
Abstract
Aims/hypothesis
We studied the role of protein degradation pathways in the regulation of insulin production and secretion and hypothesised that autophagy regulates proinsulin degradation, thereby modulating beta cell function.
Methods
Proinsulin localisation in autophagosomes was demonstrated by confocal and electron microscopy. Autophagy was inhibited by knockdown of autophagy-related (ATG) proteins and using the H+-ATPase inhibitor bafilomycin-A1. Proinsulin and insulin content and secretion were assessed in static incubations by ELISA and RIA.
Results
Confocal and electron microscopy showed proinsulin localised in autophagosomes and lysosomes. Beta-Atg7 −/− mice had proinsulin-containing sequestosome 1 (p62 [also known as SQSTM1])+ aggregates in beta cells, indicating proinsulin is regulated by autophagy in vivo. Short-term bafilomycin-A1 treatment and ATG5/7 knockdown increased steady-state proinsulin and hormone precursor chromogranin A content. ATG5/7 knockdown also increased glucose- and non-fuel-stimulated insulin secretion. Finally, mutated forms of proinsulin that are irreparably misfolded and trapped in the endoplasmic reticulum are more resistant to degradation by autophagy.
Conclusions/interpretation
In the beta cell, transport-competent secretory peptide precursors, including proinsulin, are regulated by autophagy, whereas efficient clearance of transport-incompetent mutated forms of proinsulin by alternative degradative pathways may be necessary to avoid beta cell proteotoxicity. Reduction of autophagic degradation of proinsulin increases its residency in the secretory pathway, followed by enhanced secretion in response to stimuli.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Orci et al showed three decades ago that beta cell insulin stores are regulated by crinophagy: granules fuse directly with large vacuolar lysosomes to generate crinophagic bodies, where granule contents are degraded [1]. Insulin may also reach lysosomes via autophagosomes that engulf cytosolic components containing secretory granules (macroautophagy), or by lysosomal engulfment of a single granule (microautophagy). It was recently reported that, in contrast to most mammalian cells, in beta cells, starvation induced lysosomal degradation of nascent insulin granules triggering lysosomal recruitment and activation of mechanistic target of rapamycin, complex 1 (mTORC1), thereby suppressing autophagy [2]. This would reduce insulin secretion in fasting; indeed, stimulation of autophagy enhanced secretion in starved beta cells. Starvation is an extreme situation; therefore, the relevance of these findings for beta cell physiology is unclear.
The stimulated beta cell confronts a high protein burden as proinsulin biosynthesis rates reach 106 molecules/min, ∼50% of total protein synthesis [3]. Proinsulin rapidly enters the endoplasmic reticulum (ER) and undergoes oxidative folding. Misfolded proinsulin may be degraded via a process called ER-associated degradation (ERAD), which includes translocation to the cytosolic side of the ER membrane, followed by ubiquitination and delivery to proteasomes for degradation [4]. Properly folded proinsulin is transported to the cis-Golgi, then moves forward between multiple cisternae to the trans-Golgi network (TGN), where cisternae disintegrate to produce carriers that convey proteins to the plasma membrane, secretory granules, endosomes and lysosomes [5].
Autophagy directs cytoplasmic components to lysosomes for degradation [6]. Double-membrane vesicles (autophagosomes) sequester cytosolic constituents, including misfolded proteins and damaged organelles, then fuse with lysosomes where the material is degraded and recycled. This pathway is tightly regulated by multiple proteins encoded by autophagy genes [7–9]. Beta cell Atg7−/− mice exhibit islet degeneration, impaired insulin secretion and glucose intolerance, indicating that autophagy is essential for beta cell wellbeing [10, 11]. Alterations in autophagy have been described in animal models of diabetes and in beta cells from type 2 diabetic patients [12, 13]. Beyond ubiquitous roles, autophagy also regulates tissue-specific functions [12]. Herein, we studied the regulation of beta cell insulin-like peptides (ILPs) by autophagy and its effects on insulin secretion.
Methods
Islet isolation, cell culture and reagents
Pancreases were obtained from 16-week-old beta cell Atg7−/− mice; fed blood glucose levels were ∼12 mmol/l [14]. Islets were isolated from C57BL6 mice by collagenase injection to the bile duct. Animal use was approved by the Institutional Animal Care and Use Committee of Hebrew University. INS-1E and BON-1 cell lines were grown as previously described [15, 16]. Additional information regarding the strain of mice and the sources of cell lines and human islets is provided in electronic supplementary material (ESM) Methods. Bafilomycin-A1, MG132, lactacystin, brefeldin-A, trehalose and diazoxide were obtained from Sigma-Aldrich (Rehovot, Israel), trans-activator of transcription (Tat)-scrambled and Tat-beclin-1 were from Merck Millipore (Billerica, MA, USA).
RNA interference causing protein knockdown
INS-1E cells were transfected with small interfering (si)RNA or scrambled siRNA using Jetprime transfection reagent (Polyplus Transfection, Illkirch-Graffenstaden, France). Specific anti-rat Atg5 and Atg7 siRNA smartpool sequences were ordered from Dharmacon (Lafayette, LA, USA). Cells were harvested 48 h following transfection.
