
Abstract
A recent unexpected development of bone biology is that bone is an endocrine organ regulating a growing number of physiological processes. One of the functions regulated by bone through the hormone osteocalcin is glucose homeostasis. In this overview, we will explain why we hypothesized that bone mass and energy metabolism should be subjected to a coordinated endocrine regulation. We will then review the experiments that revealed the endocrine function of osteocalcin and the cell biology events that allow osteocalcin to become a hormone. We will also illustrate the importance of this regulation to understand whole-body glucose homeostasis in the physiological state and in pathological conditions. Lastly, we will mention epidemiological and genetic evidence demonstrating that this function of osteocalcin is conserved in humans.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The hypothesis that bone is an endocrine organ regulating, among other functions, energy metabolism arises from a conceptual view of bone biology. We interpreted the fact that bone is the only tissue that contains a cell type, the osteoclast, whose only function is to destroy or to resorb mineralized bone matrix as being of fundamental importance. By definition, because it occurs daily in a tissue that covers a large surface in our body, this active destruction of mineralized bone requires energy and the quantity of energy that this active destruction of mineralized bone requires is proportional to the surface occupied by bone; this energetic requirement is probably very high. Moreover, bone resorption does not occur in isolation but in the context of a biphasic physiological function called bone modeling during childhood and bone remodeling during adulthood [1]. In this function, bone resorption is followed by bone formation, a cellular process that relies on the daily synthesis of proteins; hence, a cellular process that also requires energy [2]. This is why we have hypothesized that bone modeling and remodeling have to be linked to the regulation of energy metabolism. Importantly, this view of bone biology that infers a coordinated regulation of bone mass accrual and energy metabolism is supported by clinical observations. For instance, when food, i.e., energy intake, is severely decreased during childhood, there is an arrest of growth; when this situation develops in adults, there is bone loss [3–13].
An Unexpected Experimental Observation
The hypothesis that there may be a coordinated regulation of bone mass accrual and energy metabolism would probably not have been tested any further if it was not for unexpected experimental evidence. Osteocalcin is an osteoblast-specific gene encoding a secreted protein that was identified in the late 1970s but whose functions in the pre-model organism era of biology were unknown [14–16]. Hypothesizing, probably naively and as it turned out wrongly, that it may regulate bone mineralization, we generated 20 years ago Osteocalcin-null mice [17, 18]. The first result that we obtained was certainly disappointing to us since bone mineralization is essentially normal in the absence of Osteocalcin [18, 19]. That is, not to say, however, that osteocalcin has no functions or that the Osteocalcin−/− mice had no phenotype. Indeed, every time that we sacrificed Osteocalcin−/− mice, we made the same observation: they had visibly more visceral fat than wild-type littermates. Since osteocalcin is only made in osteoblasts, these observations inferred from the onset that bone might be an endocrine organ and that one hormone it secretes, osteocalcin, somehow affects fat mass. It is this experimental observation that, when confronted to the conceptual view of bone (re)modeling and the clinical observations presented above, led us to propose that there is a coordinated regulation, endocrine in nature, of bone mass, energy metabolism, and reproduction. An inference of fundamental importance of this hypothesis is that bone should be an endocrine organ and not just a recipient of hormonal intake. This latter tenet of the hypothesis was consistent with the phenotypic abnormalities seen in Osteocalcin−/− mice.
