
Abstract
Aims/hypothesis
Glucosamine, generated during hyperglycaemia, causes insulin resistance in different cells. Here we sought to evaluate the possible role of endoplasmic reticulum (ER) stress in the induction of insulin resistance by glucosamine in skeletal muscle cells.
Methods
Real-time RT-PCR analysis, 2-deoxy-d-glucose (2-DG) uptake and western blot analysis were carried out in rat and human muscle cell lines.
Results
In both rat and human myotubes, glucosamine treatment caused a significant increase in the expression of the ER stress markers immunoglobulin heavy chain-binding protein/glucose-regulated protein 78 kDa (BIP/GRP78 [also known as HSPA5]), X-box binding protein-1 (XBP1) and activating transcription factor 6 (ATF6). In addition, glucosamine impaired insulin-stimulated 2-DG uptake in both rat and human myotubes. Interestingly, pretreatment of both rat and human myotubes with the chemical chaperones 4-phenylbutyric acid (PBA) or tauroursodeoxycholic acid (TUDCA), completely prevented the effect of glucosamine on both ER stress induction and insulin-induced glucose uptake. In both rat and human myotubes, glucosamine treatment reduced mRNA and protein levels of the gene encoding GLUT4 and mRNA levels of the main regulators of the gene encoding GLUT4 (myocyte enhancer factor 2 a [MEF2A] and peroxisome proliferator-activated receptor-γ coactivator 1α [PGC1α]). Again, PBA or TUDCA pretreatment prevented glucosamine-induced inhibition of GLUT4 (also known as SLC2A4), MEF2A and PGC1α (also known as PPARGC1A). Finally, we showed that overproduction of ATF6 is sufficient to inhibit the expression of genes GLUT4, MEF2A and PGC1α and that ATF6 silencing with a specific small interfering RNA is sufficient to completely prevent glucosamine-induced inhibition of GLUT4, MEF2A and PGC1α in skeletal muscle cells.
Conclusions/interpretation
In this work we show that glucosamine-induced ER stress causes insulin resistance in both human and rat myotubes and impairs GLUT4 production and insulin-induced glucose uptake via an ATF6-dependent decrease of the GLUT4 regulators MEF2A and PGC1α.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The endoplasmic reticulum (ER) is the principal site of protein synthesis, and together with the Golgi apparatus it facilitates transport and release of correctly folded proteins. Under conditions of cellular stress leading to an impairment of ER function, proteins are unable to fold properly and accumulate in the ER lumen. It is because of these unfolded or misfolded proteins that the ER has evolved a coping system known as the unfolded protein response (UPR) [1, 2].
Cellular stresses that may elicit UPR activation include glucose and energy deprivation, increased protein synthesis, inhibition of protein glycosylation and imbalance of ER calcium levels [3, 4]. In mammalian cells, at least four functionally distinct responses have been identified and three ER-resident transmembrane proteins have been described as primary sensors and transducers of the UPR: the double-stranded RNA-activated protein kinase-like ER kinase (PERK), inositol requiring-1, and activating transcription factor 6 (ATF6) [5–7]. The first response, mediated by PERK, is translational attenuation, to reduce the load of new protein synthesis and prevent further accumulation of unfolded proteins [8]. The second response is upregulation of genes encoding ER chaperone proteins such as the immunoglobulin heavy chain-binding protein/glucose-regulated protein 78 kDa (BIP/GRP78) and the glucose-regulated protein 94 kDa, to increase the ER protein-folding capacity [7, 9]. The third response is transcriptional activation of genes involved in the degradation of misfolded protein in the ER by the ubiquitin–proteasome system, called ER-associated degradation [10]. The fourth response is apoptosis, which occurs when severe and prolonged ER stress impairs ER functions, to protect the organism by eliminating the damaged cells [4]. ER stress plays an important role in several human diseases, including type 2 diabetes; indeed, recent studies reported that ER stress is involved in both pancreatic beta cell dysfunction [11–13] and peripheral insulin resistance [14, 15].
While the consequences of ER stress have been widely studied in adipose tissue and liver, ER stress in skeletal muscle, the major site of glucose disposal, has not received equal attention. The hexosamine biosynthetic pathway (HBP) is a minor glucose metabolic pathway that metabolises ∼3% of glucose entering the cell, and the final product of this pathway, UDP-N-acetylglucosamine, as other nucleotide hexosamines, is used in the ER as substrate for protein glycosylation [16, 17]. Although quantitatively using a small fraction of glucose, HBP is an important contributor to the insulin-resistant state. Several studies have shown, indeed, that chronic exposure to glucosamine (GlcN), a precursor of the HBP, impairs insulin responsiveness, thus contributing to the formation of an insulin-resistant state in cultured human skeletal muscle cells and rat adipocytes [17] as well as in vivo [18]. However, the precise mechanisms by which GlcN induces insulin resistance have not been conclusively established in these studies.
Methods
Materials
DMEM, FBS, FCS, l-glutamine and BSA were from Invitrogen (Paisley, UK). Thapsigargin (Thap), GlcN, 4-phenylbutyric acid (PBA) and N-acetyl-cysteine (NAC), were from Sigma-Aldrich (St Louis, MO, USA). Mn(III)tetrakis(4-benzoic acid) porphyrin chloride (MnTBAP) and tauroursodeoxycholic acid (TUDCA) were from Calbiochem (San Diego, CA, USA). Other reagents were as follows: Ultroser G (Pall Biosepra, Cergy, France), 2-deoxy-d-[14C]glucose (2-DG) (Perkin Elmer, Waltham, MA, USA), insulin (Novo Nordisk, Bagsværd, Denmark), GLUT4 (Abcam, Cambridge, UK), myocyte enhancer factor 2 a (MEF2A), peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α), eukaryotic translation initiation factor 2α (eIF2α) and phospho-eIF2α antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA), small interfering RNA (siRNA) and siPORT NeoFX Transfection Agent (Applied Biosystems, Carlsbad, CA, USA)
Cell culture procedures and 2-DG uptake
Human cell cultures from lean individuals were established as described previously [19–21] (Table 1). Cells were cultured in DMEM supplemented with 2% (vol./vol.) FCS, 2% (vol./vol.) Ultroser G and antibiotics. Human myotubes were allowed to differentiate under physiological conditions of insulin (25 pmol/l) and glucose (5.5 mmol/l) for 4 days. L6 rat skeletal muscle myoblasts were grown in DMEM supplemented with 10% (vol./vol.) FBS, 2 mmol/l l-glutamine and antibiotics. L6 myotubes were allowed to differentiate as described previously [22]. 2-DG uptake was measured as reported previously [23].
