
Abstract
Bone remodelling, which maintains bone mass constant during adulthood, is an energy-demanding process. This, together with the observation that the adipocyte-derived hormone leptin is a major inhibitor of bone remodelling, led to the hypothesis that bone cells regulate energy metabolism through an endocrine mechanism. Studies to test this hypothesis identified osteocalcin, a hormone secreted by osteoblasts, as a positive regulator of insulin secretion, insulin resistance and energy expenditure. Remarkably, insulin signalling in osteoblasts is a positive regulator of osteocalcin production and activation via its ability to indirectly enhance bone resorption by osteoclasts. In contrast, leptin is a potent inhibitor of osteocalcin function through its effect on the sympathetic tone. Hence, osteocalcin is part of a complex signalling network between bone and the organs more classically associated with the regulation of energy homeostasis, such as the pancreas and adipose tissue. This review summarises the molecular and cellular bases of the present knowledge on osteocalcin biology and discusses the potential relevance of osteocalcin to human metabolism and pathology.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biological processes in all cells and organs require energy; bone and the specific cell types overseeing its growth and homeostasis show no exception to this rule. In fact, bone is a particularly energy-expensive organ because it constantly destroys and regenerates itself through a process termed bone remodelling. To fulfil this function, bone hosts two functionally antagonistic cell populations: osteoclasts, which resorb mineralised bone extracellular matrix; and osteoblasts, which deposit new matrix that eventually becomes mineralised. This succession of destruction and formation occurs constantly throughout the skeleton, requiring a constant supply of energy to both cell populations.
Given that the skeleton is one of the largest organs in mammals, one can easily appreciate how the bone remodelling process is metabolically expensive for the body. This observation raises an important physiological question: Is bone remodelling worth its high energetic cost? Looking at the sedentary lifestyle of 21st century man, bone remodelling does not appear to be an essential function. If anything, it is a problem-causing process, since its dysregulation causes one of the most frequent degenerative diseases of the Western hemisphere, osteoporosis. However, if one considers the larger scope of vertebrate evolution, bone remodelling gains considerable credit. By maintaining bone integrity, in particular by preventing microfractures caused by mechanical strains, bone remodelling is key to preserving mobility. In ancient times, mobility meant being able to feed, fight and flee dangers…in other words, survive. From this broader perspective, the energy cost of bone remodelling becomes justifiable since it contributes to the survival of the species. The high cost associated with bone remodelling then raises the possibility that bone cells may have a means of regulating the flow of energy supply/energy storage to fulfil their metabolic needs. Furthermore, when energy is scarce one can logically conceive that there should be mechanisms to restrict bone growth. Studies designed to test these hypotheses have led to the identification of osteocalcin, a novel hormone secreted and activated by bone cells, which is at the core of the cross-talk between bone remodelling and glucose metabolism.
Osteoblasts secrete osteocalcin, a hormone regulating glucose homeostasis
To identify putative bone-derived signals that could influence energy homeostasis, mutant mouse models lacking genes encoding osteoblast-specific secreted molecules were systematically analysed for abnormalities in glucose handling or other metabolic dysregulations [1]. This phenotyping screen identified mice deficient in osteocalcin, a small peptide uniquely secreted by osteoblasts that is present in the bone matrix and in the blood. Indeed, osteocalcin-deficient mice are hyperglycaemic, hypoinsulinaemic, have low beta cell mass, decreased insulin sensitivity, increased fat mass and decreased energy expenditure [1]. Conversely, the subcutaneous infusion of recombinant osteocalcin into wild-type mice causes an increase in blood insulin levels, enhances glucose tolerance and improves insulin sensitivity [2]. From a mechanistic point of view, islet perifusion assays showed that osteocalcin acts as an insulin secretagogue [3]. Gene expression analyses of islets or cultured beta cell lines further demonstrated that osteocalcin directly enhances the expression of the insulin genes as well as those encoding cyclin-dependent kinase 4 (Cdk4), cyclin D1 (Ccnd1) and D2 (Ccnd2) [1–3]. These findings explain the positive effect that osteocalcin has on both insulin production and beta cell proliferation. Likewise, in vitro and ex vivo analyses demonstrated that osteocalcin signals directly to adipocytes, where it promotes the expression of the gene encoding adiponectin (Adipoq) [1, 2]. Although a positive effect of osteocalcin on the expression of the genes for forkhead box protein A2 (Foxa2) in the liver and of acyl-CoA dehydrogenase (Mcad) in muscle has been reported, it is not yet known whether it signals directly to these two cell types [1, 2, 4]. Likewise, it is not clear how osteocalcin enhances insulin sensitivity and energy expenditure.

