
Abstract
Aims/hypothesis
The study evaluated whether glucagon secretion is regulated by changes in insulin sensitivity under normal conditions.
Materials and methods
A total of 155 healthy women with NGT (aged 53–70 years) underwent a glucose-dependent arginine-stimulation test for evaluation of glucagon secretion. Arginine (5 g) was injected i.v. under fasting conditions (plasma glucose 4.8±0.1 mmol/l) and after raising blood glucose concentrations to 14.8±0.1 and 29.8±0.2 mmol/l. The acute glucagon response (AGR) to arginine during the three glucose levels (AGR1, AGR2, AGR3) was estimated, as was the suppression of baseline glucagon by the increased glucose. All women also underwent a 2-h euglycaemic–hyperinsulinaemic clamp study for estimation of insulin sensitivity.
Results
Insulin sensitivity was normally distributed, with a mean of 73.2±29.3 (SD) nmol glucose kg−1 min−1/pmol insulin l−1. When relating the variables obtained from the arginine test to insulin sensitivity, insulin resistance was associated with increased AGR and with increased suppression of glucagon levels by glucose. For example, the regression between insulin sensitivity and AGR2 was r=−0.38 (p<0.001) and between insulin sensitivity and suppression of glucagon levels by 14.8 mmol/l glucose r=0.36 (p<0.001). Insulin sensitivity also correlated negatively with insulin secretion; multivariate analysis revealed that changes in insulin sensitivity and insulin secretion were independently related to changes in glucagon secretion.
Conclusions/interpretation
The body adapts to insulin resistance by increasing the glucagon response to arginine and by increasing the suppression of glucagon levels by glucose. Hence, not only the islet beta cells but also the alpha cells seem to undergo compensatory changes during the development of insulin resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It is known that type 2 diabetes is associated with insulin resistance and defective insulin secretion [1–3]. It is, however, also known that type 2 diabetes is associated with elevated levels of glucagon [4–6] and defective suppression of glucagon secretion [4, 7, 8]. Also IGT is associated with impaired suppression of glucagon secretion [9–11]. The subsequent relative hyperglucagonaemia is probably of importance for postprandial hyperglycaemia in diabetes due to increased hepatic glucose production [5, 6, 12]. In fact, Shah et al. [13] have demonstrated that also in the absence of insulin resistance, glucose tolerance is critically dependent on normal suppression of glucagon secretion. Hence, in addition to defective insulin secretion, defective regulation of glucagon secretion is a hallmark of the islet dysfunction accompanying type 2 diabetes.
The defective regulation of glucagon secretion in type 2 diabetes, which is associated with insulin resistance, raises the issue of the relationship between glucagon secretion and insulin resistance. It is well known that insulin resistance is adaptively accompanied by increased insulin secretion (review, see [3]). In a previous study we observed that glucagon secretion is also inversely related to insulin sensitivity [10]. From the results, however, it was not possible to establish whether the body adapts to insulin resistance by increased glucagon secretion or by defective suppression of glucagon secretion. We therefore examined in a large number of women with NGT the relationship between insulin sensitivity and (1) basal and stimulated glucagon levels, and (2) suppression of glucagon secretion by glucose.
Subjects and methods
Subjects
A total of 155 women with NGT were included in the study. They were recruited from a health-screening study in Malmö, Sweden, on the basis of NGT [14]. All subjects were healthy and none was taking any medication known to affect carbohydrate metabolism. Thirty-four women were taking oestrogens. All women underwent a standardised 75-g, 2-h OGTT. The test was undertaken after an overnight fast and the subjects spent the 2 h in a semi-recumbent position. All subjects had normal values of fasting and 2-h glucose (Table 1).
Glucagon secretion
Glucagon secretion was determined with i.v. arginine stimulation at three glucose levels (fasting, 14 and >25 mmol/l) as previously described [15, 16]. After an overnight fast, i.v. catheters were inserted into antecubital veins in both arms. One arm was used for the infusion of glucose and the other arm for intermittent sampling. The sampling catheter was kept patent by slow infusion of 0.9% saline when not in use. Baseline samples were taken at −5 and −2 min. A maximally stimulating dose of arginine hydrochloride (5 g) was then injected i.v. over 45 s. Samples were taken at 2, 3, 4 and 5 min. A variable-rate 20% glucose infusion was then initiated to raise and maintain blood glucose at 13–15 mmol/l. Blood glucose was determined every 5 min at the bedside and the glucose infusion adjusted to reach the desired blood glucose level in 20–25 min. New baseline samples were taken, then arginine (5 g) was again injected and new 2, 3, 4 and 5 min samples were taken. A 2.5-h resting period was then allowed, whereafter new baseline samples were obtained and a high-speed (900 ml/h) 20% glucose infusion during 25–30 min was used to raise blood glucose to >28 mmol/l, as determined at the bedside. At this blood glucose level, new baseline samples were taken and arginine (5 g) injected followed by final 2, 3, 4 and 5 min samples.