Immunofluorescence
Paraffin sections were rehydrated and antigens retrieved. The antibodies used are detailed in ESM Methods.
Live cell imaging
A Zeiss LSM-710 confocal microscope with an incubator (37°C and 5% CO2) was used (Carl Zeiss, Oberkochen, Germany). Cells were transfected with the following constructs: Lamp1-egfp [17], Lc3 (also known as Map1lc3a)-mCherry [17], p62 (also known as Sqstm1)-mCherry [18], B4galt1-mTurqoise2 (mTurqoise-Golgi) (plasmid number 36205; Addgene, Cambridge, MA, USA) [19], wild-type (WT) proinsulin-egfp, WT proinsulin-mCherry, Akita proinsulin-egfp [20] and G(B23)V proinsulin-egfp [21]. See ESM Methods for further details.
Immunogold electron microscopy
Mouse pancreases were stained with primary polyclonal guinea pig anti-mouse (pro)insulin (1:200, Dako) and secondary donkey anti-guinea pig gold conjugate (Jackson ImmunoResearch). See ESM Methods for further details.
Western blotting
Protein levels were assessed using antibodies against different autophagy proteins, enhanced green fluorescent protein (eGFP) and chromogranin A (CgA). See ESM Methods for further details.
Insulin, proinsulin and CgA content and secretion
Insulin and CgA response to secretagogues were evaluated by static incubations, followed by cell extraction and measurements of proinsulin and insulin or CgA in extracts and the medium. See ESM Methods.
Oxygen consumption
Oxygen consumption was measured in INS-1E cells using V7 plates in a XF24 respirometer (Seahorse Bioscience, Billerica, MA, USA) as described previously [22]. See ESM Methods for further details.
Statistical analysis
Data are means ± SEM. Differences between multiple groups were analysed by one-way ANOVA with post hoc Sidak or Bonferroni corrected two-tailed t test. Two-tailed paired Student’s t test was used to compare differences between two groups, unless otherwise indicated. One-sample Student’s t test was used to validate statistical differences in experiments expressing data as relative to control. p < 0.05 was considered significant. No data was excluded from the analysis.
Results
Proinsulin degradation by lysosomes
INS-1E beta cells and mouse or human islets were incubated for 1–2 h with bafilomycin-A1, an established vacuolar H+-ATPase inhibitor. Surprisingly, short-term (≤120 min) treatment with 100 nmol/l bafilomycin-A1 increased the steady-state proinsulin content both in INS-1E cells and islets (Fig. 1a–c). In mouse islets, proinsulin was increased by ∼50%, accompanied by increased total ILP content. In INS-1E cells and human islets there was a smaller increase in proinsulin, without affecting insulin content (Fig. 1a–f). Proinsulin increase after short-term bafilomycin-A1 treatment was unexpected considering its abundance in beta cells, suggesting that substantial amounts of proinsulin are degraded in lysosomes. This cannot be explained by inhibition of secretion, as 2 h treatment of islets with bafilomycin-A1 at 11.1 mmol/l glucose did not decrease insulin or proinsulin secretion. Furthermore, inhibition of proinsulin secretion by diazoxide failed to increase islet proinsulin (ESM Fig. 1). In mouse islets, bafilomycin-A1 similarly increased proinsulin content at 5.5–22 mmol/l glucose, suggesting that lysosomal degradation of proinsulin is not affected by ambient glucose (ESM Fig. 2). Bafilomycin-A1 increased both proinsulin and insulin; thus, the effect on proinsulin content cannot be explained by perturbation of proinsulin conversion to insulin.