Osteocalcin is activated via its decarboxylation
Osteocalcin is secreted by osteoblasts following multiple post-translational modifications. These modifications include excision of a pre-pro peptide and the vitamin K-dependent γ-carboxylation of glutamic residues into Gla residues (hence osteocalcin’s other name of ‘bone Gla protein’) [5, 6]. These Gla residues confer high-affinity binding to hydroxyapatite, the mineral present in bone, explaining the high concentration of carboxylated osteocalcin found attached to the bone matrix [5]. However, both fully carboxylated and undercarboxylated forms of osteocalcin are present in the blood, raising the question of which is the biologically active form. In vitro experiments on isolated islets and primary adipocytes first revealed that fully carboxylated osteocalcin is inactive, while uncarboxylated osteocalcin is active [1]. It was then shown that recombinant (i.e. uncarboxylated) osteocalcin enhances insulin production and sensitivity when infused into wild-type mice, further indicating that this is an active form of the hormone [2]. Additional evidence was provided by the analysis of an apparently unrelated mouse model carrying mutations in Esp, a gene encoding embryonic stem cell specific phosphatase, an osteoblast-enriched protein tyrosine phosphatase. Esp-deficient mice show the mirror image phenotype of mice lacking the genes encoding osteocalcin (Ocn). They are lean, have increased energy expenditure, increased glucose tolerance and enhanced insulin sensitivity [1]. This metabolic phenotype was linked to an increase in the level of undercarboxylated osteocalcin (ucOC) in serum [1, 7, 8]. Although Esp is expressed in embryonic stem cells, Sertoli cells and osteoblasts [9], it is the ESP activity in osteoblasts that negatively regulates the decarboxylation of osteocalcin because mice deficient in Esp, whether globally or specifically in osteoblasts, present the same metabolic abnormalities [1]. Taken together, these studies indicate that the bioactive form of osteocalcin is undercarboxylated and that ESP acts as an inhibitor of osteocalcin decarboxylation.

The decarboxylation of osteocalcin is dependent on bone resorption
Osteocalcin is produced by osteoblasts in a fully carboxylated form, implying that osteocalcin needs to be decarboxylated to become active [5, 6]. However, there is no known extracellular or circulating γ-decarboxylase, suggesting that this process occurs through another mechanism. The elucidation of how ESP activity in osteoblasts is able to inhibit osteocalcin decarboxylation revealed this mechanism.
In addition to their metabolic abnormalities, the Esp-deficient mice also show an increase in bone resorption [7]. Co-culture assays determined that this is due to overactivity of the osteoclasts, which is prompted by the decreased secretion of osteoprotegerin (OPG)—a major inhibitor of osteoclast function—by Esp-deficient osteoblasts [10]. The significance of these findings for osteocalcin biology derives from two observations originally made at least two decades ago. The first is that carboxylated osteocalcin bound to the mineralised bone matrix via its Gla residues can be released upon resorption of this matrix by osteoclasts [11–14]. The second observation is that Gla residues can be decarboxylated when exposed to acid pH [15], and that bone resorption precisely involves acidification of the bone matrix. Accordingly, fully carboxylated osteocalcin incubated at a pH of 4.5, the acidity generated at the bone surface by osteoclasts during bone resorption [16], becomes sufficiently undercarboxylated to induce gene expression in beta cells [7]. Furthermore, media conditioned by osteoclasts that have been seeded onto devitalised bone matrix can stimulate insulin gene expression in beta cells, similar to recombinant osteocalcin [7]. These two in vitro observations suggested that bone resorption is simultaneously responsible for osteocalcin activation and for its release from the mineralised bone matrix, and this was confirmed in vivo using multiple mouse models in addition to pharmacological assays [7].
