
Abstract
Aims/hypothesis
Beta cell failure is caused by loss of cell mass, mostly by apoptosis, but also by simple dysfunction (decline of glucose-stimulated insulin secretion, downregulation of specific gene expression). Apoptosis and dysfunction are caused, at least in part, by lipoglucotoxicity. The mechanisms implicated are oxidative stress, increase in the hexosamine biosynthetic pathway (HBP) flux and endoplasmic reticulum (ER) stress. Oxidative stress plays a role in glucotoxicity-induced beta cell dedifferentiation, while glucotoxicity-induced ER stress has been mostly linked to beta cell apoptosis. We sought to clarify whether ER stress caused by increased HBP flux participates in a dedifferentiating response of beta cells, in the absence of relevant apoptosis.
Methods
We used INS-1E cells and murine islets. We analysed the unfolded protein response and the expression profile of beta cells by real-time RT-PCR and western blot. The signal transmission pathway elicited by ER stress was investigated by real-time RT-PCR and immunofluorescence.
Results
Glucosamine and high glucose induced ER stress, but did not decrease cell viability in INS-1E cells. ER stress caused dedifferentiation of beta cells, as shown by downregulation of beta cell markers and of the transcription factor, pancreatic and duodenal homeobox 1. Glucose-stimulated insulin secretion was inhibited. These effects were prevented by the chemical chaperone, 4-phenyl butyric acid. The extracellular signal-regulated kinase (ERK) signal transmission pathway was implicated, since its inhibition prevented the effects induced by glucosamine and high glucose.
Conclusions/interpretation
Glucotoxic ER stress dedifferentiates beta cells, in the absence of apoptosis, through a transcriptional response. These effects are mediated by the activation of ERK1/2.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pancreatic beta cell failure is increasingly recognised as central to progression of type 2 diabetes. Different insults are implicated in beta cell failure. Results obtained in rodent models of the disease, and in cultured rodent and human islet cells show that dyslipidaemia (lipotoxicity) and hyperglycaemia (glucotoxicity) negatively affect beta cell function and mass in human type 2 diabetes [1, 2].
One component of beta cell failure in type 2 diabetes is loss of beta cell mass. Increased apoptosis is, in turn, an important factor contributing to beta cell loss [3]. The other component of beta cell failure is beta cell dysfunction, represented by inhibition of glucose-stimulated insulin secretion (GSIS) and by downregulation of beta cell-specific genes resulting in beta cell dedifferentiation. Several mechanisms have been implicated in glucotoxicity: the generation of reactive oxygen species (ROS) [4, 5], the activation of endoplasmic reticulum (ER) stress [6–8] and an increase in the hexosamine biosynthetic pathway (HBP) flux [9]. However, these mechanisms are greatly interconnected. For example, an increased HBP flux causes hyper-O-glucosamine acylation (GlcNAc) of nuclear and cytoplasmic proteins [10], oxidative stress [11] and ER stress. This last mechanism has been demonstrated in cells other than beta cells [12].
Therefore, it is not clear whether increased HBP flux causes ER stress in beta cells. Moreover, while it is well established that ROS play a role in glucotoxicity-induced beta cell dedifferentiation [11, 13], the notion that glucotoxicity-induced ER stress participates in beta cell dedifferentiation has not been established yet. Rather, ER stress has been mostly associated with beta cell apoptosis [14, 15], linking its effect to loss of beta cell mass, as is the case for lipotoxicity-induced ER stress [16, 17]. Beta cells are particularly susceptible to ER stress, since they synthesise and secrete large quantities of a single protein. Proinsulin synthesis represents 30–50% of the total protein synthesis of the beta cell [18, 19]. In addition, glucose stimulates proinsulin translation [20], as well as increasing the stability of pre-proinsulin and transcription of the insulin gene [21, 22], further increasing the protein load. Therefore, chronic hyperglycaemia causes persistent activation of the unfolded protein response (UPR), beta cell failure and apoptosis. Recently, however, dedifferentiation has been identified as a new response to ER stress. ER stress dedifferentiates chondrocytes, downregulating collagen II and aggrecan [23]. These results were confirmed in vivo in transgenic mice that express mutant collagen X and in which ER stress produces a chondrodysplasia phenotype [24]. We have also shown that following ER stress, thyroid cells dedifferentiate, losing the expression of thyroid-specific genes and of their transcription factors [25].
In this study we sought to clarify whether increased HBP flux causes ER stress in beta cells, and whether this ER stress leads to apoptosis and participates in a dedifferentiating response of beta cells.
Methods
Materials
Media, sera and antibiotics were purchased from Invitrogen (Paisley, UK). Chemicals were from Sigma-Aldrich (St Louis, MO, USA). Glucosamine was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Insulin was measured by radioimmunoassay (Rat Insulin RIA Kit; Linco Research, St Louis, MO, USA). Antibodies were anti-β-actin (monoclonal; Sigma), anti-insulin (Cell Signaling, Danvers, MA, USA), and anti-binding protein (BIP)/glucose-regulated protein 78 (GRP78), anti-phospho extracellular signal-regulated kinase (ERK)1/2, anti-total ERK1/2 and anti-GLUT2 (Santa Cruz). Collagenase P was from Roche Applied Science (Penzberg, Germany).
Cell culture and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
The clonal beta cell line INS-1E was used between passages 54 and 95. INS-1E cells were cultured in a humidified atmosphere containing 5% (vol./vol.) CO2 in complete medium composed of RPMI 1640 supplemented with 5% (vol./vol.) heat-inactivated FCS, 1 mmol/l sodium pyruvate, 50 μmol/l 2-mercaptoethanol, 2 mmol/l glutamine, 10 mmol/l HEPES, 100 U/ml penicillin and 100 μg/ml streptomycin. The maintenance culture was split once a week and cells were seeded at 3 × 106 cells/75 cm2 in Falcon bottles (BD Biosciences Labware, Franklin Lakes, NJ, USA). The potential presence of mycoplasma was regularly checked using a photometric enzyme immunoassay (Roche, Penzberg, Germany). For most experiments, INS-1E were seeded at 2 × 105 cells/ml in Falcon 24 well plates and used 4 to 5 days thereafter, with one medium change on day 3 or 4. Cell viability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously reported [26].