Insulin sensitivity
Insulin sensitivity was determined with the euglycaemic–hyperinsulinaemic clamp [17]. After an overnight fast, i.v. catheters were inserted into antecubital veins in both arms. One arm was used for infusion of glucose and insulin. The contralateral arm was used for intermittent sampling, and the catheter was kept patent with a slow infusion of 0.9% saline. A primed constant infusion of insulin (Actrapid 100 U/ml; Novo Nordisk, Bagsvaerd, Denmark) with a constant infusion rate of 0.28 nmol m−2 body surface min−1 was started. After 4 min, a variable-rate 20% glucose infusion was added and its infusion rate was adjusted manually throughout the clamp procedure to maintain the blood glucose level at 5.0 mmol/l. Blood glucose was determined every 5 min. Samples for analysis of the achieved insulin concentrations were taken at 60 and 120 min.
Analyses
Blood glucose concentration was determined at the bedside by the glucose dehydrogenase technique (Hemocue, Ängelholm, Sweden) during the euglycaemic–hyerpinsulinaemic clamp and with an Accutrend (Boehringer Mannheim Scandinavia, Bromma, Sweden) during the arginine test. Blood samples for glucagon, insulin and glucose from the arginine study and for insulin from the clamp study were immediately frozen and serum or plasma frozen at −20°C. Serum glucagon and insulin were analysed with a double-antibody RIA technique. Samples for analysis of glucagon were obtained in prechilled test tubes containing 0.084 ml EDTA (0.34 mol/l) and aprotinin (250 kallikrein-inhibiting U/ml blood; Bayer, Leverkusen, Germany). For the assay, guinea-pig anti-human glucagon antibodies specific for pancreatic glucagon, 125I-glucagon as tracer and glucagon standard were used (Linco Research, St Charles, MO, USA). For insulin assay, guinea-pig anti-human insulin antibodies, mono-125I-Tyr-human insulin and human insulin standard were used (Linco). Plasma glucose concentrations were analysed using the glucose oxidase method. All samples were analysed in duplicate.
Calculations and statistics
Data are presented as means±SEM unless otherwise stated. For the determination of glucagon secretion, the acute glucagon response (AGR) to arginine was determined as the mean of the 2- to 5-min samples minus the mean prestimulus glucagon concentration at fasting (AGR1), at 13–15 mmol/l (AGR2) and at >28 mmol/l glucose (AGR3). The slope between AGR1 and AGR2 was calculated as a measure of the alpha cell sensitivity of glucose to suppress arginine-stimulated glucagon secretion. Also the maximal AGR suppression was calculated (AGR3 minus AGR1), as was the suppression by glucose of basal glucagon levels at 13–15 mmol/l glucose (baseline suppression1) and at >28 mmol/l (baseline suppression2). Finally, the rapidity of the glucagon response to arginine was calculated as the increase in glucagon levels at min 2 after arginine as a per cent of the maximal increase at fasting (rapidity1), 13–15 mmol/l glucose (rapidity2) and at >28 mmol/l glucose (rapidity3). Conversely, for insulin levels, the acute insulin response (AIR) to arginine at fasting glucose (AIR1), at 13–15 mmol/l glucose (AIR2) and at >28 mmol/l glucose (AIR3) were determined. Finally, for the calculation of insulin sensitivity, a steady-state condition was assumed during the second hour of the clamp. Insulin sensitivity was calculated as the glucose infusion rate/kg body weight during the second hour divided by the mean of the insulin levels at 60 and 120 min during the clamp (i.e. nmol glucose kg−1 min−1/pmol insulin l−1). Statistical analyses were performed with the SPSS for Windows system (SPSS, Chicago, IL, USA). Differences in responses to arginine at different glucose levels were estimated by a paired t-test. Pearson’s product moment correlation coefficients were obtained to estimate linear correlation among variables. Linear multiple regression was used to assess the independent effect of several variables; the stepwise forward method was used.
Results
Study group characteristics
Table 1 shows the characteristics of the 155 women participating in the study. They were all healthy and had normal values of fasting and 2-h glucose during an OGTT.