Real-time RT-PCR and western blot analysis
Total RNA extraction, cDNA synthesis and real-time RT-PCR analysis were performed as described previously [24]. Primer sequences are in Electronic supplementary material (ESM) Table 1. Cell lysates and immunoblotting were carried out as described previously [22]. Antibodies against GLUT4, phospho-eIF2α and eIF2α were used for detection of proteins.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed as reported [25]. Vehicle- or reagent-treated myotubes were fixed with 1% (vol./vol.) formaldehyde at 37°C. The fixed cells were lysed in a SDS lysis buffer (1% [wt/vol.] SDS, 10 mmol/l EDTA and 50 mmol/l TRIS–HCl, pH 8.1), incubated on ice, and sonicated to shear DNA. Sheared chromatin samples were taken as input control or used for immunoprecipitation with anti-MEF2A, anti-PGC1α or non-immune antibodies. DNA fragments were recovered and were subjected to real-time RT-PCR amplification by using specific primers for the analysed regions.
Atf6 siRNA-mediated knockdown
Cells were transfected with 5 nmol/l of siRNA negative control and Atf6 siRNA (GCUUGUCAGUCACGAAAGAtt) and antisense (UCUUUCGUGACUGACAAGCag) according to the manufacturer’s recommendations and processed 48 h after transfection.
Statistical procedures
Data were analysed with Statview software (Abacus Concepts, Piscataway, NJ, USA) by one-factor analysis of variance. p < 0.05 was considered statistically significant.
Results
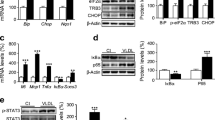
To investigate the role of GlcN in ER stress induction, differentiated L6 skeletal muscle cells were treated with different concentrations of GlcN. The classic ER stress inducer Thap, an inhibitor of sarcoplasmic/ER calcium-transporting ATPases, was used as a control of ER stress induction [2]. In L6 myotubes, Thap induced a 17-fold increase of the chaperone Bip/grp78 (also known as Hspa5) mRNA (Fig. 1a), indicating that our cellular model was sensitive to ER stress. Bip/grp78 mRNA levels were increased also by GlcN, with a maximal expression observed at 10 mmol/l GlcN for 24 h (Fig. 1b). Interestingly, pretreatment of cells with azaserine, a non-specific but commonly used inhibitor of the glutamine:fructose-6-phosphate amidotransferase (GFAT) [26, 27], the rate-limiting enzyme of the HBP, prevented high glucose (HG)-induced ER stress (ESM Fig. 1a). In addition, treatment of cells with the peptide O-acetylglucosamine-β-N-acetylglucosaminidase inhibitor O-(2-acetamido-2-deoxy-d-glucopyranosylidenamino)N-phenylcarbamate (PUGNAc), did not increase Bip/grp78 mRNA levels, suggesting that enhanced O-linked glycosylation was not responsible for the induction of ER stress (ESM Fig. 1a). To evaluate whether GlcN-induced ER stress could be mediated by oxidative stress, L6 myotubes were pretreated with two anti-oxidants, the glutathione precursor NAC and the superoxide dismutase mimetic MnTBAP. Pretreatment of cells with both NAC and MnTBAP did not affect GlcN-induced Bip/grp78 mRNA increase, suggesting that GlcN-induced ER stress was not dependent on oxidative stress in skeletal muscle cells (Fig. 1b). Then we evaluated the effects of different concentrations of GlcN on ATP intracellular levels. GlcN depleted the ATP pool only at the highest concentrations, suggesting that GlcN-induced ER stress was not dependent on ATP depletion in skeletal muscle cells (ESM Fig. 2). As expected, xylose did not induce a Bip/grp78 mRNA increase, even at high concentrations (Fig. 1b), thus excluding an osmotic stress effect caused by treatments. Time course analysis with 7.5 mmol/l GlcN showed that Bip/grp78 expression was significantly increased as early as 2 h after the treatment and was elevated up to 16 h (Fig. 1c). To investigate whether chemical chaperones could prevent GlcN-induced ER stress, we analysed Bip/grp78 mRNA in L6 myotubes treated with GlcN in the presence of either 10 mmol/l PBA, a low molecular weight non-specific chemical chaperone known to stabilise protein conformation and to improve ER folding capacity [28], or 5 mmol/l TUDCA, a bile acid derivative that also modulates ER function [28]. Both PBA and TUDCA almost completely prevented the effect of GlcN on Bip/grp78 mRNA (Fig. 1d). These data suggest that GlcN is able to induce ER stress in skeletal muscle cells and that this effect is prevented by chemical chaperones.