The oc/oc mice are a spontaneously arising mutant strain in which resorption is abrogated by an inability to generate the acid pH required for bone matrix digestion [17]. Analysis of these mutant mice showed that they are glucose intolerant, hypoinsulinaemic and have reduced blood levels of ucOC [7]. Importantly, the introduction one allele of the oc mutation into Esp-deficient mice normalises their levels of ucOC, as well as their metabolic phenotype [7]. The same normalisation can be observed in Esp-deficient mice treated with alendronate, an anti-resorptive agent belonging to the bisphosphonate class of drugs that is widely used for the treatment of osteoporosis [7, 18]. Together, these experiments demonstrate that the increase in bone resorption observed in mice lacking Esp is indeed at the origin of their osteocalcin-dependent metabolic phenotype. More generally, they show that bone resorption is a key mechanism promoting osteocalcin bioactivity, and that this arm of bone remodelling is also involved in regulating whole body glucose homeostasis. This explains why mice fed a high-fat diet and treated with a pro-resorptive agent show the same metabolic improvement as mice treated with recombinant osteocalcin itself [7].

Osteocalcin function is regulated by insulin and leptin
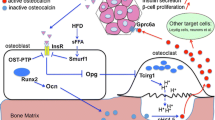
A basic principle of endocrinology is that hormonal regulation is under tight positive and negative control. The activity of osteocalcin is no exception to this rule. At the present time, two hormones that are known to control key aspects of energy homeostasis have been shown to respectively promote and inhibit osteocalcin production and/or activity (Fig. 1).

Schematic representation of the cross-talk between bone and energy metabolism. Carboxylated osteocalcin is secreted by osteoblasts and binds to the bone matrix. It is then released and activated through decarboxylation during bone resorption. Released undercarboxylated, i.e. active, osteocalcin reaches pancreatic beta cells to increase their proliferation and enhance the production and release of insulin. In turn, insulin stimulates osteocalcin production by osteoblasts and favours its activation through an osteoblast-dependent activation of bone resorption. In contrast, leptin secreted by adipocytes and signalling to brainstem neurons inhibits the production of brain serotonin, which itself is a negative regulation of sympathetic output, and this results in an increase of sympathetic tone. The sympathetic tone is a negative regulator of bone resorption, and this leptin-dependent regulatory mechanism inhibits osteocalcin activation
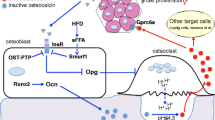
The first of these two hormones is insulin. Indeed, the insulin receptor (InsR) is a substrate of the phosphatase activity of ESP and, accordingly, mice lacking InsR specifically in osteoblasts are glucose intolerant and hypoinsulinaemic [7, 19]. From a mechanistic point of view, it was demonstrated that InsR signalling in osteoblasts has a dual and positive action on osteocalcin biology. One the one hand, InsR induces Ocn gene expression in osteoblasts by repressing the negative activity that the nuclear factor Twist2 exerts on Runx2, a transcriptional master gene of osteoblast differentiation and a major regulator of Ocn expression [19, 20]. On the other hand, through the phosphorylation-mediated destabilisation of FOXO1, InsR signalling in osteoblasts reduces the ability of FOXO1 to activate the Opg promoter and thereby decreases the secretion of this inhibitor of osteoclast function by these cells (Fig. 2) [7, 21]. In response to this stimulation of bone resorption, osteocalcin is released from the bone matrix and activated via decarboxylation. Thus, in a feed-forward loop, insulin signalling in osteoblasts promotes the production of active osteocalcin by bone cells, which in turn enhances insulin production and release by beta cells (Fig. 1).