Identification of Osteocalcin as a Hormone Regulating Insulin Secretion
An additional and serendipitous observation made 10 years later further suggested that the osteoblast is an endocrine cell type regulating energy metabolism and, more specifically, glucose metabolism. We had generated mice lacking a tyrosine phosphatase expressed only in osteoblasts and Sertoli cells of the testis, hence its name, osteoblast testis-specific protein tyrosine phosphatase (OST-PTP) encoded by a gene termed Esp [20, 21]. Importantly, whether the gene was deleted in all cells or in osteoblasts only, Esp−/− mice exhibited the same phenotype made of hypoglycemia, hyperinsulinemia, and increased glucose utilization by peripheral tissues [22••]. Moreover, mice lacking Esp in all cells or in osteoblasts only had much less visceral fat. These findings were established in an unambiguous manner that the osteoblast was an endocrine cell type regulating one particular aspect of energy metabolism: glucose homeostasis. However, since OST-PTP is not a secreted protein, these observations also implied the existence of another molecule, presumably a hormone, made by osteoblasts and regulating glucose homeostasis. The fact that Esp−/− mice had a low fat mass phenotype, i.e., a phenotype that was exactly the mirror image of what was observed in Osteocalcin−/− mice, led us to test whether OST-PTP could inhibit osteocalcin function. This revived our interest in the hypothetical endocrine function of osteocalcin but, this time, with a more defined and testable hypothesis.
The demonstration that osteoblasts are endocrine cells stimulating insulin secretion and that this function was fulfilled by osteocalcin came from a classical cell biology experiment [22••]. Indeed, a co-culture of mouse osteoblasts and mouse pancreatic islets resulted in an increase in Insulin expression in islets. Several controls indicated that this was demonstrating a specific function of the osteoblast. For instance, when this co-culture experiment was performed using a filter allowing transfer of small molecules but preventing cell-cell contact, the increase of Insulin expression in islets co-cultured with osteoblasts was still observed. In contrast, this effect was specific of osteoblasts since the closest relative to an osteoblast, a fibroblast, could not enhance Insulin expression in pancreatic islets. Third, osteoblasts did not increase the expression of any other hormones synthesized by pancreatic islets. Last but not least, when this experiment was repeated with Osteocalcin−/− osteoblasts instead of wild-type (WT) ones, the favorable effect of osteoblasts on Insulin expression was virtually abolished although not completely. Conversely, when Esp−/− osteoblasts were used in this assay, the increase in Insulin expression was significantly greater than when islets were co-cultured with WT osteoblasts. The notion that osteocalcin was an osteoblast-derived hormone regulating insulin secretion was further strengthened by showing that forced expression of Osteocalcin in COS cells conferred to these cells an ability to induce insulin secretion in a co-culture assay that they did not have otherwise.
Thus, these cell biology experiments demonstrated that osteoblasts are endocrine cells regulating Insulin expression, identified osteocalcin as an osteoblast-derived hormone responsible of this function, and revealed the existence of a genetic pathway taking place in osteoblasts and in which Esp inhibits, through mechanism that had to be uncovered, the ability of osteoblasts to favor Insulin expression in pancreatic islets. All these conclusions were verified in vivo through genetic means.
Going back to an in vivo analysis, we then showed that Osteocalcin−/− mice fed a normal chow were hyperglycemic and hypoinsulinemic [22••]. A glucose-stimulated insulin secretion test (GSIS) showed that insulin secretion was decreased in the absence of Osteocalcin, whereas it was increased in the absence of Esp. Consistent with this observation, β-cell mass, β-cell area, and insulin content were decreased in Osteocalcin−/− mice and increased in Esp−/− mice. A glucose tolerance test (GTT) showed that Osteocalcin−/− mice were glucose intolerant, in part, because of a decrease in Insulin expression. Again, Esp−/− mice had exactly the opposite phenotype. Lastly and although it is not directly relevant to the topic of this review that focuses only on the bone-pancreas crosstalk, an insulin tolerance test (ITT) and euglycemic hyperinsulinemia clamp analysis showed that Osteocalcin−/− mice were resistant to insulin signaling in several peripheral tissues while Esp−/− mice were more sensitive to insulin signaling than WT mice.