Real-time RT-PCR
Total RNA was isolated from INS-1E cells and islets using a kit (RNeasy; Qiagen Sciences, Valencia, CA, USA). For real-time RT-PCR analysis, 1 μg RNA was reverse-transcribed using Superscript II Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). PCRs were analysed using SYBR Green mix (Invitrogen). Reactions were performed using Platinum SYBR Green quantitative PCR Super-UDG (BioRad, Hercules, CA, USA) and a real-time PCR detection system (iCycler IQ multicolour; BioRad). All reactions were performed in triplicate and cyclophilin was used as an internal standard. The primer sequences are shown in the electronic supplementary material (ESM) Table 1.
Insulin secretion
GSIS was tested in INS-1E cells between passages 54 and 95. Before the experiments, cells were maintained for 2 h in glucose-free medium. The cells were then washed twice and preincubated for 30 min at 37°C in glucose-free KRB HEPES buffer (KRBH, 135 mmol/l CaCl2, 3.6 mmol/l KCl, 5 mmol/l NaHCO3, 0.5 mmol/l NaH2PO4, 0.5 mmol/l MgCl2, 1.5 mmol/l CaCl2 and 10 mmol/l HEPES, pH 7.4). BSA (0.1%, wt/vol.) was added as an insulin carrier. Next, cells were washed once with glucose-free KRBH and then incubated for 30 min in KRBH and stimuli as indicated. Incubation was stopped on ice and the supernatant fractions collected for insulin secretion, which was measured by RIA using rat insulin as standard.
Western blot
Western blots were carried out as previously reported [27].
Immunofluorescence
Immunofluorescence experiments were performed as previously reported [25].
Islet isolation and ex vivo insulin secretion
Islets were isolated from 6-month-old C57Bl/6J mice. Animals were killed by cervical dislocation, the fur was soaked with ethanol and the abdomen was opened. The pancreas was inflated by KRBH injection and excised. The excised pancreas was washed twice with KRBH and digested with collagenase in a water bath (37°C), shaken by hand for 5–8 min. The digested pancreas was treated with Dnase I. The islets were handpicked under a stereo-microscope and cultured for 24 h in complete RPMI 1640 medium. For glucose-induced insulin release, 20 size-matched islets were preincubated at 37°C for 30 min in KRBH and then incubated for 1 h in a shaking water bath at 37°C; incubation was with 500 μl KRBH medium containing 2.8 mmol/l glucose or 2.8 mmol/l glucose plus 16.7 mmol/l glucose. Islets were then pelleted and supernatant fractions collected for measurement of insulin secretion. Insulin concentrations were determined by RIA. Experiments involving animals were conducted in accordance with the Principle of Laboratory Care.
Statistical procedures
All data are presented as mean±SE. Statistical differences were determined by one-way or two-way ANOVA as appropriate, and Bonferroni’s post hoc testing was performed when applicable. A value of p < 0.05 was considered to be significant.
Results
Glucosamine and glucose induce ER stress in INS-1E cells
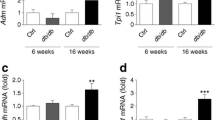
The induction of mRNA encoding the diagnostic UPR marker Bip/Grp78 was examined by real-time RT-PCR after exposure to various concentrations of glucosamine (2.5, 5.0, 7.5 and 10.0 mmol/l; Fig. 1a) and glucose (25 mmol/l; Fig. 2a) for 24 h. Glucosamine dose-dependently induced Bip mRNA, with a maximal effect at 7.5 and 10.0 mmol/l (Fig. 1a), and a similar effect at 24 and 48 h (Fig. 1b). As a control for the osmotic effects of these treatments, cells were exposed to 5.0 and 25 mmol/l xylose, which showed no effect (Fig. 1a). As a positive control, INS-1E cells were treated with the widely used inductor of ER stress, tunicamycin. Interestingly, tunicamycin showed a quantitatively similar effect to that of glucosamine (Fig. 1c). Accordingly, BIP protein was induced to a similar degree by glucosamine and tunicamycin (Fig. 1d).