INS-1E cells (a, d) and mouse islets (b, e) were treated for 1 h (islets) and 2 h (INS-1E cells) at 11.1 mmol/l glucose with or without bafilomycin-A1 (100 nmol/l) followed by islet extraction and measurement of proinsulin by ELISA and total ILP by RIA. Human islets (c, f) were incubated for 1 h in CMRL-1066 medium at 5.5 mmol/l glucose with or without bafilomycin-A1 followed by measurement of proinsulin and insulin (INS-1E n = 6 in triplicates; mouse islets n = 4–6 in quadruplicates). (g, h) INS-1E cells were transfected with proinsulin-egfp construct, then treated for 2 h or 4 h with bafilomycin-A1 (100 nmol/l), lactacystin (10 μmol/l) or MG132 (50 μmol/l). The proinsulin level was analysed by western blotting for eGFP (n = 3–4 at 2 h and n = 5–7 at 4 h). (i, j) INS-1E cells were incubated with cycloheximide for 4 h with or without bafilomycin-A1 or lactacystin. Proinsulin-eGFP content was assessed by western blotting; grey line, control; solid black line, bafilomycin-A1; dashed black line, lactacystin; n = 4. Representative experiments (g, i) and quantifications (h, j) are shown (n = 4). *p < 0.05 and **p < 0.01. Baf A1, bafilomycin-A1; C-pep, C-peptide; Ctrl, control; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PI, proinsulin
To gain mechanistic information, we used proinsulin constructs tagged with various fluorescent markers. Enhanced GFP or mCherry was inserted into the C-peptide domain of proinsulin [20]. This enabled monitoring of proinsulin and C-peptide levels without interference from fully processed insulin, devoid of tag. We studied the effects of bafilomycin-A1 and proteasome inhibitors (MG-132 and lactacystin) on proinsulin levels and degradation in INS-1E cells (Fig. 1g–j). Bafilomycin-A1 increased the proinsulin-eGFP level, thus adequately reflecting changes in proinsulin concentrations measured by ELISA. Proteasome inhibitors had a small effect on proinsulin in INS-1E cells, consistently smaller than that of bafilomycin-A1 (Fig. 1g, h). Treatment of islets with lactacystin for 2 h was associated with decreased, rather than increased, proinsulin content (ESM Fig. 3), further suggesting that lysosomes are the main regulators of proinsulin degradation. To exclude interference with changes in translation rate, beta cells were treated with the protein biosynthesis inhibitor cycloheximide; bafilomycin-A1 again increased proinsulin at 2 and 4 h, while there was a small, non-significant effect of lactacystin (Fig. 1i, j). Bafilomycin-A1 may impair the acidification of young secretory granules, where proinsulin is processed to insulin; this may inhibit proinsulin-to-insulin conversion, thereby increasing proinsulin content. To exclude this, we analysed the bafilomycin-A1 effect on C-peptide-eGFP expression in INS-1E cells treated with cycloheximide (Fig. 1i, ESM Fig. 4). Bafilomycin-A1 induced a parallel increase in proinsulin-eGFP and C-peptide-eGFP, indicating that proinsulin conversion to insulin was not impaired.
Collectively, these findings indicate that the bafilomycin-A1 effect on beta cell proinsulin is mediated via inhibition of protein degradation, rather than impairment of proinsulin processing or insulin secretion, and that lysosomal degradation of proinsulin dominates degradation by the proteasome.
Lysosomal degradation of proinsulin is via autophagy
Proteins may reach the lysosome through multiple pathways including, but not limited to, autophagy. We have previously shown that in beta cell lines and islets autophagic flux is rapid, evident by high turnover of light chain microtubule-associated protein 3 (LC3), vesicle-associated form (LC3-ΙΙ) [23]. Consistently, bafilomycin-A1, but not the proteasome inhibitor, robustly increased LC3-ΙΙ, along with proinsulin-eGFP (and C-peptide-eGFP) expression (ESM Fig. 5). To clarify whether proinsulin is delivered to lysosomes via autophagy, we performed imaging studies and knocked down autophagy proteins. INS-1E cells were co-transfected with mCherry- or eGFP-labelled proinsulin and the (auto)lysosome markers Lamp1-egfp or p62 -mCherry and then treated with or without bafilomycin-A1 (Fig. 2a, b). At baseline, proinsulin-mCherry and lysosomal-associated membrane protein 1 (LAMP1)-eGFP colocalisation was relatively scant. Inhibition of lysosome activity resulted in accumulation of proinsulin-mCherry in lysosomes at 30 min, suggesting that proinsulin turnover is rapid.


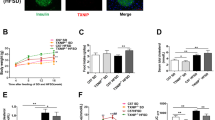
(a, b) INS-1E beta cells were transfected with WT proinsulin labelled with mCherry or eGFP and with the autolysosome markers LAMP-1-eGFP or with p62-mCherry. After 24 h, cells were treated with bafilomycin-A1 for different times, followed by confocal microscope imaging. (b) Quantification of proinsulin/p62 colocalisation (n = 3). (c, d) INS-1E cells were transfected with proinsulin-egfp or proinsulin-mCherry and p62-mCherry and then treated with 50 nmol/l rapamycin for 16 h. (d) Quantification of proinsulin/p62 colocalisation (n = 3). (e) Cells were incubated with cycloheximide for 2 h with or without 50 nmol/l rapamycin and proinsulin content was analysed by ELISA (n = 3 in triplicates); p = 0.059. (f, g) INS-1E cells were treated overnight with 100 mmol/l trehalose or with Tat-beclin1 (10 μmmol/l) for 3.5 h. (f) Autophagy was assessed by western blotting for LC3-II in cells treated with or without bafilomycin-A1 (n = 3). Stimulation of autophagy is shown by increased accumulation of LC3-II in the presence of bafilomycin-A1. (g) Proinsulin content was analysed by ELISA (n = 3 in triplicates). (h) INS-1E cells were transfected with proinsulin-egfp, Lc3-mCherry and B4galt1-mTurquoise2 (mTurquoise-Golgi) constructs, and treated with and without bafilomycin-A1. Proinsulin and B4GALT1 localisation in autophagosomes (LC3-eGFP+ punctae) was analysed by confocal microscope. Scale bars, 10 μm. *p < 0.05, **p < 0.01 and ***p < 0.001. Baf A1, bafilomycin-A1; Ctrl, control; PI, proinsulin
We have shown that the mTORC1 inhibitor rapamycin stimulates autophagy in beta cells [23]. Rapamycin increased the localisation of proinsulin-eGFP/mCherry in p62+ vesicles (Fig. 2c, d); this was associated with a small, borderline significant, decrease in proinsulin level (Fig. 2e). The autophagy stimulators Tat-beclin1 and trehalose also decreased proinsulin level in INS-1E cells (Fig. 2f, g). Together, these findings suggest that proinsulin is assigned for lysosomal degradation through autophagy.