Regulation of osteocalcin activation by ESP and insulin receptor signalling in osteoblasts. Osteocalcin is both released from the bone matrix and activated by the resorption activity of the osteoclasts, a process inhibited by OPG. The expression of the gene encoding OPG in osteoblasts is positively regulated by FOXO1, which is inactivated by insulin receptor signalling (green arrows). ESP (or PTP1B, its functional counterpart in humans) inhibits this process through dephosphorylation of the insulin receptor (blue arrows)
How is this feed-forward regulation counterbalanced? Studies investigating this identified a second hormone, leptin, as a negative regulator of osteocalcin activity. A large body of work has shown that both arms of bone remodelling, resorption and formation, are negatively controlled by leptin, an adipocyte-specific hormone that appeared during evolution at the same time as a remodelling skeleton [22]. To exert its functions, leptin binds to receptors expressed on brainstem neurons where it inhibits the synthesis of serotonin, a positive regulator of appetite and an inhibitor of catecholamine production that signals to hypothalamic neurons [23] (Fig. 1). Sympathetic output then inhibits bone formation by signalling through the β2 adrenergic receptor (Adrβ2), which is expressed by osteoblasts, although it does not decrease the expression of Ocn in these cells [3, 24]. In contrast, Adrβ2 signalling increases the production of RANKL, a cytokine exerting the opposite function of OPG on osteoclasts, and thus stimulates the proliferation and function of these cells (i.e. bone resorption) [4, 25]. Although this latter regulation should favour osteocalcin activation, the fact that Adrβ2 signalling also strongly favours Esp expression, and thus enhances OPG secretion via the InsR/FOXO1 pathway, offsets this effect [3, 7]. Accordingly, Adrβ2-deficient mice have low serum levels of ucOC and are glucose intolerant and insulin resistant [3]. These data established that leptin, through its stimulation of sympathetic output, negatively regulates osteocalcin activity.

Clinical relevance of the osteocalcin-mediated regulation of energy metabolism
That the role of osteocalcin in glucose metabolism and its modes of regulation were uncovered using mouse models raised concerns that this endocrine system could be different or even non-existent in humans. A fast growing number of studies, however, have now established that multiple aspects of osteocalcin biology are similar in rodents and humans. Indeed, levels of circulating osteocalcin have been inversely correlated with BMI, fat mass and plasma glucose levels in adult or elderly men and women of diverse ethnicities [26–29]. Additional studies have linked low osteocalcin to impaired levels of HbA1c, fasting insulin and insulin resistance (estimated by the HOMA of insulin resistance HOMA-IR) in adult men and women, irrespective of whether they have diabetes [28, 30–33]. In contrast, serum osteocalcin has been positively correlated with the HOMA of beta cell function (HOMA-%B) in diabetic patients, before and after glycaemic control [33, 34]. Interestingly, plasma levels of osteocalcin are higher in women with gestational diabetes and positively correlate with the disposition index during pregnancy, but return to normal postpartum [35]. This observation suggests that raising serum osteocalcin levels could be part of the adaptive process initiated to counter glucose intolerance during gestational diabetes [35].
Levels of circulating osteocalcin have also been associated with a number of lipid abnormalities. For instance, serum osteocalcin levels are inversely correlated with levels of adipocyte-specific fatty acid-binding protein (A-FABP) in a mixed population, and with levels of HDL-cholesterol in men, and are positively correlated with total adiponectin levels in post-menopausal women [27, 31, 34]. In addition, an association of decreased levels of osteocalcin with premature myocardial infarction in young patients or with coronary heart disease in older individuals have been reported [34, 36].