Glucosamine induces ER stress but does not affect cell viability in INS-1E cells. a INS-1E cells were treated with the indicated concentrations of glucosamine (GlcN) or xylose, or (b, d, e) with 7.5 mmol/l glucosamine or (c, d, e) 0.5 μg/ml tunicamycin (Tun) for times as indicated. Bip/Grp78 (a–c) and Chop (e) mRNA was determined by real-time RT-PCR analysis of total RNA isolated from INS-1E, using cyclophilin as internal standard. mRNA levels in treated cells are relative expression units (REU) to those in control cells. Values are mean±SD; n = 6; *p < 0.05, **p < 0.01 and ***p < 0.001. d INS-1E cells treated with 0.5 μg/ml tunicamycin or 7.5 mmol/l glucosamine for the indicated times were solubilised and equal amounts of protein (40 μg/sample) were analysed by western blotting using BIP/GRP78- or β-actin-specific antibodies, with BIP and actin from the same gel; n = 3. f MTT assay for cell viability (48 h treatment) in INS-1E cells; results are percentage of the control (n = 3)

High glucose induces ER stress and UPR through activation of the hexosamine pathway in INS-1E cells. High glucose-/glucosamine (GlcN)-induced ER stress is reversed by the chemical chaperone PBA, but not by the antioxidant NAC. a INS-1E cells were pre-treated or not with 5 μmol/l azaserine (Aza) for 20 min, or (a–e) with 2.5 mmol/l PBA or (a–c) 1 mmol/l NAC for 24 h, followed by 24 h culture in the presence of glucose, PUGNAc or GlcN (a) as indicated, or of 7.5 mmol/l glucosamine (b–e) or 0.5 μg/ml tunicamycin (Tun) (d, e) for 24 h. Bip/Grp78 (a, b), Chop (c), spliced Xbp1 (d) and Atf6 (e) mRNA in treated cells was determined by real-time RT-PCR analysis using cyclophilin as internal standard and is quantified in relative expression units (REU) vs control cells; values mean±SD; n = 5; *p < 0.05, **p < 0.01, ***p < 0.001
Glucosamine (and tunicamycin) also increased the mRNA levels of other ER stress markers, such as Chop (Fig. 1e), spliced (active) Xbp1 (Fig. 2d) and Atf6 (Fig. 2e), suggesting that the three major branches of the UPR were activated by glucosamine. However, glucosamine did not decrease viability of INS-1E cells, suggesting that apoptosis was not significantly activated (Fig. 1f). Tunicamycin had an effect similar to that of glucosamine on Chop, spliced Xbp1 and Atf6 mRNA (Figs 1e and 2d, e).
The ability of azaserine or 6-diazo-5-oxo-norleucine (not shown), which are potent inhibitors of glutamine:fructose-6-phosphate amidotransferase (GFAT), to attenuate glucose- but not glucosamine-induced ER stress (Fig. 2a) suggests that elevated concentrations of glucose caused ER stress through a glucosamine intermediate. Glucosamine can promote the O-GlcNAc modification of intracellular proteins. O-GlcNAc levels can also be increased by treatment of cells with O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate (PUGNAc), an inhibitor of O-GlcNAcase. However, PUGNAc treatment did not promote ER stress (Fig. 2a). Thus, we cannot conclude that glucosamine causes ER stress via increased protein O-GlcNAc levels, although this mechanism cannot be ruled out.
Chemical chaperones are a group of compounds that can stabilise protein conformation and improve ER folding capacity. We sought to test whether ER stress induced by glucosamine and high glucose in beta cells is, in fact, alleviated by chemical chaperones. As shown in Fig. 2a–e, 4-phenyl butyric acid (PBA) was able to inhibit activation of glucosamine-, high glucose- (and tunicamycin-) induced ER stress markers. Therefore, PBA could be used to investigate the causal relationship between glucosamine-induced ER stress and beta cell dedifferentiation (see below).
To evaluate whether glucosamine- and high glucose-induced ER stress could be mediated by oxidative stress, INS-1E cells were pretreated with the glutathione precursor N-acetylcysteine (NAC) (Fig. 2a–c). NAC was not able to inhibit the glucosamine- and high glucose-induced increase of Bip and Chop mRNA, suggesting that glucosamine- and high glucose-induced ER stress was not dependent on oxidative stress in INS-1E beta cells.
Glucosamine-induced ER stress inhibits differentiation of INS-1E
To examine the effects of HBP activation on beta cell function, we examined the expression of two beta cell markers, Glut2 and Ins1. Glucosamine treatments, which are able to trigger ER stress, downregulated the expression of Glut2 (Fig. 3a, b) and Ins1 (Fig. 3c). To gain insights into the mechanism of downregulation of Glut2 and Ins1, we analysed the expression of a transcriptional regulator of both genes, the homeobox transcription factor Pdx1. As shown in Fig. 3d, Pdx1 was downregulated by glucosamine treatment. These results suggest that Glut2 and Ins1 were transcriptionally downregulated by glucosamine-induced ER stress and that the downregulation of Pdx1 participated in this effect.

Effects of glucosamine on beta cell-associated gene expression and glucose-induced insulin secretion in INS-1E cells. a–d INS-1E cells were treated with 7.5 mmol/l glucosamine (GlcN) for times indicated or (e) for 24 h. Glut2 (a), Ins1 (c) and Pdx1 (d) mRNA was determined by real-time RT-PCR analysis of total RNA isolated from INS-1E cells, using cyclophilin as internal standard. mRNA levels in treated cells are quantified as relative expression units (REU) vs control cells; values are mean±SD; n = 5. b Total proteins (40 μg/sample) were analysed by western blotting using GLUT2- or β-actin-specific antibodies (n = 4). e Glucosamine inhibited the insulin secretory response to 30 min incubation with 20 mmol/l glucose. This inhibition was prevented by pretreatment with 2.5 mmol/l PBA for 24 h. GSIS was assayed by RIA in the culture medium; values are mean±SD; n = 4. **p < 0.01 and ***p < 0.001
To test the functionality of the beta cell following glucosamine-induced ER stress, we measured GSIS in control and glucosamine-treated INS-1E cells. In comparison with untreated cells, glucosamine-treated cells had a significantly reduced GSIS (Fig. 3e), showing that glucosamine alters the insulin secretory response of normal beta cells to glucose.
The glucosamine-induced dedifferentiation of INS-1E cells is suppressed by the chemical chaperone PBA
To establish a causal relationship between glucosamine-induced ER stress and the decreased differentiation of INS-1E cells, we used pre-treatment with PBA, which alleviated the glucosamine-induced ER stress (Fig. 2b–e). PBA reversed the effect of glucosamine on Glut2 mRNA (Fig. 4a), Ins1 mRNA (Fig. 4b), Pdx1 mRNA (Fig. 4c) and GSIS (Fig. 3e). These results indicate that glucosamine-induced ER stress causally decreases differentiation of INS-1E cells. Since oxidative stress is another important determinant of beta cell damage, we checked for its involvement in glucosamine-induced ER stress. However, pre-treatment with NAC did not prevent the glucosamine-induced downregulation of beta cell markers (Fig. 4a–c).