Autophagosome maturation involves conversion of LC3, soluble cytosolic form (LC3-І) to LC3-ІI, which appears as dots (punctae) on fluorescence microscopy. Proteins destined for different cellular domains are sorted in the TGN [24]; β1,4-galactosyltransferase 1 (B4GALT1) is an integral membrane protein of this compartment. Beta cells were transfected with constructs expressing LC3-mCherry, proinsulin-eGFP and B4GALT1-mTurquoise, and analysed by confocal microscopy. LC3-mCherry colocalised with proinsulin-eGFP and B4GALT1-mTurqoise in autophagic punctae (Fig. 2h), indicating that proinsulin is present in autophagosomes containing TGN-derived membrane proteins. Treatment with brefeldin-A, which prevents protein transport from ER to Golgi, abolished the lysosomal degradation of proinsulin (ESM Fig. 6), further suggesting that proinsulin channelling for autophagic degradation occurs downstream to the ER.
Autophagy related (ATG)5 and ATG7 are required for autophagosome elongation and maturation [7]. Partial depletion of both ATGs impaired autophagy, as shown by decreased LC3-ІІ expression (Fig. 3a–e). This resulted in an ∼50% increase in beta cell proinsulin at 3.3–22.2 mmol/l glucose, without affecting insulin content (ESM Fig. 7, Fig. 3f–h). This cannot be explained by inhibition of proinsulin secretion, as increased proinsulin content in autophagy-deficient INS-1E cells was accompanied by increased proinsulin secretion (see Fig. 4).

INS-1E cells were transfected with scrambled siRNA or with siRNA directed against Atg5 and Atg7 for 48 h. (a–c) Western blotting for ATGs (n = 3). (d, e) Autophagy was assessed by western blotting for LC3-II in cells treated with or without bafilomycin-A1; quantification of LC3-II in bafilomycin-A1-treated cells is shown in (e) (n = 3). (f, g) Proinsulin and total ILP measured by ELISA and RIA in control and ATG5/7-knockdown cells (n = 3 in triplicates); (h) proinsulin/total ILP ratio. *p < 0.05, **p < 0.01 and ***p < 0.001. Baf A1, bafilomycin-A1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PI, proinsulin; Scr, scrambled

The effects of ATG5/7 knockdown and stimulators of autophagy on proinsulin and insulin secretion. INS-1E cells were transfected with scrambled siRNA or siRNA directed against Atg5 and Atg7 for 48 h. (a) Glucose-dependent stimulation of total ILP and proinsulin secretion. Control (squares) and ATG5/7-knockdown cells (circles) were incubated at 3.3, 11.1 and 22.2 mmol/l glucose for 1 h. Black symbols, ILP; white symbols, proinsulin (n = 3 in duplicates). (b, c) ILP and proinsulin secretion was assessed by static incubations at 3.3 mmol/l glucose and after stimulation with 16.7 mmol/l glucose (n = 3 in triplicates), 30 mmol/l KCl (n = 3) and 10 μmol/l glibenclamide (n = 3). (b) Basal and stimulated ILP secretion; (c) stimulated proinsulin secretion; and (d) proinsulin/ILP ratio. (e, f) INS-1E cells were treated with Tat-beclin1 (10 μmol/l) for 3.5 h (e) or overnight with 100 mmol/l trehalose (f); insulin secretion assessed by static incubation at 16.7 mmol/l glucose. *p < 0.05, **p < 0.01 and ***p < 0.001. PI, proinsulin; Scr, scrambled; si, siAtg5/7; Stim, stimulated
Together, our findings show that proinsulin is assigned for lysosomal degradation through autophagy, and suggest that this occurs at the TGN.
Effects of inhibiting autophagy on proinsulin and insulin secretion
Autophagy deficiency induced by ATG5/7 knockdown did not affect basal insulin secretion. By contrast, it increased glucose-stimulated insulin secretion (Fig. 4a), especially at submaximal glucose (11.1 mmol/l). ATG5/7 knockdown increased in parallel insulin and proinsulin secretion in response to glucose, plasma membrane depolarisation by KCl and the sulfonylurea glibenclamide (Fig. 4a–c). In autophagy-deficient cells, the relative increase in proinsulin secretion exceeded that of insulin (Fig. 4d), suggesting that processing of rescued proinsulin to insulin was incomplete. ATG5/7 knockdown did not affect mitochondrial respiration (ESM Fig. 8). Inhibition of autophagy increased insulin secretion by KCl and glibenclamide, further suggesting that its effects on secretion are not mediated via modulation of mitochondrial metabolism. Consistent with the hypothesis that autophagy inhibits insulin secretion, we found that Tat-beclin1 decreased glucose-stimulated insulin secretion (Fig. 4e). Trehalose also decreased insulin secretion, although the difference was not significant (Fig. 4f).