GlcN induces BIP/GRP78 in L6 myotubes. a–d Bip/grp78 mRNA was determined by real-time RT-PCR analysis of total RNA isolated from myotubes, using Gapdh as internal standard. mRNA levels in treated cells are relative expression units (REU) to those in control (C; mean ± SD; n = 8). ***p < 0.001. a L6 cells were treated with 0.5 µmol/l Thap for 30 min, followed by 24 h without Thap. b L6 cells were pretreated or not with 5 mmol/l NAC or with 1 mg/ml MnTBAP for 2 h and then cultured in the presence of GlcN or xylose, as indicated, for 24 h. c Time course of Bip/grp78 mRNA in L6 cells cultured with 7.5 mmol/l GlcN for the indicated times. d L6 cells were pretreated or not for 1 h with 10 mmol/l PBA or 5 mmol/l TUDCA and then treated with 7.5 mmol/l GlcN for 24 h
To verify whether GlcN was able to induce UPR activation, L6 myotubes were treated with GlcN or Thap and different markers of UPR activation were analysed. The mRNA levels of the gene (Xbp1) encoding the spliced active form of the X-box binding protein-1 (XBP-1; Xbp1 s) increased significantly following both Thap (Fig. 2a) and GlcN treatments (Fig. 2b). Time course experiments showed that the increase of Xbp1 s peaked at 8 h of GlcN treatment and remained higher than basal level up to 24 h (Fig. 2b). In addition, also the mRNA levels of the genes encoding the total form of XBP-1 (Xbp1 t) and ATF6 (Atf6) were significantly increased upon both Thap (Fig. 2c,e) and GlcN treatments (Fig. 2d,f). Furthermore, phosphorylation of eIF2α was evident as early as 30 min after both GlcN and Thap treatment, and it persisted up to 24 h following GlcN treatment (Fig. 2g). These data suggest that GlcN caused UPR activation in L6 myotubes.

GlcN induces UPR activation in L6 myotubes. a–g L6 cells were treated with 0.5 µmol/l Thap for 30 min, followed by 24 h without Thap, or with 7.5 mmol/l GlcN for the indicated times (a–f). Xbp1 s (a, b), Xbp1 t (c, d) and Atf6 (e, f) mRNAs in treated cells are relative expression units (REU) to those in control (C; mean ± SD; n = 7) and were determined by real-time RT-PCR analysis, using Gapdh as internal standard. *p < 0.05; ***p < 0.001. g L6 cells treated with 0.5 µmol/l Thap for 30 min, followed by 24 h without Thap (left), or with 7.5 mmol/l GlcN (right) for the indicated times, were solubilised and equal amounts of proteins (80 µg per sample) were analysed by western blotting using phospho-eIF2α Ser51 and eIF2α specific antibodies (n = 5)
To investigate the effect of GlcN-induced ER stress on the insulin sensitivity of skeletal muscle cells, insulin-induced glucose uptake was evaluated in L6 myotubes treated with GlcN for 24 h. GlcN treatment reduced the capability of L6 cells to take up the glucose analogue 2-DG upon insulin stimulation, compared with control cells (Fig. 3a). Similar results were obtained when cells were treated with Thap (data not shown) and HG (ESM Fig. 1b). To verify the hypothesis that GlcN and HG may impair glucose uptake in L6 cells through ER stress induction, we analysed insulin-induced glucose uptake in cells treated with either GlcN or HG in the presence of PBA or TUDCA. Interestingly, both PBA and TUDCA prevented GlcN (Fig. 3a) and HG (ESM Fig. 1b) effects on insulin-stimulated glucose uptake, suggesting that ER stress caused insulin resistance in skeletal muscle cells.

Effect of ER stress on 2-DG uptake and Glut4 expression in L6 myotubes. a L6 cells were pretreated or not for 1 h with 10 mmol/l PBA or 5 mmol/l TUDCA and then treated with 7.5 mmol/l GlcN for 24 h. 2-DG uptake was measured following 30 min of insulin stimulation (mean ± SD; n = 5). **p < 0.01; ***p < 0.001. b, c Glut4 mRNA was determined by real-time RT-PCR analysis of total RNA isolated from myotubes, using Gapdh as internal standard. mRNA levels in treated cells are relative expression units (REU) to those in control (mean ± SD; n = 5). ***p < 0.001. b Time course of Glut4 mRNA in L6 cells treated with 7.5 mmol/l GlcN for the indicated times. c Glut4 mRNA was detected in L6 cells pretreated or not with 10 mmol/l PBA for 1 h, and then treated with 7.5 mmol/l GlcN for 24 h. d L6 cells pretreated or not with 10 mmol/l PBA for 1 h, and then treated with 7.5 mmol/l GlcN for 24 h, were solubilised and equal amounts of proteins (80 μg/sample) were analysed by western blotting using GLUT4 specific antibody (n = 5). A representative autoradiograph is shown. **p < 0.01
We then evaluated GlcN effects on the expression of GLUT4, since it is the main glucose transporter responsible for insulin-mediated glucose uptake in muscle [29]. Time course analysis showed that Glut4 (also known as Slc2a4) mRNA levels were significantly decreased as early as 6 h after GlcN treatment, and were reduced by about 50% after 16 h of treatment (Fig. 3b). Glut4 mRNA levels did not show any significant variation when L6 myotubes were treated with GlcN in the presence of PBA (Fig. 3c). In addition, the GlcN-dependent decrease of Glut4 mRNA expression was paralleled by a similar reduction of GLUT4 protein levels and this was also prevented by pretreatment with PBA (Fig. 3d). As for GlcN, both Thap (data not shown) and HG treatment (ESM Fig. 1c), induced a significant decrease of both GLUT4 protein and mRNA levels compared with control cells. These data indicated that GlcN and HG reduced Glut4 expression through the induction of ER stress.