The demonstration that Esp acts upstream of osteocalcin and inhibits its endocrine functions was provided by a genetic epistasis experiment. We hypothesized that if the reason why Esp−/− mice are able to secrete more insulin following a glucose challenge and are more tolerant to glucose than WT mice is because they have higher circulating osteocalcin levels, then removing one copy of Osteocalcin from these Esp−/− mice should normalized osteocalcin circulating levels, insulin secretion, and glucose tolerance. This prediction was verified by the analysis of Esp−/−;Osteocalcin+/− mice that have a normal glucose homeostasis and normal circulating osteocalcin levels. In other words, Esp−/− mice are a mouse model of gain of osteocalcin function. This turned out to be quite important, as it provided an internal control for all subsequent experiments addressing one function or another of osteocalcin. In each case, we confronted a loss-of-function model, the Osteocalcin−/− mice, and a gain-of-function model, the Esp−/− mice, that should have the opposite phenotype. In closing, we must clearly reiterate that osteocalcin regulation of glucose metabolism is not synonymous of bone only origin of diabetes. It is simply a broadening of our understanding of the regulation of glucose metabolism and of bone biology as a whole.
A Post-Translational Modification Modulates Osteocalcin Bioactivity
The demonstration that osteocalcin is a hormone that regulates insulin secretion and glucose homeostasis raises novel questions, for instance: How does osteocalcin act to regulate insulin secretion and glucose tolerance; in other words, what is its receptor? Can we extend these mouse-based findings to human? Two other questions were more specific to osteocalcin biology. Given that osteocalcin is subjected to a post-translational modification, a gamma carboxylation of some glutamate residues, what exactly is the mechanism whereby OST-PTP, the gene product of Esp, inhibits osteocalcin endocrine functions and which form of osteocalcin is fulfilling its endocrine functions? For obvious reasons that have to do with experimental simplicity, this latter question was the first one to be answered.
Osteocalcin is carboxylated on three glutamine acid residues within the osteoblasts before being released into the bone extracellular matrix; however, both the carboxylated and uncarboxylated forms of osteocalcin can be found in the general circulation [23]. Since the gamma carboxylase enzyme responsible of this post-translational modification is not expressed in bacteria, the use of recombinant, bacterially produced osteocalcin, allowed to address this aspect of osteocalcin biology. Only recombinant, i.e., uncarboxylated osteocalcin, induces Insulin expression in pancreatic islets, thus indicating that it is the uncarboxylated form of osteocalcin that is acting as a hormone [22••, 24•]. Consistent with this notion, this form of osteocalcin is significantly more abundant in the serum of Esp−/− mice than in the serum of WT mice [25].
Identification of Insulin Signaling in Osteoblasts as a Mean to Regulate Osteocalcin Bioactivity
How could Esp that encodes an intracellular enzyme regulate the activity of a secreted molecule like osteocalcin? This was a more burning question considering that OST-PTP is a tyrosine phosphatase, but neither osteocalcin nor the enzymes responsible of its carboxylation are phosphorylated [26••]. We then considered the possibility that OST-PTP could dephosphorylate, i.e., inactivate, the insulin receptor in osteoblasts. An implication of this hypothesis is that insulin should be a positive regulator of osteocalcin bioactivity; in other words, insulin signaling in osteoblasts should be necessary for whole-body glucose homeostasis in animals fed a normal diet.
Biochemical and genetic evidence gathered in cells and in vivo verified that OST-PTP dephosphorylates the insulin receptor in osteoblasts [26••]. As inferred by these data and as hypothesized, mice lacking the Insulin receptor only in osteoblasts displayed, when fed a normal chow, a decrease in circulating levels of the active form of osteocalcin, a decrease in insulin secretion, glucose intolerance, and insulin resistance. Molecularly, insulin signaling in osteoblasts inhibits expression of Osteoprotegerin (Opg), an inhibitor of osteoclast differentiation, and as a result favors osteoclastic bone resorption. This is important because bone resorption is a process that requires an acidification of the bone extracellular matrix and a low pH is the only known means to decarboxylate proteins outside cells. This led us to show that the low pH of the resorption lacunae is necessary for decarboxylating and activating osteocalcin. Hence, insulin and osteocalcin are locked in a feed-forward regulatory loop and insulin signaling in osteoblasts is necessary for whole-body glucose homeostasis in mice fed a normal chow [26••, 27].