Collectively, our findings show that autophagy restrains insulin secretion; its inhibition enables proinsulin accumulation in secretory granules, followed by processing and increased secretion in response to stimuli.
Regulation of proinsulin by autophagy in vivo
The intracellular localisation of ILP was studied by electron microscopy (EM). Pancreatic sections were stained by immunogold labelling for insulin using an antibody that recognises all ILPs, including proinsulin. As expected, ILPs were abundant in secretory granules and were not observed in non-beta cells (ESM Fig. 9), indicating specific staining. ILP was observed in lysosomes containing single secretory granules suggestive of microautophagy, and in crinophagic bodies containing single secretory granules (Fig. 5a), as well as multigranular bodies, which contain secretory granule core-like material (not shown). In addition, we observed double-membrane structures, likely autophagosomes, containing dispersed ILPs, not in the form of insulin granules (Fig. 5a). To further show that proinsulin is regulated by autophagy in vivo, we immunostained pancreatic sections from beta-Atg7−/− mice for proinsulin and for p62. Life-long ATG7 deficiency results in beta cell degeneration, insulin deficiency and glucose intolerance. Beta-Atg7−/− mice were depleted of insulin and contained p62+ ubiquitinated protein aggregates [10, 11]. We found that islets were also depleted of proinsulin (Fig. 5b); despite this, proinsulin was found in part of the p62+ aggregates, suggesting that it serves as substrate for autophagy also in vivo.

(a) EM analysis of ILP localisation in beta cells. Immunogold labelling was performed using an antibody that recognises all ILP on pancreatic sections from non-diabetic mice. Boxes with a solid line show double-membrane organelles, likely autophagosomes, containing dispersed ILP. The box with the dashed line shows a lysosome engulfing a secretory granule and secretory granule core-like material. Scale bars, 500 nm. (b) Pancreatic sections of beta-Atg7 +/+ and Atg7 −/− mice stained for proinsulin and p62. Scale bars, 20 μm
Autophagy regulates CgA in endocrine cells
We studied whether autophagy also regulates other residents of the secretory pathway. CgA belongs to a family of large acidic secretory proteins found in most neuroendocrine tissues, including beta cells, stored with peptide hormones in secretory vesicles [25]. CgA is a large precursor peptide that, like proinsulin, undergoes proteolytic processing during its routing to and storage in secretory vesicles [26]. Inhibition of autophagy by ATG5/7 knockdown or treatment with bafilomycin-A1 markedly increased CgA expression in INS-1 beta cells, as well as in human pancreatic neuroendocrine cells (BON-1), whereas inhibition of proteasomal degradation had no effect (Fig. 6a–d). Increased CgA expression in BON-1 cells was accompanied by a small but significant increase in CgA secretion (Fig. 6e), thus resembling proinsulin regulation by autophagy.

The effects of inhibiting autophagic degradation by bafilomycin-A1, MG132 and ATG5/7 knockdown on CgA expression in INS-1E beta cells (a–c) and BON-1 cells (d, e). Cells were treated with or without bafilomycin-A1 (a, c), MG132 (c) or transfected with scrambled siRNA or Atg5/7 siRNA (b, d) followed by measurement of CgA in extracts by western blotting (n = 6). (e) The effects of ATG5/7 knockdown on CgA secretion in BON-1 cells (n = 3). *p < 0.05. Baf A1, bafilomycin-A1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HSP90, heat shock protein 90; Scr, scrambled
Irreparably mutated forms of proinsulin are refractory to autophagic degradation
The Akita proinsulin is trapped in the ER [23, 27]. Akita proinsulin labelled with eGFP was transfected to INS-1E cells and its assignment for autophagic degradation analysed by confocal microscopy and western blotting (Fig. 7). Intriguingly, Akita proinsulin was not found in autophagic punctae, including in cells in which autophagy was stimulated by rapamycin. Consistently, Akita proinsulin was not degraded in lysosomes, as shown by the failure of bafilomycin-A1 to increase Akita proinsulin-eGFP expression (Fig. 7b–e). The G(B23)V proinsulin mutated form also generates ER stress; however, a small portion of this form may reach secretory granules [21]. In contrast to WT cells, bafilomycin-A1 failed to increase G(B23)V proinsulin expression; similarly, inhibition of the proteasome did not affect the steady-state proinsulin content (Fig. 7d, e).