More generally, obese individuals have been shown to have lower osteocalcin levels than non-obese controls, and type 2 diabetic individuals have lower plasma osteocalcin than non-diabetic individuals [26, 27, 37]. Higher levels of osteocalcin were also associated with a lower odds ratio of developing metabolic syndrome in black and in white non-Hispanic individuals [29, 38]. A case–control study actually identified coding variants in the fourth exon of human OCN that appeared to correlate with type 2 diabetes in African-American patients [39]. However, this finding should be interpreted cautiously as recent genome-wide association studies of large populations, including one specifically performed on an African-American cohort, did not report an association between variants in the human OCN gene and fasting blood glucose or other traits of diabetes [40, 41].
More recently, studies have begun to assess whether ucOC might be the active form of this hormone in human metabolism, as it is in mice. It has long been known that multiple proteolytic products and undercarboxylated forms of osteocalcin are present in human serum [42, 43]. However, the lack of homogeneity and established specificity of commercially available assays used to detect ucOC considerably limit the study of ucOC [43–46]. Yet, it was recently shown that ucOC levels are correlated with several metabolic variables. Indeed, higher levels of ucOC are associated with higher insulin secretion, as measured by HOMA-%B, and higher levels of high-molecular-weight adiponectin in healthy children, while lower concentrations of ucOC are associated with impaired fasting glucose and impaired glucose tolerance in children [47, 48]. It has also been reported that, in adult men, serum ucOC is associated with enhanced beta cell function and negatively correlated with fat mass, fasting plasma glucose and HbA1c [30, 49]. Two additional observations support a metabolic role for ucOC in humans by showing that the mechanisms controlling osteocalcin decarboxylation are conserved between humans and rodents. First, although there is no functional Esp gene in humans, the role of ESP in controlling OPG expression is fulfilled in human osteoblasts by another phosphatase, PTP1B (Fig. 2) [7]. Second, patients with impaired bone resorption, the mechanism responsible for osteocalcin decarboxylation in mice, display a decrease in postprandial insulin serum levels [7].
A number of studies are beginning to test the dynamics of osteocalcin production as a marker of energy and/or glucose metabolism in interventional studies. For example, it was recently shown that significant weight loss causes a decrease in HOMA-IR as well as an increase in osteocalcin levels in obese children [37]. In non-diabetic adults, weight loss associated with diet and exercise also increased osteocalcin levels in parallel with a decrease in visceral fat and improved insulin sensitivity [50]. Interestingly, acute aerobic, but not power exercise could increase serum ucOC in obese individuals [51].

Therapeutic potential of osteocalcin in the treatment of type 2 diabetes
Mouse genetic data have established osteocalcin as a positive regulator of insulin production and insulin sensitivity. There is also a significant body of clinical correlations between low levels of osteocalcin and various measures associated with diabetes. These combined data suggest that raising serum osteocalcin levels could be a novel therapeutic avenue to prevent or treat diabetes.
At the present time, the therapeutic potential of osteocalcin has only been tested in vivo in two mouse models of metabolic dysregulation. In a first set of experiments, recombinant osteocalcin or vehicle was infused subcutaneously into mice fed a high-fat diet. The mice receiving osteocalcin gained significantly less weight, were less glucose intolerant and were less insulin resistant than mice receiving vehicle [2]. Osteocalcin-treated mice also showed normalised triacylglycerol levels. In a second experiment, mice were pre-treated with gold thioglucose, a chemical destroying neurons of the arcuate nuclei that control satiety, and thus developed hyperphagia. While vehicle-treated mice gained weight and became glucose intolerant and insulin resistant, osteocalcin-treated mice remained normoglycaemic and had normal response to glucose and insulin [2]. In both cases, the beneficial effects of osteocalcin therapy occurred without a change in appetite in the treated mice, a feature that is consistent with the lack of effect of osteocalcin on the expression of the gene encoding leptin or production of the protein [1, 2]. Although of very limited scope, the successful outcome of these studies is a strong incentive to further evaluate the therapeutic potential of osteocalcin.