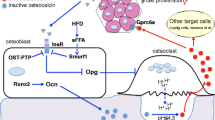
If this novel regulatory axis between bone and pancreas is relevant, one would expect that bone might contribute to whole-body insulin resistance in mice fed a high fat diet (HFD). As anticipated, in mice fed a HFD, bones become insulin resistant; this leads to a decrease in the circulating levels of the active form of osteocalcin and, thereby, a decrease in insulin secretion and sensitivity (Fig. 1) [28•]. Accordingly, mice lacking one copy of either the Insulin receptor in osteoblasts only or of Osteocalcin, even though they do not demonstrate any metabolic abnormalities when fed a normal diet, develop a more severe insulin resistance when fed a HFD due to a significantly lower circulating levels of the active form of osteocalcin compared to control mice fed the same HFD. Conversely, transgenic mice overexpressing the Insulin receptor in osteoblasts only are partially protected from whole-body insulin resistance when fed a HFD. At the molecular level, insulin resistance in osteoblasts develops because the increase in circulating levels of free saturated fatty acids that it generates favors the ubiquitination of the insulin receptor in osteoblasts through the E3 ubiquitin ligase Smurf1. These data illustrate, in a pathological condition, the importance of the osteocalcin in regulation of glucose homeostasis.

Regulation of glucose homeostasis by the bone-pancreas axis. Active osteocalcin is undercarboxylated osteocalcin, and inactive osteocalcin is carboxylated osteocalcin
Identification of Gprc6a as an Osteocalcin Receptor in Mice and in Humans
By and large, most published clinical studies have reported a correlation between osteocalcin circulating levels and glucose homeostasis in humans as well [29–40]. These studies, because of their correlative nature, had to be interpreted cautiously and needed a more direct confirmation; this came once the osteocalcin receptor had been identified.
Like most hormones, osteocalcin regulates several biological processes, one of them being the synthesis of testosterone by Leydig cells in the testis [41••]. Through the study of this particular aspect of osteocalcin biology, we identified a specific receptor for this hormone, a GPCR, called Gprc6a [42]. Of note, the notion that Gprc6a might be a receptor for osteocalcin has been proposed previously [43]. Gprc6a is expressed in Leydig cells of the testes and in β-cells of the pancreatic islets. Genetic evidence gathered in mice showed that Gprc6a is needed for osteocalcin regulation of insulin secretion and expression and pancreatic β-cell proliferation [44•] and that this function makes use of one particular transcription factor downstream of Gprc6a, CREB (unpublished data).
The male fertility phenotype of the Osteocalcin−/− mice resembled closely a human condition called peripheral testicular failure [45–48]. This was an incentive to search for mutation either in Osteocalcin or in Gprc6a in patients affected by this disease. This search identified the same missense mutation in one of the transmembrane domains in two unrelated patients [49•]. This mutation acts in cell-based assay and in vivo as a dominant negative mutation. Remarkably for the purpose of this review, both patients had an abnormal glucose tolerance. These results provided the first genetic evidence that GPRC6A is a receptor for osteocalcin in human as it is in mice. More importantly, they provided evidence that osteocalcin fulfills the same endocrine functions in humans as it does in mice.
Perspective
As it is often the case in biology, the data presented above raised more questions than they answered. The more specialized and immediate questions have to do with osteocalcin signaling in β-cells and with the mechanism whereby osteocalcin favors glucose utilization in peripheral tissues. A more general question, although difficult to address, is to understand the rationale for bone to regulate glucose homeostasis and the other functions that it regulates. Attached to this latter question is the interrogation that has never been experimentally addressed until now of the functions of glucose itself in osteoblasts. If we now look beyond osteocalcin and if osteoblasts are to be bona fide endocrine cells, then, it is likely that they secrete more hormones than we know. Conceivably, some of these as-yet unidentified hormones may regulate other aspects of energy metabolism. More generally, we as a field will need to provide a verifying rationale for the existence of the endocrine function of the bone.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance•• Of major importance
Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289:1508–14.