INS-1E cells were transfected with Akita proinsulin-egfp (a–e) and G(B23)V proinsulin-egfp constructs (d, e) followed by assessment of mutated proinsulin localisation (a) and degradation (b–e). (a) Akita proinsulin-eGFP and p62-mCherry colocalisation following treatment with or without rapamycin or bafilomycin-A1. Scale bars, 10 μm. (b, c) Beta cells expressing proinsulin-eGFP were treated with or without bafilomycin-A1 at 3.3 and 22.2 mmol/l glucose followed by western blotting for LC3-II and eGFP. Quantification of Akita proinsulin expression is shown in (c); n = 3. (d, e) Beta cells expressing WT proinsulin and the mutated Akita and G(B23)V forms were treated with and without bafilomycin-A1 or lactacystin and analysed by western blotting. Quantification is shown in (e); n = 3. *p < 0.05. Baf/Baf A1, bafilomycin-A1; G3.3, glucose 3.3 mmol/l; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Lac, lactacystin; PI, proinsulin; Scr, scrambled; UT, untreated
Collectively, our findings show that irreparably mutated forms of proinsulin are trapped in the ER, are not transported to autophagosomes and are refractory to lysosomal degradation.
Discussion
Our study suggests a previously unappreciated role for autophagy in beta cell proinsulin handling and insulin secretion. We show that a substantial amount of proinsulin is rapidly delivered to autophagosomes and directed to lysosomal degradation. Quantitatively, the impact of this pathway on proinsulin level is robust, as short-term inhibition of lysosomal degradation increased steady-state proinsulin content quite significantly. This was entirely unexpected, considering the background abundance of proinsulin in the beta cell. Indeed, while stimulation of beta cells with high glucose may induce up to a 25-fold increase in proinsulin biosynthesis [3], the steady-state proinsulin level nevertheless remains unchanged. Thus, the increase in proinsulin content following inhibition of lysosomal degradation shown here is indeed striking. Bafilomycin-A1 increased proinsulin by inhibiting its degradation; the changes in steady-state proinsulin level were not due to inhibition of its conversion to insulin or to reduction of exocytosis. Bafilomycin-A1 may indeed affect vesicular pH, thereby impairing proinsulin processing and secretion; however, such alterations cannot account for the observed changes in proinsulin level. Bafilomycin-A1 did not affect cumulative insulin secretion; moreover, inhibition of insulin secretion by diazoxide failed to increase proinsulin content. Increased proinsulin in bafilomycin-A1-treated cells and mouse islets was accompanied by a parallel increase in C-peptide, indicating that proinsulin was converted to insulin. Most importantly, genetic disruption of autophagy similarly increased proinsulin content, further suggesting that inhibition of lysosomal degradation is the main cause of the increase in beta cell proinsulin.
Several lines of evidence suggest that the canonical macroautophagy is the central degradation mechanism regulating proinsulin: EM revealed dispersed, non-granular ILP located in autophagosome-like structures and confocal microscopy showed proinsulin localisation in LC3+ and p62+ punctae and in protein aggregates that appear in autophagy-deficient beta-Atg7 knockout mice. Moreover, knockdown of key ATGs increased, whereas genetic and pharmacologic stimulators of autophagy (Tat-beclin 1, rapamycin and trehalose) decreased, proinsulin content.
CgA, a precursor of secretory hormones in different neuroendocrine cells, is also robustly degraded by autophagy, suggesting that autophagy is a key regulatory pathway of hormone precursors in endocrine cells. It has previously been shown that certain residents of the secretory pathway in beta cells, e.g. pancreatic prohormone convertases, are not degraded by autophagy [28], suggesting that the autophagic degradation of complex peptides like proinsulin and CgA is a selective process.
The degradation of insulin and proinsulin by lysosomes appears dissimilar. Insulin granule degradation is mainly mediated via microautophagy and crinophagy, whereas proinsulin degradation is mediated via macroautophagy. Pharmacological and genetic inhibition of autophagy increased proinsulin content without affecting insulin, hence the importance of lysosomal proinsulin degradation greatly outweighs that of insulin. It has recently been suggested that insulin granules may undergo autophagy-independent lysosomal degradation to keep insulin secretion low and avoid hypoglycaemia in the fasting state [2]. Stimulation of autophagy by Tat-beclin1 increased insulin secretion under fasting conditions and in response to high glucose; however, the mechanisms involved are not clear. We found that in beta cells autophagic flux is rapid, rather than being suppressed, both in KRB-HEPES and rich medium. Strikingly, increasing the proinsulin content by genetic disruption of autophagy was associated with a parallel increase in proinsulin and insulin secretion, whereas stimulation of autophagy inhibited insulin secretion, indicating that autophagy restrains, rather than stimulates, secretion. Our findings are in agreement with Pearson et al, who also found that inhibiting autophagy enhanced insulin secretion [29]. This may suggest that inhibition of proinsulin export to autophagosomes extends proinsulin residency in the secretory pathway followed by packaging into secretory granules, processing and secretion.