PBA, but not NAC reversed the dedifferentiating effect of glucosamine. INS-1E cells were pre-treated or not with 2.5 mmol/l PBA or 1 mmol/l NAC for 24 h and then cultured in the presence of 7.5 mmol/l glucosamine (GlcN) or 20 μmol/l PUGNAc for 24 h. a Glut2, (b) Ins1 and (c) Pdx1 mRNA was determined by real-time RT-PCR analysis of total RNA isolated from INS-1E cells, using cyclophilin as internal standard. mRNA levels in treated cells are quantified as relative expression units (REU) vs control cells; values are mean±SD; n = 3; *p < 0.05, **p < 0.01, ***p < 0.001
Since chemical chaperones prevented glucosamine-induced ER stress and glucosamine-induced downregulation of Ins1, Glut2 and Pdx1 mRNA, we sought to determine whether PUGNAc, which was unable to induce ER stress, was also unable to induce beta cell dedifferentiation. As shown in Fig. 4a–c, treatment of INS-1E cells with PUGNAc did not cause dedifferentiation, thus strengthening the notion that ER stress was the cause of loss of differentiation.
The glucosamine-induced dedifferentiation of INS-1E cells is mediated by activation of the mitogen-activated protein kinase/ERK kinase–ERK pathway
To obtain insights into the signal transmission pathway linking glucosamine-induced ER stress and beta cell dedifferentiation, we analysed the effect of specific inhibitors of pathways emanating from the stressed ER [28]. We began by using SB203580 to inhibit p38 mitogen-activated protein kinase (MAPK). SB203580 pretreatment had no effect on glucosamine-induced Ins1 and Pdx1 mRNA downregulation evaluated by real-time RT-PCR (not shown). Next, we used U0126, a specific inhibitor of MAPK/ERK kinase (MEK)1/2. In this case, U0126 not only completely reversed the effect of glucosamine, but even increased mRNA expression of Ins1, Glut2 and Pdx1 in the control condition (Fig. 5a). Therefore, it appears that MEK1/2, probably through phosphorylation of ERK1/2, exerts an inhibitory effect on Ins1 and Glut2 expression under basal conditions and following glucosamine treatment. In addition to this inhibitory effect, MEK1/2 also seems to contribute to decreased expression of Pdx1.

Role of phosphorylation of ERK 1/2 on glucosamine-induced dedifferentiation in INS-1E cells. INS-1E cells were pretreated or not with 10 μmol/l U0126 for 1 h (a, b, d, e) and then treated (a–f) with 7.5 mmol/l glucosamine (GlcN) for 24 h. a mRNA was determined by real-time RT-PCR analysis of total RNA isolated from INS-1E cells, using cyclophilin as internal standard. mRNA levels in treated cells are quantified as relative expression units (REU) vs control cells; values are mean±SD; n = 3; *p < 0.05, **p < 0.01, ***p < 0.001. b–d Cells were solubilised and equal amounts of proteins (40 μg/sample) were analysed by western blotting using antibodies to phosphorylated (P)-ERK 1/2 and ERK 1/2 (b, c), and (d) to BIP/GRP78 or β-actin (n = 5). e Glucose-stimulated insulin release was assayed by RIA in the culture medium; values are mean±SD; n = 4; *p < 0.05, **p < 0.01. f INS-1E cells were stained with antibodies (Ab) against insulin (green). Following glucosamine treatment, the signal for insulin decreased, compared with untreated control cells. However, pre-treatment for 1 h with 10 μmol/l U0126 not only prevented the decrease, but also increased insulin levels above those of control. g Quantification of three different experiments was performed with the software package of a confocal microscope (LSM 510 META; Zeiss, Oberkochen, Germany) *p < 0.05, **p < 0.01
Next, we sought to determine whether glucosamine treatments activated ERK1/2 and whether U0126 prevented such activation. As shown in Fig. 5b, c, glucosamine induced an approximately twofold increase in phosphorylated ERK1/2 at 24 h (the effect being evident as early as 6 h and lasting at least 48 h, data not shown). PBA blocked ERK activation by glucosamine. U0126 not only prevented the increase of phosphorylated ERK1/2, but also inhibited the basal levels of activation of ERK1/2. Notably, BIP induction by glucosamine was not affected by U0126 pretreatment, indicating that U0126 did not prevent ER stress (Fig. 5d). Moreover, U0126 almost completely restored glucosamine-inhibited GSIS (Fig. 5e). Finally, immunofluorescence experiments confirmed that U0126 was not only able to prevent the downregulation of total insulin levels (the antibodies used react with insulin and proinsulin), but also to increase this level above the control value (Fig. 5f–g).
High glucose-induced ER stress displays similar effects to glucosamine-induced ER stress
As shown in Fig. 6, high glucose-induced ER stress was also able to dedifferentiate INS-1E cells (Glut2, Ins1 and Pdx1 mRNA). These effects were reversed by azaserine, PBA and U0126, suggesting that they were dependent on increased HBP flux, ER stress and ERK activation, respectively; however, the effects were not reversed by NAC, suggesting that they were independent of oxidative stress. GSIS was inhibited by high glucose, an effect that was reversed by azaserine, PBA and U0126, but not by NAC (Fig. 6d).