Conclusion
The identification of osteocalcin as a novel hormone regulating glucose metabolism and the notion that bone is an endocrine organ opens the door to a new field of investigation. Indeed, not only does the biology of this new hormone need to be explored further, but the existence of additional interactions between bone, pancreas and most likely other organs also need to be identified. From this perspective, the identification of the osteocalcin receptor will likely be a pivotal step.
Additional studies are also necessary to evaluate the impact of the bone metabolism connection in humans. For example, it was recently shown that postmenopausal osteoporotic women treated with alendronate, which inhibits bone resorption, have lower levels of osteocalcin [45]. It would therefore be important to test whether anti-resorptive treatments, which are the main therapeutic approach to osteoporosis at the present time, might in some cases have a negative effect on glucose tolerance. Likewise, anticoagulating agents that affect the function of vitamin K, and therefore the γ-carboxylation of osteocalcin [8], could potentially have an unrecognised effect on glucose metabolism. The view that osteocalcin is only a marker of bone formation should also be revisited since, at least in some cases, variations in its serum levels might reflect a transient metabolic adaptation rather than a primary effect on bone formation. In line with this notion, there was a negative association of osteocalcin levels with impaired fasting glucose and impaired glucose tolerance in overweight children but not with bone mineral content [48]. More generally, analysis of the effects of some glucose-lowering agents on the production or activation of osteocalcin may provide novel insights on their mechanism of action.
Abbreviations
- HOMA-%B:
-
HOMA of beta cell function
- HOMA-IR:
-
HOMA of insulin resistance
- InsR:
-
Insulin receptor
- ucOC:
-
Undercarboxylated osteocalcin
- OPG:
-
Osteoprotegerin
References
Lee NK, Sowa H, Hinoi E et al (2007) Endocrine regulation of energy metabolism by the skeleton. Cell 130:456–469
Ferron M, Hinoi E, Karsenty G, Ducy P (2008) Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci USA 105:5266–5270
Hinoi E, Gao N, Jung DY et al (2008) The sympathetic tone mediates leptin’s inhibition of insulin secretion by modulating osteocalcin bioactivity. J Cell Biol 183:1235–1242
Yoshizawa T, Hinoi E, Jung DY et al (2009) The transcription factor ATF4 regulates glucose metabolism in mice through its expression in osteoblasts. J Clin Invest 119:2807–2817
Hauschka PV, Lian JB, Cole DE, Gundberg CM (1989) Osteocalcin and matrix Gla protein: vitamin K-dependent proteins in bone. Physiol Rev 69:990–1047
Price PA (1989) Gla-containing proteins of bone. Connect Tissue Res 21:51–57, discussion 57–60
Ferron M, Wei J, Yoshizawa T et al (2010) Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 142:296–308
Ferron M, Wei J, Yoshizawa T, Ducy P, Karsenty G (2010) An ELISA-based method to quantify osteocalcin carboxylation in mice. Biochem Biophys Res Commun 397:691–696
Dacquin R, Mee PJ, Kawaguchi J et al (2004) Knock-in of nuclear localised β-galactosidase reveals that the tyrosine phosphatase Ptprv is specifically expressed in cells of the bone collar. Dev Dyn 229:826–834
Teitelbaum SL, Ross FP (2003) Genetic regulation of osteoclast development and function. Nat Rev Genet 4:638–649
Price PA, Williamson MK, Epstein DJ (1981) Specific tritium incorporation into γ-carboxyglutamic acid in proteins. The pH dependence of γ-proton exchange. J Biol Chem 256:1172–1176
