesh Candra, TO and the overall help by Mr. Ram Chandra in the work is duly acknowledged. Rakhi Agarwal is grateful to UGC, India for the award of Junior Research Fellowship (JRF).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Agarwal, R., Behari, J.R. Effect of Selenium Pretreatment in Chronic Mercury Intoxication in Rats. Bull Environ Contam Toxicol 79, 306–310 (2007). https://doi.org/10.1007/s00128-007-9226-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00128-007-9226-3