High glucose-induced ER stress displays similar effects to those of glucosamine-induced ER stress. INS-1E cells were pre-treated or not with 5 μmol/l azaserine (Aza) for 20 min, 10 μmol/l U0126 for 1 h, and 2.5 mmol/l PBA and 1 mmol/l NAC for 24 h, followed by 24 h culture in the presence of 25 mmol/l glucose. mRNA for Glut2 (a), Ins1 (b) and Pdx1 (c) was determined by real-time RT-PCR analysis of total RNA isolated from INS-1E cells, using cyclophilin as internal standard. mRNA levels in treated cells are quantified as relative expression units (REU) vs control cells; values are mean±SD; n = 3. d GSIS was assayed by RIA in the culture medium; values are mean±SD; n = 4. 7*p < 0.05, **p < 0.01, ***p < 0.001
Glucosamine induces ER stress and dedifferentiates primary mouse islets in a manner that is dependent on ER stress and ERK
Next, we sought to determine whether the dedifferentiating effect of glucosamine was also present in primary mouse islets. To this end, we first evaluated whether glucosamine was able to induce ER stress in this system. As shown in Fig. 7a, b, 7.5 mmol/l glucosamine induced Bip and Chop mRNAs with a time course similar to that seen in INS-1E cells. Thus, at 24 h the effect of glucosamine was already maximal (Fig. 7a, b). Next, we investigated the effect of glucosamine on beta cell genes Glut2 and Ins1. In a manner that was reciprocal to Bip and Chop mRNA induction, Glut2 and Ins1 mRNA downregulation was maximal as early as 24 h (Fig. 7c, d), again mimicking the results obtained in INS-1E cells. Also in primary islets, the inhibitory effect of glucosamine on Glut2 and Ins1 mRNA is likely to have been transcriptional, as glucosamine also downregulated Pdx1 mRNA (Fig. 7e). Moreover, GSIS was reduced in islets treated with glucosamine, suggesting that there was also a functional defect in the secretion machinery of islets (Fig. 7f). Finally, these effects were dependent on phosphorylation of ERK, since they were reversed by pretreatment with U0126 (Fig. 7g, h).

Effects of glucosamine in isolated mice islets. Islets isolated from 6-month-old mice were pre-treated or not with 2.5 mmol/l PBA for 24 h (f) or with 10 μmol/l U0126 for 1 h (g, h), followed by treatment for 1 day (a–h) with 7.5 mmol/l glucosamine (GlcN). a Bip/Grp78, (b) Chop, (c, g) Glut2, (d, g) Ins1 and (e, g) Pdx1 mRNA was determined by real-time RT-PCR analysis of total RNA isolated from mice islets, using cyclophilin as internal standard. mRNA levels in treated cells are quantified as relative expression units (REU) vs control; values are mean±SD; n = 4; *p < 0.05, **p < 0.01 and ***p < 0.001. f, h GSIS was examined over 30 min with batches of 20 islets, comparing 3.3 mmol/l with 20 mmol/l (+) glucose. Islets were subsequently collected by centrifugation at 1500 g and supernatant fractions assayed for insulin content by RIA. Values are mean±SD; n = 3; **p < 0.01
Discussion
In this study, we show that glucosamine and high glucose induced ER stress in INS-1E cells and in murine islets. As a result, the expression of beta cells markers was decreased and GSIS was inhibited. These deleterious consequences of glucosamine and high glucose treatment were fully prevented by the chemical chaperone PBA, but not by the antioxidant NAC. The effect of glucosamine- and high glucose-induced ER stress on beta cell differentiation and function appears to be mediated by the MEK–ERK pathway.
The declining function and mass of the pancreatic beta cells is central to the progression of type 2 diabetes. While it is well established that dyslipidaemia and hyperglycaemia contribute to beta cell dysfunction [1, 2], the molecular mechanisms implicated are far less clear. In particular, the adverse action of glucose on beta cells is much less well understood, being slower and more subtle than that of fatty acids, which cause strong ER stress and subsequent apoptosis. High glucose has been reported to cause generation of ROS [4, 5], activation of ER stress [6–8] and increase in the HBP flux [9]. Under physiological conditions, only 1–3% of intracellular glucose enters the hexosamine pathway; however, the flux increases with glucose concentration [9]. Increased HBP flux, in turn, causes hyper-O-GlcNAc of proteins [10], oxidative stress [11] and ER stress, although this last mechanism has been demonstrated in cells other than beta cells [12, 29].
We have demonstrated that glucose and glucosamine promote ER stress in beta cells. From our data, we cannot conclude that glucosamine causes ER stress via increased protein O-GlcNAc levels, although this mechanism cannot be ruled out. In fact, data from the literature support the existence of mechanisms that are dependent on [30] and independent of [31] protein O-GlcNAc. Our results also suggest that a glucosamine intermediate is involved in ER stress induction, as inhibition of glutamine amidotransferases, including GFAT, by azaserine blocked ER stress induction by high glucose but not by glucosamine.
ER stress is recognised as an important determinant of type 2 diabetes, and is a central feature of peripheral insulin resistance, acting by inhibiting insulin receptor signalling. However, ER stress also plays an important role in the beta cell failure that precipitates type 2 diabetes. Beta cells, like plasma cells and thyroid cells, have a high protein load [32, 33], synthesising large quantities of (a single) protein. Proinsulin represents up to 20% of the total mRNA and 30–50% of the total protein synthesis of the beta cell [18, 19]. These percentages increase further, when considering only cargo (secretory and membrane) proteins synthesised by the beta cell. In addition, glucose stimulates proinsulin translation [20], and increases the stability of pre-proinsulin and transcription of the insulin gene [21, 22], further increasing the protein load. Therefore, beta cells are highly susceptible to ER stress.