Price PA, Poser JW, Raman N (1976) Primary structure of the γ-carboxyglutamic acid-containing protein from bovine bone. Proc Natl Acad Sci USA 73:3374–3375
Gundberg CM, Weinstein RS (1986) Multiple immunoreactive forms of osteocalcin in uremic serum. J Clin Invest 77:1762–1767
Ivaska KK, Hentunen TA, Vaaraniemi J, Ylipahkala H, Pettersson K, Vaananen HK (2004) Release of intact and fragmented osteocalcin molecules from bone matrix during bone resorption in vitro. J Biol Chem 279:18361–18369
Engelke JA, Hale JE, Suttie JW, Price PA (1991) Vitamin K-dependent carboxylase: utilization of decarboxylated bone Gla protein and matrix Gla protein as substrates. Biochim Biophys Acta 1078:31–34
Silver IA, Murrills RJ, Etherington DJ (1988) Microelectrode studies on the acid microenvironment beneath adherent macrophages and osteoclasts. Exp Cell Res 175:266–276
Scimeca JC, Franchi A, Trojani C et al (2000) The gene encoding the mouse homologue of the human osteoclast-specific 116-kDa V-ATPase subunit bears a deletion in osteosclerotic (oc/oc) mutants. Bone 26:207–213
Fisher JE, Rogers MJ, Halasy JM et al (1999) Alendronate mechanism of action: geranylgeraniol, an intermediate in the mevalonate pathway, prevents inhibition of osteoclast formation, bone resorption, and kinase activation in vitro. Proc Natl Acad Sci USA 96:133–138
Fulzele K, Riddle RC, DiGirolamo DJ et al (2010) Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 142:309–319
Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89:747–754
Puigserver P, Rhee J, Donovan J et al (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1–PGC-1α interaction. Nature 423:550–555
Wei J, Ducy P (2010) Co-dependence of bone and energy metabolisms. Arch Biochem Biophys 503:35–40
Yadav VK, Oury F, Suda N et al (2009) A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell 138:976–989
Takeda S, Elefteriou F, Levasseur R et al (2002) Leptin regulates bone formation via the sympathetic nervous system. Cell 111:305–317
Elefteriou F, Ahn JD, Takeda S et al (2005) Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature 434:514–520
Kindblom JM, Ohlsson C, Ljunggren O et al (2009) Plasma osteocalcin is inversely related to fat mass and plasma glucose in elderly Swedish men. J Bone Miner Res 24:785–791
Lee YJ, Lee H, Jee SH et al (2010) Serum osteocalcin is inversely associated with adipocyte-specific fatty acid-binding protein in the Korean metabolic syndrome research initiatives. Diabetes Care 33:e90
Pittas AG, Harris SS, Eliades M, Stark P, Dawson-Hughes B (2009) Association between serum osteocalcin and markers of metabolic phenotype. J Clin Endocrinol Metab 94:827–832
Saleem U, Mosley TH Jr, Kullo IJ (2010) Serum osteocalcin is associated with measures of insulin resistance, adipokine levels, and the presence of metabolic syndrome. Arterioscler Thromb Vasc Biol 30:1474–1478
Hwang YC, Jeong IK, Ahn KJ, Chung HY (2009) The uncarboxylated form of osteocalcin is associated with improved glucose tolerance and enhanced beta-cell function in middle-aged male subjects. Diabetes Metab Res Rev 25:768–772
Kanazawa I, Yamaguchi T, Yamamoto M et al (2009) Serum osteocalcin level is associated with glucose metabolism and atherosclerosis parameters in type 2 diabetes mellitus. J Clin Endocrinol Metab 94:45–49
Im JA, Yu BP, Jeon JY, Kim SH (2008) Relationship between osteocalcin and glucose metabolism in postmenopausal women. Clin Chim Acta 396:66–69
Bao YQ, Zhou M, Zhou J et al (2011) Relationship between serum osteocalcin and glycemic variability in type 2 diabetes. Clin Exp Pharmacol Physiol 38:50–54