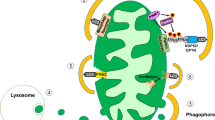
Intriguingly, the Akita mutated form of proinsulin, which is trapped in the ER and cannot be delivered to the Golgi and secretory granules [20, 30, 31], was resistant to lysosomal degradation; this may suggest that ER exit to the secretory pathway is essential for proinsulin degradation via autophagy to occur. Indeed, treatment with brefeldin-A, which prevents ER exit, abolished the lysosomal degradation of WT proinsulin. We found that proinsulin-containing autophagosomes expressed the TGN protein B4GALT1, hence we suggest that proinsulin delivery to autophagosomes takes place at the TGN. Further studies are required to uncover the precise location and the molecular mechanisms of selective proinsulin delivery to autophagosomes.
What is the biological rationale for proinsulin degradation by autophagy? In endocrine cells such as beta cells, intense hormone biosynthesis and flow through the ER–Golgi, along with highly dynamic changes in secretion, rationalises the need for an effective post-ER degradation pathway that clears surplus hormone precursors in the resting state. It may be speculated that this may function as a ‘buffering’ system aimed to prevent cargo overload at the Golgi, which may saturate the convertases with consequent prohormone oversecretion. Hence, autophagy can be viewed as a secretory pathway checkpoint that regulates hormone secretion.
The importance of autophagy for quality control is demonstrated by the fact that its disruption results in beta cell stress, cellular degeneration and impaired insulin secretion [10, 11], which may promote the progression from insulin resistance to diabetes [14]. Recently, it was shown that impaired autophagy led to accumulation of human islet amyloid polypeptide (hIAPP) and exacerbated hIAPP-induced beta cell toxicity [32–34]. Hence, inhibiting autophagy might be a double-edged sword: increasing insulin secretion in the short term, at the expense of ER stress and consequent beta cell degeneration and insulin deficiency in the long term.
Overall, we show that autophagy is a post-ER pathway regulating proinsulin levels and insulin secretion. The Golgi network may function as a bifurcation node determining proinsulin fate in the beta cell, assigning it either for further processing and secretion, or for degradation. Reduction of autophagy increases proinsulin retention in the secretory pathway, resulting in enhanced insulin secretion. The autophagic degradation of WT proinsulin has important pathophysiological implications for the development of insulin deficiency in diabetes and highlights the potential for novel therapeutic strategies to manipulate proinsulin clearance as a means to enhance insulin secretion in type 2 diabetes.
Abbreviations
- ATG:
-
Autophagy related
- B4GALT1:
-
β1,4-Galactosyltransferase 1
- CgA:
-
Chromogranin A
- eGFP:
-
Enhanced green fluorescent protein
- EM:
-
Electron microscopy
- ER:
-
Endoplasmic reticulum
- ILP:
-
Insulin-like peptide
- LAMP1:
-
Lysosomal-associated membrane protein 1
- LC3:
-
Light chain microtubule-associated protein 3
- LC3-I:
-
LC3, soluble cytosolic form
- LC3-II:
-
LC3, vesicle-associated form
- mTORC1:
-
Mammalian target of rapamycin complex 1
- p62 (SQSTM1):
-
Sequestosome 1
- si(RNA):
-
Small interfering (RNA)
- Tat:
-
Trans-activator of transcription
- TGN:
-
Trans-Golgi network
- WT:
-
Wild type
References
Orci L, Ravazzola M, Amherdt M et al (1984) Insulin, not C-peptide (proinsulin), is present in crinophagic bodies of the pancreatic B cell. J Cell Biol 98:222–228
Goginashvili A, Zhang Z, Erbs E et al (2015) Insulin granules. Insulin secretory granules control autophagy in pancreatic beta cells. Science 347:878–882
Schuit FC, In't Veld PA, Pipeleers DG (1988) Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc Natl Acad Sci U S A 85:3865–3869
Brodsky JL (2012) Cleaning up: ER-associated degradation to the rescue. Cell 151:1163–1167
Day KJ, Staehelin LA, Glick BS (2013) A three-stage model of Golgi structure and function. Histochem Cell Biol 140:239–249
Cecconi F, Levine B (2008) The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell 15:344–357
Klionsky DJ (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8:931–937
Levine B, Mizushima N, Virgin HW (2011) Autophagy in immunity and inflammation. Nature 469:323–335
Green DR, Levine B (2014) To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157:65–75
Ebato C, Uchida T, Arakawa M et al (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 8:325–332
Jung HS, Chung KW, Won Kim J et al (2008) Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 8:318–324
Stienstra R, Haim Y, Riahi Y, Netea M, Rudich A, Leibowitz G (2014) Autophagy in adipose tissue and the beta cell: implications for obesity and diabetes. Diabetologia 57:1505–1516
Masini M, Bugliani M, Lupi R et al (2009) Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 52:1083–1086