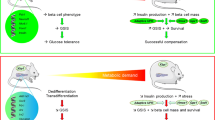
The ER stress induced by high glucose/glucosamine is mild, in contrast to that induced by fatty acids and Ca2+-perturbing drugs. Accordingly, we did not detect a significant effect of glucosamine on the viability of INS-1E cells, while ER stress induced by fatty acids [17] and the Ca2+-perturbing drug cyclopiazonic acid [34] is followed by massive apoptosis. However, the activation of HBP flux, even in the absence of relevant apoptosis, has deleterious effects on beta cells, causing ER stress and inhibiting beta cell differentiation. High glucose-/glucosamine-induced ER stress and beta cell dedifferentiation are causally linked, as demonstrated by the effect of PBA. Indeed, pretreatment with PBA prevented ER stress and dedifferentiation of beta cells. In general, adaptation or apoptosis are believed to be the major outcomes of ER stress. However, dedifferentiation has recently been identified as a new response to ER stress. It has been shown that ER stress dedifferentiates chondrocytes, downregulating collagen II and aggrecan [23]. In vivo, in transgenic mice expressing mutant collagen X, ER stress altered chondrocyte differentiation [24]. Chondrocytes survive ER stress, but terminal differentiation is interrupted. Thyroid cells subjected to ER stress downregulate thyroid-specific markers and their transcriptional factors [25]. Dedifferentiation may constitute a new tactic for survival, since cells avoid energy expenditure for the expression of genes that, in this condition, are unnecessary or even dispensable. Moreover, in all cited cases [23–25], as well as in the case of beta cells (this study), the differentiation genes encode cargo proteins, resulting in a reduction of ER-specific protein load. This represents a selective and long-term downregulation, which is temporally distinct from the general and short-term shut-off of protein synthesis elicited by (PKR)-like endoplasmic reticulum kinase (PERK) [35]. The mechanism of this downregulation is, at least in part, transcriptional, since downregulation of Ins1 and Glut2 is accompanied by the coordinate downregulation of Pdx1.
Our results indicate that ER stress induced by HBP flux triggers signalling via the MEK–ERK pathway to elicit dedifferentiation of beta cells. It is well known that glucose regulates insulin transcription. Acute exposure of beta cells to high glucose stimulates insulin transcription, while chronic exposure to high glucose results in inhibition of the insulin gene promoter activity. Both effects are mediated by phosphorylation of ERK1/2 [36]. Downstream of ERK1/2 there are several factors that bind to the insulin gene promoter to enhance transcription in response to glucose [37]. Pancreatic and duodenal homeobox 1 (PDX1) and beta cell E-box transcriptional activator 2 (BETA2) synergistically activate insulin gene transcription [38]. v-Maf musculoaponeurotic fibrosarcoma oncogene family, protein A (avian) (MAFA) also contributes to glucose responsiveness [39]. Negative regulators of insulin promoter activity include Jun and the CCAAT/enhancer binding protein β (C/EBP-β). These transcription factors are increased in beta cells during prolonged exposure to low and high glucose, respectively [40, 41]. ERK1 and 2 are activated in pancreatic beta cells by stimulatory concentrations of glucose, this activation being Ca2+-dependent [42, 43]. A rise in intracellular Ca2+ is secondary to glucose metabolism and cell depolarisation. Moreover, activation of ERK1/2 by glucose is sensitive to inhibitors of calmodulin and the class 2B Ca2+/calmodulin-dependent phosphatase, calcineurin [44]. Hence, calcineurin is an upstream regulator of the ERK1/2 pathway in pancreatic beta cells. More recently a possible mechanism linking glucose, Ca2+ and ERK activation has been elucidated. The protein phosphatase calcineurin selectively dephosphorylates kinase suppressor of Ras 2 (KSR2) in response to Ca2+ signals, regulating KSR2 localisation and ERK scaffold activity [45].
However, besides Ca2+- and calcineurin-mediated mechanisms, our study suggests that ER stress may play a role. This may occur specifically during chronic hyperglycaemia. Thus, it is known that chronic challenge of islets with high glucose causes a reduction of Ca2+ influx induced by glucose and diazoxide compared with control islets and, moreover, does not lead to detectable changes in the intracellular Ca2+ concentration [46]. Therefore, in chronic hyperglycaemia, the Ca2+-mediated mechanisms of ERK activation are likely to be much less involved. Instead, prolonged exposure of beta cells to high glucose or glucosamine induces ER stress. Following the stress, ERK is activated through inositol-requiring 1 (IRE1)-dependent mechanisms [27]. This long term ERK activation may inhibit proinsulin transcription by the already described, mainly post-translational mechanisms. BETA2, PDX1, MAFA, NFAT and C/EBP-β are ERK1/2 substrates [36, 47, 48]. The latter three associate with the insulin gene promoter in an ERK1/2-dependent manner [36]. Finally, the transactivating activities of BETA2 and PDX1 depend at least partly on ERK1/2 activity [49]. Moreover, the present study suggests that, in addition to those known mechanisms in the regulation of insulin transcription, inhibition (probably transcriptional) of an important beta cell transcriptional factor, PDX1, may also play a role.
Interestingly, U0126 not only prevented the glucosamine stimulation of ERK1/2 phosphorylation, but also inhibited the phosphorylation of ERK1/2 present in the basal condition. This effect is paralleled by prevention of the inhibitory effect of glucosamine on Ins1 and Pdx1 mRNA, and on total insulin protein (as detected by immunofluorescence), and also by an increase of these mRNAs and total insulin protein levels above those present in basal conditions. It has been proposed recently that cells with a high protein load, such as beta cells, have a level of ‘physiological’ UPR activation that balances the high protein load with the folding capability [50]. It is conceivable that this ‘physiological’ UPR causes a certain level of ERK activation. This, in turn, may basally inhibit insulin transcription. In this model, insulin load and ‘physiological’ UPR activation exert a feedback loop at the level of insulin transcription, in addition to the loop at the translational level, which is operated by PERK–eukaryotic translation initiation factor 2A (eIF2α).