Actinomycin D treatment did not further decrease Glut4 mRNA upon GlcN treatment, suggesting that GlcN-induced ER stress determined a transcriptional inhibition of Glut4 in skeletal muscle cells, without affecting its mRNA stability (data not shown). To gain further insight into the mechanisms leading to Glut4 reduced transcription, we analysed the expression of genes relevant to Glut4 transcriptional regulation by real-time RT-PCR analysis. Time course experiments showed that GlcN caused a significant reduction of Mef2a mRNA expression as early as 6 h after the treatment, with a 60% reduction at 16 h (Fig. 4a). Similarly, MEF2A coactivator Pgc1α (also known as Ppargc1a) was reduced by 50% after 16 h of treatment compared with control cells (Fig. 4c). As for GlcN, both Thap (data not shown) and HG treatment (ESM Fig. 1c) induced a significant decrease of Mef2a and Pgc1α expression levels. L6 cells treated with GlcN in the presence of PBA exhibited no differences in the mRNA levels of both Mef2a (Fig. 4b) and Pgc1α (Fig. 4d) compared with control cells, confirming that also Mef2a and Pgc1α reduced expression was dependent on GlcN-induced ER stress. To establish whether the reduction of Mef2a and Pgc1α expression induced by ER stress was paralleled by a reduced binding of these two proteins to the GLUT4 promoter, we performed ChIP and re-ChIP experiments in L6 cells treated with GlcN. MEF2A binding to GLUT4 promoter showed a 60% decrease upon GlcN treatment compared with control cells (Fig. 4e). Similarly, PGC1α indirect binding to GLUT4 promoter measured by re-ChIP assay was reduced by 40% upon GlcN treatment compared with control cells (Fig. 4f). These data indicate that GlcN-induced ER stress causes the transcriptional inhibition of Glut4, at least in part by reducing both Mef2a and Pgc1α mRNA levels and their binding to the GLUT4 promoter.

Effect of ER stress on Mef2a and Pgc1α mRNA expression and GLUT4 promoter binding in L6 myotubes. a–d Mef2a and Pgc1α mRNAs were determined by real-time RT-PCR analysis, using Gapdh as internal standard. mRNA levels in treated cells are relative expression units (REU) to those in control (mean ± SD; n = 4). *p < 0.05; **p < 0.01; ***p < 0.001. a Time course of Mef2a mRNA in L6 cells treated with 7.5 mmol/l GlcN for the indicated times. b Mef2a mRNA was detected in L6 cells pretreated or not with 10 mmol/l PBA for 1 h, and then treated with 7.5 mmol/l GlcN for 24 h. c Time course of Pgc1α mRNA in L6 cells treated with 7.5 mmol/l GlcN for the indicated times. d Pgc1α mRNA was detected in L6 cells pretreated or not with 10 mmol/l PBA for 1 h, and then treated with 7.5 mmol/l GlcN for 24 h. e, f Soluble chromatin was prepared from L6 cells vehicle treated or treated with 7.5 mmol/l GlcN for 24 h as described in Methods and immunoprecipitated with MEF2A antibody (e) or re-immunoprecipitated with PGC1α antibody (f). Immunoprecipitates were then amplified by real-time RT-PCR analysis using specific primers for the analysed regions (mean ± SD; n = 3). ***p < 0.001
To understand the mechanisms involved in the transcriptional inhibition of Glut4, Mef2a and Pgc1α by ER stress, we sought to evaluate the role of Atf6, a gene whose expression has been reported to be upregulated during ER stress and may cause inhibition of gene expression via upregulation of the orphan nuclear receptor small heterodimer partner (SHP) in pancreatic beta cells [30]. To test the hypothesis that the overexpression of ATF6 is sufficient to impair GLUT4 expression, we generated L6 cell lines stably overexpressing Atf6 (Fig. 5a). In Atf6-overexpressing cells, Glut4 mRNA levels were reduced by 60% in basal condition (Fig. 5b), and were further decreased by 75% upon GlcN treatment (Fig. 5b). Atf6 overexpression also induced similar decreases of Mef2a and Pgc1a expression compared with control cells (data not shown). Treatment with a specific siRNA for Atf6 significantly inhibited the mRNA level of Atf6 in both untreated and GlcN-treated cells (Fig. 5c). As expected, the transfection of cells with an siRNA for a non-eukaryotic gene, used as negative control, did not affect the upregulation of Atf6 induced by GlcN (Fig. 5c). In addition, the siRNA for Atf6 completely prevented GlcN-induced downregulation of Glut4, Mef2a and Pgc1α (Fig. 5d). L6 cells were also transfected with an siRNA for Shp (also known as Nr0b2). As expected, the treatment with the siRNA for Shp significantly inhibited the mRNA level of Shp in both untreated and GlcN-treated cells (ESM Fig. 3a); by contrast, it was not able to prevent the GlcN-induced downregulation of Glut4, Mef2a and Pgc1α (ESM Fig. 3b). These data indicate that the activation of Atf6 is responsible for the impairment of Glut4 expression during GlcN-induced ER stress through a mechanism independent of Shp activation.

Role of ATF6 on GlcN-induced ER stress in L6 myotubes. a, b L6 cells were stably transfected with Atf6 cDNA. Atf6 (a) and Glut4 (b) mRNAs in clones are relative expression units (REU) to those in untransfected cells (L6) and were determined by real-time RT-PCR analysis using Gapdh as internal standard (mean ± SD; n = 3). ***p < 0.001. a Atf6 mRNA was detected in several clones. Two clones (termed clones Cl.a and Cl.d) overexpressing Atf6 by 2.5- and 3.5-fold were further characterised. b Glut4 mRNA was detected in L6 cells untransfected or stably transfected with Atf6 cDNA and treated with 7.5 mmol/l GlcN for 24 h. c, d L6 cells were transiently transfected with Atf6 siRNA or with a negative control siRNA, and then treated with 7.5 mmol/l GlcN for 24 h. Forty-eight hours after transfection, total RNA was extracted from transfected and non-transfected cells. mRNAs for Atf6 (c), Glut4, Mef2a and Pgc1α (d) in treated cells are relative expression units (REU) to those in control cells and were determined by real-time RT-PCR analysis using Gapdh as internal standard (mean ± SD; n = 3). ***p < 0.001
Finally, to evaluate GlcN effects on human skeletal muscle cells we used cultured human skeletal muscle cells that display several features of mature skeletal muscle and that have been previously used for studies of muscle metabolism [20]. In differentiated human muscle cells, GlcN induced a significant increase of both BIP/GRP78 and ATF6 mRNA levels (Fig. 6a, b), indicating that also the human skeletal muscle cells were sensitive to GlcN-induced ER stress. In addition, pretreatment with PBA completely prevented the increase of both BIP/GRP78 and ATF6 mRNAs observed upon GlcN treatment (Fig. 6a, b), confirming in human skeletal muscle cells the results obtained in the rat cells. In human myotubes, GlcN treatment reduced the expression of GLUT4 MEF2A and PGC1α mRNAs (Fig. 6c–e), and completely inhibited the uptake of 2-DG upon insulin stimulation compared with control cells (Fig. 6f). Furthermore, human myotubes treated with GlcN in the presence of PBA exhibited no differences in the mRNA expression of both GLUT4 and its upstream regulators MEF2A and PGC1α (Fig. 6c–e) and a rescue of insulin-induced 2-DG uptake (Fig. 6f) compared with control cells. Thus, GlcN-induced ER stress impairs insulin sensitivity also in human skeletal muscle, at least in part by inhibiting GLUT4, MEF2A and PGC1α expression.