Karsenty G, Kronenberg HM, Settembre C. Genetic control of bone formation. Annu Rev Cell Dev Biol. 2009;25:629–48.
Mika C, Holtkamp K, Heer M, Gunther RW, Herpertz-Dahlmann B. A 2-year prospective study of bone metabolism and bone mineral density in adolescents with anorexia nervosa. J Neural Transm. 2007;114:1611–8.
Audi L, Vargas DM, Gussinye M, Yeste D, Marti G, Carrascosa A. Clinical and biochemical determinants of bone metabolism and bone mass in adolescent female patients with anorexia nervosa. Pediatr Res. 2002;51:497–504.
Soyka LA, Grinspoon S, Levitsky LL, Herzog DB, Klibanski A. The effects of anorexia nervosa on bone metabolism in female adolescents. J Clin Endocrinol Metab. 1999;84:4489–96.
Jacoangeli F, Zoli A, Taranto A, Staar Mezzasalma F, Ficoneri C, Pierangeli S, et al. Osteoporosis and anorexia nervosa: relative role of endocrine alterations and malnutrition. Eating Weight Disorders: EWD. 2002;7:190–5.
Misra M, Miller KK, Bjornson J, Hackman A, Aggarwal A, Chung J, et al. Alterations in growth hormone secretory dynamics in adolescent girls with anorexia nervosa and effects on bone metabolism. J Clin Endocrinol Metab. 2003;88:5615–23.
Misra M, Katzman DK, Cord J, Manning SJ, Mendes N, Herzog DB, et al. Bone metabolism in adolescent boys with anorexia nervosa. J Clin Endocrinol Metab. 2008;93:3029–36.
Misra M, Klibanski A. Bone metabolism in adolescents with anorexia nervosa. J Endocrinol Investig. 2011;34:324–32.
Misra M, Klibanski A. Anorexia nervosa, obesity and bone metabolism. Pediatric Endocrinol Rev: PER. 2013;11:21–33.
Fazeli PK, Klibanski A. Bone metabolism in anorexia nervosa. Curr Osteoporosis Reports. 2014;12:82–9.
Himes JH. Bone growth and development in protein-calorie malnutrition. World Rev Nutr Diet. 1978;28:143–87.
Faridi MM, Ansari Z, Bhargava SK. Imprints of protein energy malnutrition on the skeleton of children. J Trop Pediatr. 1984;30:150–3.
Hauschka PV, Lian JB, Gallop PM. Direct identification of the calcium-binding amino acid, gamma-carboxyglutamate, in mineralized tissue. Proc Natl Acad Sci U S A. 1975;72:3925–9.
Price PA, Otsuka AA, Poser JW, Kristaponis J, Raman N. Characterization of a gamma-carboxyglutamic acid-containing protein from bone. Proc Natl Acad Sci U S A. 1976;73:1447–51.
Price PA, Poser JW, Raman N. Primary structure of the gamma-carboxyglutamic acid-containing protein from bovine bone. Proc Natl Acad Sci U S A. 1976;73:3374–5.
Desbois C, Hogue DA, Karsenty G. The mouse osteocalcin gene cluster contains three genes with two separate spatial and temporal patterns of expression. J Biological Chem. 1994;269:1183–90.
Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, et al. Increased bone formation in osteocalcin-deficient mice. Nature. 1996;382:448–52.
Murshed M, Schinke T, McKee MD, Karsenty G. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J Cell Biol. 2004;165:625–30.