Abbreviations
- BETA2:
-
Beta cell E-box transcriptional activator 2
- BIP:
-
Binding protein
- C/EBP-β:
-
CCAAT/enhancer binding protein β
- ER:
-
Endoplasmic reticulum.
- ERK:
-
Extracellular signal-regulated kinase
- GFAT:
-
Glutamine:fructose-6-phosphate amidotransferase
- GlcNAc:
-
Glucosamine acylation
- GRP78:
-
Glucose-regulated protein 78
- GSIS:
-
Glucose-stimulated insulin secretion
- HBP:
-
Hexosamine biosynthetic pathway
- KRBH:
-
KRB HEPES
- KSR2:
-
Kinase suppressor of Ras 2
- MAFA:
-
v-Maf musculoaponeurotic fibrosarcoma oncogene family, protein A (avian)
- MAPK:
-
Mitogen-activated protein kinase
- MEK:
-
MAPK/ERK kinase
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAC:
-
N-Acetylcysteine
- PBA:
-
4-Phenyl butyric acid
- PDX1:
-
Pancreatic and duodenal homeobox 1
- PERK:
-
(PKR)-like endoplasmic reticulum kinase
- PUGNAc:
-
O-(2-Acetamido-2-deoxy-d-glucopyranosylidene) amino-N-phenylcarbamate
- ROS:
-
Reactive oxygen species
- UPR:
-
Unfolded protein response
References
Donath MY, Halban PA (2004) Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications. Diabetologia 47:581–589
Kaiser N, Leibowitz G, Nesher R (2003) Glucotoxicity and beta-cell failure in type 2 diabetes mellitus. J Pediatr Endocrinol Metab 16:5–22
Rhodes CJ (2005) Type 2 diabetes—a matter of beta-cell life and death? Science 307:380–384
Robertson RP (2004) Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet cells in diabetes. J Biol Chem 279:42351–42354
Kaneto H, Nakatani Y, Kawamori D et al (2005) Role of oxidative stress, endoplasmic reticulum stress, and c-Jun N-terminal kinase in pancreatic beta cell dysfunction and insulin resistance. Int J Biochem Cell Biol 37:1595–1608
Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL (2005) Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54:S97–S107
Elouil H, Bensellam M, Guiot Y et al (2007) Acute nutrient regulation of the unfolded protein response and integrated stress response in cultured rat pancreatic islets. Diabetologia 50:1442–1452
Eizirik DL, Cardozo AK, Cnop M (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29:42–61
Hanover JA, Lai Z, Lee G, Lubas WA, Sato SM (1999) Elevated O-linked N-acetylglucosamine metabolism in pancreatic beta-cells. Arch Biochem Biophys 362:38–45
Park J, Kwon H, Kang Y, Kim Y (2007) Proteomic analysis of O-GlcNAc modifications derived from streptozotocin and glucosamine induced beta-cell apoptosis. J Biochem Mol Biol 40:1058–1068
Kaneto H, Xu G, Song KH et al (2001) Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function through the induction of oxidative stress. J Biol Chem 276:31099–31104
Qiu W, Kohen-Avramoglu R, Mhapsekar S, Tsai J, Austin RC, Adeli K (2005) Glucosamine-induced endoplasmic reticulum stress promotes ApoB100 degradation: evidence for Grp78-mediated targeting to proteasomal degradation. Arterioscler Thromb Vasc Biol 25:571–577
Harmon JS, Stein R, Robertson RP (2005) Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic cells. J Biol Chem 280:11107–11113
Oyadomari S, Araki E, Mori M (2002) Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta cells. Apoptosis 7:335–345
Fonseca SG, Burcin M, Gromada J, Urano F (2009) Endoplasmic reticulum stress in beta cells and development of diabetes. Curr Opin Pharmacol 9:763–770
Cnop M, Ladriere L, Hekerman P et al (2007) Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta cell dysfunction and apoptosis. J Biol Chem 282:3989–3997
Cunha DA, Hekerman P, Ladrière L et al (2008) Initiation and execution of lipotoxic ER stress in pancreatic beta cells. J Cell Sci 121:2308–2318
van Lommel L, Janssens K, Quintens R et al (2006) Probe-independent and direct quantification of insulin mRNA and growth hormone mRNA in enriched cell preparations. Diabetes 55:3214–3220
Schuit FC, Kiekens R, Pipeleers DG (1991) Measuring the balance between insulin synthesis and insulin release. Biochem Biophys Res Commun 178:1182–1187
Wicksteed B, Uchizono Y, Alarcon C, McCuaig JF, Shalev A, Rhodes CJ (2007) A cis-element in the 5′ untranslated region of the preproinsulin mRNA (ppIGE) is required for glucose regulation of proinsulin translation. Cell Metab 5:221–227
Leibiger B, Moede T, Schwarz T et al (1998) Short-term regulation of insulin gene transcription by glucose. Proc Natl Acad Sci USA 95:9307–9312
German M, Ashcroft S, Docherty K et al (1995) The insulin gene promoter. A simplified nomenclature. Diabetes 44:1002–1004
Yang L, Carlson SG, McBurney D, Horton WE Jr (2005) Multiple signals induce endoplasmic reticulum stress in both primary and immortalized chondrocytes resulting in loss of differentiation, impaired cell growth, and apoptosis. J Biol Chem 280:31156–31165
Tsang KY, Chan D, Cheslett D et al (2007) Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol 5:568–585
Ulianich L, Garbi C, Treglia AS et al (2008) ER stress is associated with dedifferentiation and an epithelial-to-mesenchymal transition-like phenotype in PC Cl3 thyroid cells. J Cell Sci 121:477–486
De Vitis S, Treglia AS, Ulianich L et al (2011) Tyr phosphatase-mediated P-ERK inhibition suppresses senescence in EIA + v-raf transformed cells, which, paradoxically, are apoptosis-protected in a MEK-dependent manner. Neoplasia 13:120–130