GlcN effects in human myotubes. a–f Human myotubes were pretreated or not with 10 mmol/l PBA for 1 h, and then were treated with 7.5 mmol/l GlcN for 24 h. BIP/GRP78 (a), ATF6 (b), GLUT4 (c), MEF2A (d) and PGC1α (e) mRNAs were determined by real-time RT-PCR analysis of total RNA isolated from human myotubes, using GAPDH as internal standard. mRNA levels in treated cells are relative expression units (REU) to those in control (mean ± SD; n = 4). **p < 0.01; ***p < 0.001. f 2-DG uptake was measured following 30 min of insulin stimulation (mean ± SD; n = 5). ***p < 0.001
Discussion
Glucose toxicity may contribute to impaired insulin action in diabetes [22]. A widely accepted hypothesis regarding the mechanism responsible for glucose-induced insulin resistance is that glucose toxicity is mediated by increased flux of glucose into the HBP [17, 26, 27]. Other studies have shown that GlcN, but not HG, causes impairment of insulin-stimulated IRS-1 tyrosine phosphorylation and phosphoinositide-3-kinase activation, and that defective protein kinase B activation by insulin is associated with glucose- but not GlcN-mediated insulin resistance in adipocytes [31]. Nevertheless, studies in several model systems, including overexpression of GFAT and infusion/treatment with GlcN have confirmed that increased flux through HBP can lead to impaired glucose metabolism [27, 32]. Thus it seems likely that a role for the HBP in the development of the metabolic syndrome and insulin resistance will prevail. According to this, it has been reported that GlcN infusion in rats induces insulin resistance to glucose uptake at the level of both the whole body and skeletal muscle [33]. Furthermore, GlcN induces insulin resistance in vitro in skeletal muscle [34] and adipose cells [35], by reducing the insulin-induced glucose uptake.
Many investigators have suggested the reversible O-acetylglucosamine modification of proteins as one possible mechanism by which increased HBP activity may cause insulin resistance and diabetes complications. However, the causal relationship between increased flux through HBP and insulin resistance has not been clearly established. Recently, it has been reported that elevated GlcN levels could interfere with correct protein folding in the ER, inducing ER stress and impairment of cellular function in hepatic cells, monocytes and smooth muscle cells [36]. On the other hand, it has been described that ER stress and UPR activation may play an important role in the pathogenesis of type 2 diabetes [37], affecting both beta cell function/survival [11–13] and obesity-induced peripheral insulin sensitivity in liver and adipose tissue [15, 28]. However, little is known about the role of ER stress in the development of insulin resistance in skeletal muscle tissue.
A very recent study hypothesised a molecular convergence of activated HBP and ER stress pathways leading to insulin-resistance in L6 cells [38]. However, the causal link between the O-glycosylation pathway and the ER stress pathway in determining insulin resistance remained elusive. Indeed, cells silenced for O-linked N-acetylglucosamine transferase, the enzyme responsible for the addition of UDP-N-acetylglucosamine to Ser/Thr residues of proteins, and treated with GlcN or HG, showed improved insulin-stimulated glucose uptake without any effect on ER chaperone regulation [38].
In the present work, we suggest that ER stress may represent the molecular link between GlcN and insulin resistance in skeletal muscle cells. We show that high GlcN concentrations, as well as the ER stress inducer Thap, cause ER stress and the activation of the UPR in L6 rat skeletal muscle cells, as demonstrated by increased expression of the chaperone Bip/grp78 and of the transcription factor Atf6, the phosphorylation of eIF2α and the increase of both the expression and splicing of the transcription factor XBP-1. Furthermore, ER stress and UPR activation are induced by GlcN in a model of human skeletal muscle cells [20], suggesting that both rat and human skeletal muscle cells are sensitive to GlcN-induced ER stress. More interestingly, HG also induces ER stress in both L6 cells and human myotubes (data not shown). Pretreatment of cells with azaserine, a non-specific but commonly used inhibitor of GFAT [26, 27] prevented HG-induced ER stress, suggesting that at least in part HG levels cause ER stress through hexosamines production. In addition, others [34] and we have shown that L6 myotubes are insulin-resistant upon GlcN as well as HG treatment, as demonstrated by the significant decrease in their capability to take up the glucose analogue 2-DG upon insulin stimulation. To gain further insight into the mechanisms leading to insulin resistance, Bailey and Turner [34] evaluated GlcN-induced insulin-resistance in L6 myotubes using three different insulin-sensitive acting agents. None of these agents was able to prevent GlcN-induced insulin resistance [34], suggesting that GlcN does not impair insulin sensitivity by altering the upstream steps of insulin signalling. It has been shown that inhibition of insulin-stimulated glucose uptake by GlcN is due to intracellular ATP depletion in rat skeletal muscle [31], adipocytes [39] and chondrocytes [40]. However, in other cell types, ATP depletion by exposure to sodium azide or dinitrophenol did not mimic the effects of GlcN to induce insulin resistance [41]. Thus, ATP depletion is not the sole mechanism underlying all of the effects of GlcN. Indeed, we show that GlcN does not induce ATP depletion at the concentrations used in this study, suggesting that GlcN effects on both ER stress and insulin-resistance were not dependent on ATP depletion in skeletal muscle cells. Interestingly, we have found that pretreatment of both rat and human myotubes with PBA or TUDCA, two chemical chaperones known to prevent ER stress and the UPR activation in different cellular systems [15, 28], completely prevents the effect of GlcN and HG on both ER stress induction and insulin-induced glucose uptake, suggesting that GlcN-induced insulin-resistance is, at least in part, dependent on ER stress.