Quan W, Hur KY, Lim Y et al (2012) Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 55:392–403
Shaked M, Ketzniel-Gilad M, Cerasi E et al (2011) AMP-activated protein kinase (AMPK) mediates nutrient regulation of thioredoxin-interacting protein (TXNIP) in pancreatic beta-cells. PLoS One 6:e28804
Stålberg P, Wang S, Larsson C et al (1999) Suppression of the neoplastic phenotype by transfection of phospholipase C β 3 to neuroendocrine tumor cells. FEBS Lett 450:210–216
Falcon-Perez JM, Nazarian R, Sabatti C, Dell'Angelica EC (2005) Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J Cell Sci 118:5243–5255
Bjorkoy G, Lamark T, Brech A et al (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171:603–614
Goedhart J, von Stetten D, Noirclerc-Savoye M et al (2012) Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun 3:751
Rajan S, Eames SC, Park SY et al (2010) In vitro processing and secretion of mutant insulin proteins that cause permanent neonatal diabetes. Am J Physiol Endocrinol Metab 298:E403–E410
Wright J, Wang X, Haataja L et al (2013) Dominant protein interactions that influence the pathogenesis of conformational diseases. J Clin Invest 123:3124–3134
Wikstrom JD, Sereda SB, Stiles L et al (2012) A novel high-throughput assay for islet respiration reveals uncoupling of rodent and human islets. PLoS ONE 7:e33023
Bachar-Wikstrom E, Wikstrom JD, Ariav Y et al (2013) Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes 62:1227–1237
Kienzle C, von Blume J (2014) Secretory cargo sorting at the trans-Golgi network. Trends Cell Biol 24:584–593
Winkler H, Fischer-Colbrie R (1992) The chromogranins A and B: the first 25 years and future perspectives. Neuroscience 49:497–528
Barbosa JA, Gill BM, Takiyyuddin MA, O'Connor DT (1991) Chromogranin A: posttranslational modifications in secretory granules. Endocrinology 128:174–190
Preston AM, Gurisik E, Bartley C, Laybutt DR, Biden TJ (2009) Reduced endoplasmic reticulum (ER)-to-Golgi protein trafficking contributes to ER stress in lipotoxic mouse beta cells by promoting protein overload. Diabetologia 52:2369–2373
Zhang X, Yuan Q, Tang W, Gu J, Osei K, Wang J (2011) Substrate-favored lysosomal and proteasomal pathways participate in the normal balance control of insulin precursor maturation and disposal in beta-cells. PLoS ONE 6:e27647
Pearson GL, Mellett N, Chu KY et al (2014) Lysosomal acid lipase and lipophagy are constitutive negative regulators of glucose-stimulated insulin secretion from pancreatic beta cells. Diabetologia 57:129–139
Liu M, Hodish I, Rhodes CJ, Arvan P (2007) Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci U S A 104:15841–15846
Zuber C, Fan JY, Guhl B, Roth J (2004) Misfolded proinsulin accumulates in expanded pre-Golgi intermediates and endoplasmic reticulum subdomains in pancreatic beta cells of Akita mice. FASEB J 18:917–919
Rivera JF, Costes S, Gurlo T, Glabe CG, Butler PC (2014) Autophagy defends pancreatic beta cells from human islet amyloid polypeptide-induced toxicity. J Clin Invest 124:3489–3500
Shigihara N, Fukunaka A, Hara A et al (2014) Human IAPP-induced pancreatic beta cell toxicity and its regulation by autophagy. J Clin Invest 124:3634–3644
Kim J, Cheon H, Jeong YT et al (2014) Amyloidogenic peptide oligomer accumulation in autophagy-deficient beta cells induces diabetes. J Clin Invest 124:3311–3324
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by grants from the Israel Science Foundation to GL (ISF-347/12), the Golda Meir foundation and the European Foundation for the Study of Diabetes (EFSD) to JDW, and the National Institutes of Health (NIH-DK48280) to PA.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
GL designed the study, drafted the manuscript and approved its final version. YR and JDW contributed to the study design, acquired data, revised the intellectual content of the article and approved the final version. EB-W, NP, HZ, WQ, LH and ML acquired data, revised the intellectual content of the article and approved the final version. M-SL, PA and EC contributed to the study design, revised the intellectual content of the article and approved the final version. YR and GL are responsible for the integrity of this work as a whole.
Additional information
Yael Riahi and Jakob D. Wikstrom contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 118 kb)
ESM Fig. 1
(PDF 76.8 kb)
ESM Fig. 2
(PDF 41.7 kb)
ESM Fig. 3
(PDF 32.5 kb)
ESM Fig. 4
(PDF 44 kb)
ESM Fig. 5
(PDF 48.4 kb)
ESM Fig. 6
(PDF 31.6 kb)
ESM Fig. 7
(PDF 51.3 kb)
ESM Fig. 8
(PDF 41 kb)
ESM Fig. 9
(PDF 163 kb)
Rights and permissions
About this article

Cite this article
Riahi, Y., Wikstrom, J.D., Bachar-Wikstrom, E. et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia 59, 1480–1491 (2016). https://doi.org/10.1007/s00125-016-3868-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3868-9