Mauro LJ, Olmsted EA, Skrobacz BM, Mourey RJ, Davis AR, Dixon JE. Identification of a hormonally regulated protein tyrosine phosphatase associated with bone and testicular differentiation. J Biol Chem. 1994;269:30659–67.
Morrison DF, Mauro LJ. Structural characterization and chromosomal localization of the mouse cDNA and gene encoding the bone tyrosine phosphatase, mOST-PTP. Gene. 2000;257:195–208.
Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–69. This is an original study revealing the physiological function of osteocalcin in regulating glucose metabolism.
Poser JW, Esch FS, Ling NC, Price PA. Isolation and sequence of the vitamin K-dependent protein from human bone. Undercarboxylation of the first glutamic acid residue. J Biol Chem. 1980;255:8685–91.
Ferron M, Hinoi E, Karsenty G, Ducy P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci U S A. 2008;105:5266–70. This study demonstrated the direct physiological functions of osteocalcin toward pancreatic beta cells and adipocytes in WT mice.
Ferron M, Wei J, Yoshizawa T, Ducy P, Karsenty G. An ELISA-based method to quantify osteocalcin carboxylation in mice. Biochem Biophys Res Commun. 2010;397:691–6.
Ferron M, Wei J, Yoshizawa T, Del Fattore A, DePinho RA, Teti A, et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142:296–308. This study uncovered that insulin signaling in osteoblasts is necessary for whole-body glucose homeostasis by favoring bone resorption to activate osteocalcin.
Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, Chen D, et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142:309–19.
Wei J, Ferron M, Clarke CJ, Hannun YA, Jiang H, Blaner WS, et al. Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J Clin Invest. 2014;124:1–13. This study explored the pathogenetic contribution of the local insulin resistance in bone to the high fat diet induced insulin resistance and identified a molecular mechanism causing the bone specific insulin resistance.
Im JA, Yu BP, Jeon JY, Kim SH. Relationship between osteocalcin and glucose metabolism in postmenopausal women. Clinica Chimica Acta; Int J Clin Chem. 2008;396:66–9.
Hwang YC, Jeong IK, Ahn KJ, Chung HY. The uncarboxylated form of osteocalcin is associated with improved glucose tolerance and enhanced beta-cell function in middle-aged male subjects. Diabetes Metab Res Rev. 2009;25:768–72.
Kanazawa I, Yamaguchi T, Yamamoto M, Yamauchi M, Kurioka S, Yano S, et al. Serum osteocalcin level is associated with glucose metabolism and atherosclerosis parameters in type 2 diabetes mellitus. J Clin Endocrinol Metab. 2009;94:45–9.
Kindblom JM, Ohlsson C, Ljunggren O, Karlsson MK, Tivesten A, Smith U, et al. Plasma osteocalcin is inversely related to fat mass and plasma glucose in elderly Swedish men. J bone Min Res: Off J Am Soc Bone Min Res. 2009;24:785–91.
Zhou M, Ma X, Li H, Pan X, Tang J, Gao Y, et al. Serum osteocalcin concentrations in relation to glucose and lipid metabolism in Chinese individuals. Eur J Endocrinol/Eur Federation Endocrine Soc. 2009;161:723–9.
Hwang, Y.C., Jeong, I.K., Ahn, K.J., and Chung, H.Y. Circulating osteocalcin level is associated with improved glucose tolerance, insulin secretion and sensitivity independent of the plasma adiponectin level. Osteoporos Int. 2012;23:1337–42. doi:10.1007/s00198-011-1679-x.
Kanazawa I, Yamaguchi T, Yamauchi M, Yamamoto M, Kurioka S, Yano S, et al. Serum undercarboxylated osteocalcin was inversely associated with plasma glucose level and fat mass in type 2 diabetes mellitus. Osteoporos Int. 2011;22:187–94.