Glucose uptake into skeletal muscle is primarily mediated by GLUT4 [29]. Since it is well documented that insulin resistance in type 2 diabetes can be associated with a marked reduction in GLUT4 expression [42] and/or translocation [43], we hypothesised that GlcN-induced ER stress might affect transcription of the gene for GLUT4 in both rat and human muscle cells. Here we show that this is the case, as both GLUT4 gene mRNA and protein levels are decreased by 50% upon GlcN as well as upon Thap and HG treatment. Both the human and the rat GLUT4 gene promoter are regulated through the cooperative function of two distinct regulatory elements, domain 1 and MEF2 domain, each required for the maximal transcription of GLUT4 promoter. Domain 1 binds the transcription factor GEF (GLUT4 enhancer factor); MEF2 domain binds transcription factor isoforms MEF2A and MEF2D [42]. MEF2A and GEF associate and function together to activate GLUT4 gene transcription [44]. Little is known about GEF, whilst the role of MEF2A as the main regulator of GLUT4 gene is well documented. Indeed, MEF2A reduced activity correlates with decreased Glut4 transcription in skeletal muscle of diabetic mice and its activity is completely normalised after insulin treatment [45]. GLUT4 gene transcription can also be modulated in skeletal muscle by other proteins, such as PGC1α [46]. PGC1α is a coactivator of MEF2A. Indeed, MEF2D binds PGC1α, recruiting this transcriptional coactivator to MEF2A [42]. Moreover, PGC1α expression is reduced in skeletal muscle of prediabetic and diabetic individuals [47], and enhanced GLUT4 mRNA expression coincides with increased PGC1α mRNA in human skeletal muscle cell culture after treatment with rosiglitazone [46]. Our data, obtained in both rat and human myotubes, show that both GlcN- and HG-induced GLUT4 inhibition is paralleled by a significant decrease of both MEF2A and PGC1α gene mRNA expression, indicating that GlcN-induced GLUT4 inhibition is exerted very likely at the transcriptional level. Furthermore, mRNA stability of those genes does not appear to be affected by GlcN-dependent ER stress (data not shown). These observations were confirmed by ChIP and re-ChIP experiments, showing a reduced binding of both MEF2A and PGC1α to Glut4 promoter. Again, these effects appear to be mediated by ER stress signalling, since PBA or TUDCA pretreatment of myotubes is able to prevent GLUT4, MEF2A and PGC1α gene inhibition following both GlcN and Thap treatments. These observations prompted us to consider the GlcN-induced insulin resistance of skeletal muscle cells as a consequence of GLUT4 inhibition and, therefore, reduced membrane translocation (data not shown). However, the contribution of additional components known to be relevant to insulin resistance caused by ER stress in adipocytes and skeletal muscle cells [15, 38], such as JUN N-terminal kinase activation, cannot be excluded. Different proteins have been described to be activated by ER stress and to play a role in ER stress-mediated transcriptional repression. Very recently, indeed, ER stress-dependent activation of ATF6 has been reported to impair insulin gene expression in INS-1 pancreatic beta cells cultured in HG conditions or treated with different ER stressors, via upregulation of SHP [30]. We demonstrate that the overexpression of Atf6 is sufficient to inhibit the expression of Glut4, Mef2a and Pgc1α both in basal conditions and upon GlcN treatment and that the silencing of Atf6 expression with a specific siRNA is sufficient to completely prevent GlcN-induced downregulation of Glut4, Mef2a and Pgc1α. This effect seems to be independent of the upregulation of SHP, since its silencing does not modify Glut4, Mef2a and Pgc1α in skeletal muscle cells upon GlcN treatment.
It has been recently shown that ATF6 is responsible for cystic fibrosis transmembrane conductance regulator transcriptional repression by binding to its promoter [48]. Our analysis of the minimal promoter region of Glut4, Mef2a and Pgc1α (GenBank accession numbers: NC_005109.2, NW_001084766.1 and NC_005113.2), using MatInspector, identified several putative binding sites for ATF6 and for other UPR regulatory factors. Further studies will be necessary to understand if ATF6 could repress Glut4, Mef2a and Pgc1α expression in our cell models through a similar mechanism.
In conclusion, in this work we show that GlcN- as well as HG-induced ER stress causes insulin resistance in both human and rat myotubes and impairs GLUT4 gene expression and insulin-induced glucose uptake via an ATF6-dependent decrease of the GLUT4 regulator genes for MEF2A and PGC1α. Interestingly, treatment with the molecular chaperones PBA and TUDCA completely prevents HG- and GlcN-induced UPR activation and restores insulin sensitivity in myotubes. These findings are particularly relevant for understanding the molecular mechanisms of glucose toxicity in skeletal muscle and of the consequences of ER stress in the pathogenesis of type 2 diabetes.