Zhou M, Ma X, Li H et al (2009) Serum osteocalcin concentrations in relation to glucose and lipid metabolism in Chinese individuals. Eur J Endocrinol 161:723–729
Winhofer Y, Handisurya A, Tura A et al (2010) Osteocalcin is related to enhanced insulin secretion in gestational diabetes mellitus. Diabetes Care 33:139–143
Goliasch G, Blessberger H, Azar D et al (2010) Markers of bone metabolism in premature myocardial infarction (≤40 years of age). Bone 48:622–626
Reinehr T, Roth CL (2010) A new link between skeleton, obesity and insulin resistance: relationships between osteocalcin, leptin and insulin resistance in obese children before and after weight loss. Int J Obes (Lond) 34:852–858
Yeap BB, Chubb SA, Flicker L et al (2010) Reduced serum total osteocalcin is associated with metabolic syndrome in older men via waist circumference, hyperglycemia, and triglyceride levels. Eur J Endocrinol 163:265–272
Das SK, Sharma NK, Elbein SC (2010) Analysis of osteocalcin as a candidate gene for type 2 diabetes (T2D) and intermediate traits in Caucasians and African Americans. Dis Markers 28:281–286
Bonnefond A, Froguel P, Vaxillaire M (2010) The emerging genetics of type 2 diabetes. Trends Mol Med 16:407–416
Ramos E, Chen G, Shriner D et al (2010) Replication of genome-wide association studies (GWAS) loci for fasting plasma glucose in African-Americans. Diabetologia 54:783–788
Cairns JR, Price PA (1994) Direct demonstration that the vitamin K-dependent bone Gla protein is incompletely gamma-carboxylated in humans. J Bone Miner Res 9:1989–1997
Vergnaud P, Garnero P, Meunier PJ, Breart G, Kamihagi K, Delmas PD (1997) Undercarboxylated osteocalcin measured with a specific immunoassay predicts hip fracture in elderly women: the EPIDOS Study. J Clin Endocrinol Metab 82:719–724
Nimptsch K, Hailer S, Rohrmann S, Gedrich K, Wolfram G, Linseisen J (2007) Determinants and correlates of serum undercarboxylated osteocalcin. Ann Nutr Metab 51:563–570
Aonuma H, Miyakoshi N, Hongo M, Kasukawa Y, Shimada Y (2009) Low serum levels of undercarboxylated osteocalcin in postmenopausal osteoporotic women receiving an inhibitor of bone resorption. Tohoku J Exp Med 218:201–205
Lee AJ, Hodges S, Eastell R (2000) Measurement of osteocalcin. Ann Clin Biochem 37:432–446
Prats-Puig A, Mas-Parareda M, Riera-Perez E et al (2010) Carboxylation of osteocalcin affects its association with metabolic parameters in healthy children. Diabetes Care 33:661–663
Pollock NK, Bernard PJ, Wenger K et al (2010) Lower bone mass in prepubertal overweight children with prediabetes. J Bone Miner Res 25:2484–2493
Kanazawa I, Yamaguchi T, Yamauchi M et al (2010) Serum undercarboxylated osteocalcin was inversely associated with plasma glucose level and fat mass in type 2 diabetes mellitus. Osteoporos Int 22:187–194
Fernandez-Real JM, Izquierdo M, Ortega F et al (2009) The relationship of serum osteocalcin concentration to insulin secretion, sensitivity, and disposal with hypocaloric diet and resistance training. J Clin Endocrinol Metab 94:237–245
Levinger I, Zebaze R, Jerums G, Hare DL, Selig S, Seeman E (2011) The effect of acute exercise on undercarboxylated osteocalcin in obese men. Osteoporos Int 22:1621–1626
Duality of interest
The author declares that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ducy, P. The role of osteocalcin in the endocrine cross-talk between bone remodelling and energy metabolism. Diabetologia 54, 1291–1297 (2011). https://doi.org/10.1007/s00125-011-2155-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2155-z