Strapazzon G, De Toni L, Foresta C. Serum undercarboxylated osteocalcin was inversely associated with plasma glucose level and fat mass in type 2 diabetes mellitus. Osteoporosis Int: J Established Result Cooperation Between Eur Foundation Osteoporosis National Osteoporosis Foundation USA. 2011;22:1643–4.
Wedrychowicz A, Stec M, Sztefko K, Starzyk JB. Associations between bone, fat tissue and metabolic control in children and adolescents with type 1 diabetes mellitus. Exp Clin Endocrinol Diabetes. 2014;122:491–5.
Levinger, I., Jerums, G., Stepto, N.K., Parker, L., Serpiello, F.R., McConell, G.K., Anderson, M., Hare, D.L., Byrnes, E., Ebeling, P.R., et al. (2014). The effect of acute exercise on undercarboxylated osteocalcin and insulin sensitivity in obese men. J Bone Miner Res. 2014;29:2571–6. doi:10.1002/jbmr.2285.
Kim GS, Jekal Y, Kim HS, Im JA, Park JY, Chu SH. Reduced serum total osteocalcin is associated with central obesity in Korean children. Obesity Res Clin Pract. 2014;8:e201–298.
Garanty-Bogacka B, Syrenicz M, Rac M, Krupa B, Czaja-Bulsa G, Walczak M, et al. Association between serum osteocalcin, adiposity and metabolic risk in obese children and adolescents. Endokrynologia Polska. 2013;64:346–52.
Oury F, Sumara G, Sumara O, Ferron M, Chang H, Smith CE, et al. Endocrine regulation of male fertility by the skeleton. Cell. 2011;144:796–809. This study identified Gprc6a as a osteocalcin receptor.
Wellendorph P, Brauner-Osborne H. Molecular cloning, expression, and sequence analysis of GPRC6A, a novel family C G-protein-coupled receptor. Gene. 2004;335:37–46.
Pi M, Faber P, Ekema G, Jackson PD, Ting A, Wang N, et al. Identification of a novel extracellular cation-sensing G-protein-coupled receptor. J Biol Chem. 2005;280:40201–9.
Wei J, Hanna T, Suda N, Karsenty G, Ducy P. Osteocalcin promotes beta-cell proliferation during development and adulthood through Gprc6a. Diabetes. 2014;63:1021–31. This study identified Gprc6a as a receptor mediating osteocalin functions in pancreatic beta cells.
Boisen KA, Main KM, Rajpert-De Meyts E, Skakkebaek NE. Are male reproductive disorders a common entity? The testicular dysgenesis syndrome. Ann N Y Acad Sci. 2001;948:90–9.
Glass AR, Vigersky RA. Testicular reserve of testosterone precursors in primary testicular failure. Fertil Steril. 1982;38:92–6.
Paduch DA. Testicular cancer and male infertility. Curr Opin Urol. 2006;16:419–27.
Winters SJ, Troen P. A reexamination of pulsatile luteinizing hormone secretion in primary testicular failure. J Clin Endocrinol Metab. 1983;57:432–5.
Oury F, Ferron M, Huizhen W, Confavreux C, Xu L, Lacombe J, et al. Osteocalcin regulates murine and human fertility through a pancreas-bone-testis axis. J Clin Invest. 2013;123:2421–33. This study reported that two human subjects with mutations in Gprc6a, a receptor for osteocalcin, demonstrated similar abnormalities in fertility as described in mice lack of osteocalcin.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
J Wei and G Karsenty both declare no conflicts of interest.
Human and Animal Rights and Informed Consent
All studies by the authors involving animal and/or human subjects were performed after approval by the appropriate institutional review boards. When required, written informed consent was obtained from all participants.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Bone and Diabetes
Rights and permissions
About this article

Cite this article
Wei, J., Karsenty, G. An Overview of the Metabolic Functions of Osteocalcin. Curr Osteoporos Rep 13, 180–185 (2015). https://doi.org/10.1007/s11914-015-0267-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-015-0267-y