Abbreviations
- 2-DG:
-
2-Deoxy-d-glucose
- ATF6:
-
Activating transcription factor 6
- BIP/GRP78:
-
Immunoglobulin heavy chain-binding protein/glucose-regulated protein 78 kDa
- ChIP:
-
Chromatin immunoprecipitation assay
- eIF2α:
-
Eukaryotic translation initiation factor 2α
- ER:
-
Endoplasmic reticulum
- GEF:
-
GLUT4 enhancer factor
- GlcN:
-
Glucosamine
- GFAT:
-
Glutamine:fructose-6-phosphate amidotransferase
- HBP:
-
Hexosamine biosynthetic pathway
- HG:
-
High glucose
- MEF2A:
-
Myocyte enhancer factor 2 a
- MnTBAP:
-
Mn(III)tetrakis(4-benzoic acid) porphyrin chloride
- NAC:
-
N-Acetyl-cysteine
- PBA:
-
4-Phenylbutyric acid
- PERK:
-
Double-stranded RNA-activated protein kinase-like ER kinase
- PGC1α:
-
Peroxisome proliferator-activated receptor-γ coactivator 1 α
- PUGNAc:
-
O-(2-Acetamido-2-deoxy-d-glucopyranosylidenamino)N-phenylcarbamate
- SHP:
-
Orphan nuclear receptor small heterodimer partner
- siRNA:
-
Small interfering RNA
- Thap:
-
Thapsigargin
- TUDCA:
-
Tauroursodeoxycholic acid
- UPR:
-
Unfolded protein response
- XBP-1s:
-
X-box binding protein-1, spliced active form
- XBP-1t:
-
X-box binding protein-1, total form
References
Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13:1211–1233
Liang SH, Zhang W, McGrath BC, Zhang P, Cavener DR (2006) PERK (eIF2alpha kinase) is required to activate the stress-activated MAPKs and induce the expression of immediate-early genes upon disruption of ER calcium homoeostasis. Biochem J 393:201–209
Mori K (2000) Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 101:451–454
Oyadomari S, Araki E, Mori M (2002) Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 7:335–345
Shi Y, Vattem KM, Sood R et al (1998) Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol 18:7499–7509
Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D (1998) Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J 17:5708–5717
Yoshida H, Haze K, Yanagi H, Yura T, Mori K (1998) Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem 273:33741–33749
Harding HP, Calfon M, Urano F, Novoa I, Ron D (2002) Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol 18:575–599
Kozutsumi MY, Segal M, Normington K, Gething MJ, Sambrook J (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332:462–464
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101:249–258
Kaneto H, Matsuoka TA, Nakatani Y et al (2005) Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J Mol Med 83:429–439
Scheuner D, Song B, McEwen E et al (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7:1165–1176
Shi Y, Taylor SI, Tan SL, Sonenberg N (2003) When translation meets metabolism: multiple links to diabetes. Endocr Rev 24:91–101
Nakatani Y, Kaneto H, Kawamori D et al (2005) Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem 280:847–851
Ozcan U, Cao Q, Yilmaz E et al (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461
Rossetti L (2000) Perspective: hexosamines and nutrient sensing. Endocrinology 141:1922–1925
Pirola L, Johnston AM, Van Obberghen E (2004) Modulation of insulin action. Diabetologia 47:170–184
Rossetti L, Hawkins M, Chen W, Gindi J, Barzilai N (1995) In vivo glucosamine infusion induces insulin resistance in normoglycemic but not in hyperglycaemic conscious rats. J Clin Invest 96:132–140
Bergstrom J (1975) Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest 35:609–616
Gaster M, Kristensen SR, Beck-Nielsen H, Schroder HD (2001) A cellular model system of differentiated human myotubes. APMIS 109:735–744
Gaster M, Schroder HD, Handberg A, Beck-Nielsen H (2001) The basal kinetic parameters of glycogen synthase in human myotube cultures are not affected by chronic high insulin exposure. Biochim Biophys Acta 1537:211–221
Cassese A, Esposito I, Fiory F et al (2008) In skeletal muscle advanced glycation end products (AGEs) inhibit insulin action and induce the formation of multimolecular complexes including the receptor for AGEs. J Biol Chem 283:36088–36099
Miele C, Riboulet A, Maitan MA et al (2003) Human glycated albumin affects glucose metabolism in L6 skeletal muscle cells by impairing insulin-induced insulin receptor substrate (IRS) signaling through a protein kinase C alpha-mediated mechanism. J Biol Chem 278:47376–47387
Ulianich L, Garbi C, Treglia AS et al (2008) ER stress is associated with dedifferentiation and an epithelial-to-mesenchymal transition-like phenotype in PC CI3 thyroid cells. J Cell Sci 121:477–486
Ungaro P, Teperino R, Mirra P et al (2008) Molecular cloning and characterization of the human PED/PEA-15 gene promoter reveal antagonistic regulation by hepatocyte nuclear factor 4alpha and chicken ovalbumin upstream promoter transcription factor II. J Biol Chem 283:30970–30979
Marshall S, Bacote V, Traxinger RR (1991) Discovery of a metabolic pathway mediating glucose induced desensitization of the glucose transport system: role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem 266:4706–4712
Buse MG (2006) Hexosamines, insulin resistance, and the complications of diabetes: current status. Am J Physiol Endocrinol Metab 290:E1–E8
Ozcan U, Yilmaz E, Ozcan L et al (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313:1137–1140
Klip A (2009) The many ways to regulate glucose transporter 4. Appl Physiol Nutr Metab 34:481–487
Seo HY, Kim YD, Lee KM et al (2008) Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases insulin gene expression via up-regulation of orphan nuclear receptor small heterodimer partner. Endocrinology 149:3832–3841
Han DH, Chen MM, Holloszy JO (2003) Glucosamine and glucose induce insulin resistance by different mechanisms in rat skeletal muscle. Am J Physiol Endocrinol Metab 285:E1267–E1272
Ciaraldi TP, Carter L, Nikoulina S, Mudaliar S, McClain DA, Henry RR (1999) Glucosamine regulation of glucose metabolism in cultured human skeletal muscle cells: divergent effects on glucose transport/phosphorylation and glycogen synthase in non-diabetic and type 2 diabetic subjects. Endocrinology 140:3971–3980