Di Jeso B, Park YN, Ulianich L et al (2005) Mixed-disulfide folding intermediates between thyroglobulin and endoplasmic reticulum resident oxidoreductases ERp57 and protein disulfide isomerase. Mol Cell Biol 25:9793–9805
Nguyên DT, Kebache S, Fazel A et al (2004) Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol Biol Cell 15:4248–4260
Raciti GA, Iadicicco C, Ulianich L et al (2010) Glucosamine-induced endoplasmic reticulum stress affects GLUT4 expression via activating transcription factor 6 in rat and human skeletal muscle cells. Diabetologia 53:955–965
Vosseller K, Wells L, Lane MD, Hart GW (2002) Elevated nucleoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3–L1 adipocytes. Proc Natl Acad Sci USA 99:5313–5318
Robinson KA, Ball LE, Buse MG (2007) Reduction of O-GlcNAc protein modification does not prevent insulin resistance in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 292:E884–E890
Di Jeso B, Ulianich L, Pacifico F et al (2003) Folding of thyroglobulin in the calnexin/calreticulin pathway and its alteration by loss of Ca2+ from the endoplasmic reticulum. Biochem J 370:449–458
Di Jeso B, Pereira R, Consiglio E, Formisano S, Satrustegui J, Sandoval IV (1998) Demonstration of a Ca2+ requirement for thyroglobulin dimerization and export to the Golgi complex. Eur J Biochem 252:583–590
Pirot P, Naamane N, Libert F et al (2007) Global profiling of genes modified by endoplasmic reticulum stress in pancreatic beta cells reveals the early degradation of insulin mRNAs. Diabetologia 50:1006–1014
Harding H, Zhang Y, Ron D (1999) Translation and protein folding are coupled by an endoplasmic reticulum resident kinase. Nature 397:271–274
Lawrence MC, McGlynn K, Park BH, Cobb MH (2005) ERK1/2-dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J Biol Chem 280:26751–26759
Ohneda K, Ee H, German M (2000) Regulation of insulin gene transcription. Semin Cell Dev Biol 11:227–233
Frayling TM, Evans JC, Bulman MP (2001) Beta cell genes and diabetes: molecular and clinical characterization of mutations in transcription factors. Diabetes 1:S94–S100
Olbrot M, Rud J, Moss LG, Sharma A (2002) Identification of beta cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci USA 99:6737–6742
Lu M, Seufert J, Habener JF (1997) Pancreatic beta cell-specific repression of insulin gene transcription by CCAAT/enhancer-binding protein beta. Inhibitory interactions with basic helix–loop–helix transcription factor E47. J Biol Chem 272:28349–28359
Inagaki N, Maekawa T, Sudo T, Ishii S, Seino Y, Imura H (1992) c-Jun represses the human insulin promoter activity that depends on multiple cAMP response elements. Proc Natl Acad Sci USA 89:1045–1049
Khoo S, Cobb MH (1997) Activation of mitogen-activating protein kinase by glucose is not required for insulin secretion. Proc Natl Acad Sci USA 94:5599–5604
Briaud I, Lingohr MK, Dickson LM, Wrede CE, Rhodes CJ (2003) Differential activation mechanisms of Erk-1/2 and p70(S6K) by glucose in pancreatic beta-cells. Diabetes 52:974–983
Arnette D, Gibson TB, Lawrence MC et al (2003) Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic beta cells. J Biol Chem 278:32517–32525
Dougherty MK, Ritt DA, Zhou M et al (2009) KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol Cell 34:652–662
Khaldi MZ, Guiot Y, Gilon P, Henquin JC, Jonas JC (2004) Increased glucose sensitivity of both triggering and amplifying pathways of insulin secretion in rat islets cultured for 1 wk in high glucose. Am J Physiol Endocrinol Metab 287:E207–E217
Piwien-Pilipuk G, Galigniana MD, Schwartz J (2003) Subnuclear localization of C/EBP is regulated by growth hormone and dependent on MAPK. J Biol Chem 278:35668–35677
Yang TT, Xiong Q, Graef IA, Crabtree GR, Chow CW (2005) Recruitment of the extracellular signal-regulated kinase/ribosomal S6 kinase signaling pathway to the NFATc4 transcription activation complex. Mol Cell Biol 25:907–920
Khoo S, Griffen SC, Xia Y, Baer RJ, German MS, Cobb MH (2003) Regulation of insulin gene transcription by extracellular-signal regulated protein kinases (ERK) 1 and 2 in pancreatic beta cells. J Biol Chem 278:32969–32977
Rutkowski DT, Hegde RS (2010) Regulation of basal cellular physiology by the homeostatic unfolded protein response. J Cell Biol 189:783–794
Acknowledgements
This work was supported by grant 2006069102_004 of MIUR to B. Di Jeso, as well as by the EFSD and the European Community’s PREPOBEDIA (201681). The financial support of Telethon, Italy to F. Beguinot is acknowledged.
Contribution statement
AL, LU, AST, CN, LP, DDL, GN, CG, FB, CM analysed the data. AL, LU, AST, CN, LP, DDL, GN, CG, FB, CM revised the article critically. BDJ conceived and designed the study, and wrote the article. All authors approved the final version of the paper to be published.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
Primer sequences (PDF 57 kb)
Rights and permissions
About this article
Cite this article
Lombardi, A., Ulianich, L., Treglia, A.S. et al. Increased hexosamine biosynthetic pathway flux dedifferentiates INS-1E cells and murine islets by an extracellular signal-regulated kinase (ERK)1/2-mediated signal transmission pathway. Diabetologia 55, 141–153 (2012). https://doi.org/10.1007/s00125-011-2315-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2315-1