Patti ME, Virkamäki A, Landaker EJ, Kahn CR, Yki-Järvinen H (1999) Activation of the hexosamine pathway by glucosamine in vivo induces insulin resistance of early postreceptor insulin signalling events in skeletal muscle. Diabetes 48:1562–1571
Bailey CJ, Turner SL (2004) Glucosamine-induced insulin resistance in L6 muscle cells. Diabetes Obes Metab 6:293–298
Heart E, Choi WS, Sung CK (2000) Glucosamine-induced insulin resistance in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 278:E103–E112
Werstuck GH, Khan MI, Femia G et al (2006) Glucosamine-induced endoplasmic reticulum dysfunction is associated with accelerated atherosclerosis in a hyperglycaemic mouse model. Diabetes 55:93–101
Sundar Rajan S, Srinivasan V, Balasubramanyam M, Tatu U (2007) Endoplasmic reticulum (ER) stress and diabetes. Indian J Med Res 125:411–424
Srinivasan V, Tatu U, Mohan V, Balasubramanyam M (2009) Molecular convergence of hexosamine biosynthetic pathway and ER stress leading to insulin resistance in L6 skeletal muscle cells. Mol Cell Biochem 328:217–224
Hresko RC, Heimberg H, Chi MM, Mueckler M (1998) Glucosamine-induced insulin resistance in 3T3-L1 adipocytes is caused by depletion of intracellular ATP. J Biol Chem 273:20658–20668
Shikhman AR, Brinson DC, Valbracht J, Lotz MK (2009) Differential metabolic effects of glucosamine and N-acetylglucosamine in human articular chondrocytes. Osteoarthritis Cartilage 17:1022–1028
Kang J, Heart E, Sung CK (2001) Effects of cellular ATP depletion on glucose transport and insulin signaling in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 280:E428–E435
Armoni M, Harel C, Karnieli E (2007) Transcriptional regulation of the GLUT4 gene: from PPAR-gamma and FOXO1 to FFA and inflammation. Trends Endocrinol Metab 18:100–107
Shepherd PR, Kahn BB (1999) Glucose transporters and insulin action: implications for insulin resistance and diabetes mellitus. N Engl J Med 341:248–257
Knight JB, Eyster CA, Griesel BA, Olson AL (2003) Regulation of the human GLUT4 gene promoter: interaction between a transcriptional activator and myocyte enhancer factor 2A. Proc Natl Acad Sci U S A 100:14725–14730
Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM (2003) An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1 α expression in muscle. Proc Natl Acad Sci USA 100:7111–7116
Al-Khalili L, Forsgren M, Kannisto K, Zierath JR, Lönnqvist F, Krook A (2005) Enhanced insulin-stimulated glycogen synthesis in response to insulin, metformin or rosiglitazone is associated with increased mRNA expression of GLUT4 and peroxisomal proliferator activator receptor gamma co-activator 1. Diabetologia 48:1173–1179
Patti ME, Butte AJ, Crunkhorn S (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100:8466–8471
Bartoszewski R, Rab A, Twitty G et al (2008) The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J Biol Chem 283:12154–12165
Acknowledgements
This work was supported by the European Foundation for the Study of Diabetes (EFSD), the European Community’s FP6 EUGENE2 (LSHM-CT-2004-512013) and PREPOBEDIA (201681), grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC), and from the Ministero dell’Università e della Ricerca Scientifica (PRIN and FIRB). The financial support of Telethon-Italy is also gratefully acknowledged. I. Lynfort provided excellent technical assistance. The Danish Medical Research Council, the Novo Nordisk Foundation and the Danish Diabetes Association are thanked for financial support.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
G. A. Raciti and C. Iadicicco contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 65 kb)
ESM Table 1
(PDF 79 kb)
ESM Fig. 1
HG effects in L6 myotubes. a L6 cells were pretreated or not with 20 μmol/l azaserine for 20 min and then cultured in the presence of 25 mmol/l glucose or 100 μmol/l PUGNAc, as indicated, for 24 h. Bip/grp78 mRNA was determined by real-time RT-PCR analysis of total RNA isolated from myotubes, using Gapdh as internal standard. mRNA levels in treated cells are relative expression units (REU) to those in control (mean ± SD; n = 3). ***p < 0.001. b L6 cells were pretreated or not for 1 h with 10 mmol/l PBA and then treated with 25 mmol/l glucose for 24 h. 2-DG uptake was measured following 30 min of insulin stimulation (mean ± SD; n = 3). *p < 0.05; **p < 0.01. c L6 cells were treated with 25 mmol/l glucose for 24 h. Glut4, Mef2a and Pgc1α mRNAs were determined by real-time RT-PCR analysis, using Gapdh as internal standard. mRNA levels in treated cells are relative to those in control (mean ± SD; n = 3). **p < 0.01 (PDF 23 kb)
ESM Fig. 2
GlcN effects on intracellular ATP concentrations in L6 myotubes. L6 cells were treated or not with different concentrations of GlcN (5, 7.5, 10, 20 and 40 mmol/l) for 24 h. ATP content in treated cells is relative to that in control cells, which is expressed as 100% (mean ± SD; n = 3). ***p < 0.001 (PDF 11 kb)
ESM Fig. 3
Inhibition of SHP did not prevent ATF6-induced downregulation of Glut4, Mef2a and Pgc1α in L6 myotubes. L6 cells were transiently transfected with Shp siRNA or with a negative control siRNA, and then treated with 7.5 mmol/l GlcN for 24 h. Forty-eight hours after transfection, total RNA was extracted from transfected and non transfected cells. mRNAs for Shp (a) and Glut4, Mef2a and Pgc1α (b) in treated cells are relative expression units (REU) to those in control cells and were determined by real-time RT-PCR analysis using Gapdh as internal standard (mean ± SD; n = 3). ***p < 0.001 (PDF 29 kb)
Rights and permissions
About this article
Cite this article
Raciti, G.A., Iadicicco, C., Ulianich, L. et al. Glucosamine-induced endoplasmic reticulum stress affects GLUT4 expression via activating transcription factor 6 in rat and human skeletal muscle cells. Diabetologia 53, 955–965 (2010). https://doi.org/10.1007/s00125-010-1676-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1676-1