Glucose-dependent arginine-stimulation test
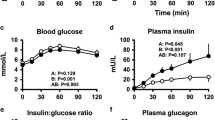
When arginine was injected i.v. after an overnight fast (plasma glucose 4.8±0.1 mmol/l) there was a sharp and rapid increase in circulating glucagon in all subjects with the peak observed after 3 min, the AGR1 being 102±3.6 ng/l (Fig. 1). After the first arginine administration, glucose was administered i.v., which raised the glucose level to 15.2±0.2 mmol/l. This reduced baseline glucagon from 64±1.4 to 48±1.4 ng/l (p<0.001). A new arginine administration resulted again in a rapid and marked increase in circulating glucagon, again with the peak level at 3 min after administration. However, the AGR2 was only 55.4±2.1 ng/l, which was a reduction from AGR1 by 44.2±1.5% (p<0.001). Finally, glucose was administered again, which raised plasma glucose to 29.8±0.3 mmol/l. This further reduced baseline glucagon to 39.3±1.1 ng/l. When arginine was injected at this higher glucose level, there was again an increase in circulating glucagon, although the increase was slower than at lower glucose levels. Thus, the increase in glucagon levels peaked at 4 min after administration. AGR3 was only 39.2±1.6 ng/l, which was equivalent to a reduction by 60.0±1.4% of AGR1 (p<0.001). Figure 1 also shows the AGRs and AIRs as functions of the plasma glucose levels. Table 2 displays the glucagon secretory characteristics of the glucose-dependent arginine-stimulation test, i.e. shows how glucagon secretion responds to a rise in circulating glucose in subjects with NGT. It is seen that glucose suppresses both baseline glucagon levels and the glucagon response to arginine. Furthermore, the rapidity of the glucagon response to arginine was suppressed by glucose, i.e. the time from arginine injection to peak of glucagon. Hence, both the baseline and stimulated glucagon secretion as well as the rapidity of the responsiveness of glucagon secretion are suppressed by glucose.

Glucagon levels before and after i.v. administration of arginine (5 g) under fasting conditions (plasma glucose 4.8±0.1 mmol/l) a or after raising the glucose levels to 15.2±0.2 mmol/l b or 29.8±0.3 mmol/l c in 155 healthy women with NGT. d, e The estimated acute glucagon response (AGR) or acute insulin response (AIR) to arginine respectively, vs the glucose levels during the glucose-dependent arginine-stimulation test. Means±SEM
Relationship between glucagon secretion and insulin sensitivity
In all women, a euglycaemic–hyperinsulinaemic clamp was undertaken to establish the insulin sensitivity. It was found (Fig. 2) that insulin sensitivity displayed a normal distribution and a broad range. To explore whether insulin sensitivity correlated with the glucagon responses during the arginine-stimulation test, linear correlation was undertaken between the insulin sensitivity M/I value during the clamp with the variables obtained from the arginine test (Table 2). It was found that there was a significant and negative correlation between insulin sensitivity and AGR (AGR1, AGR2 as well as AGR3), showing that the highest glucagon response to arginine was seen in the most insulin-resistant subjects. Also the reduction in baseline glucagon by raising the glucose levels correlated negatively with insulin sensitivity, showing that subjects with insulin resistance were those who exhibited the strongest suppressibility of baseline glucagon to glucose. In contrast, the slope of AGR or the rapidity of the glucagon response to arginine seemed uninfluenced by insulin sensitivity. To illustrate the relationship between variables with insulin sensitivity, the subjects were divided into quartiles of insulin sensitivity. Table 3 shows fasting glucose and insulin as well as BMI in these quartiles; as expected the most insulin-resistant subjects had higher insulin levels and higher BMI; however, it should be emphasised that all subjects had NGT with normal fasting glucose. Figure 2 shows that fasting glucagon, the AGR2 and the suppression of baseline glucagon levels when raising glucose to 14 mmol/l (baseline suppression1) decreased progressively as insulin sensitivity increased.

a Distribution of insulin sensitivity as determined by the euglycaemic–hyperinsulinaemic clamp in the 155 women with NGT. Other panels show b fasting glucagon, c the acute glucagon response (AGR 2 ) to i.v. arginine (5 g) after raising the glucose level to 15.2±0.2 mmol/l, and d the suppression of baseline glucagon after raising the glucose level to 15.2±0.2 mmol/l, all vs insulin sensitivity, calculated as the glucose infusion rate/kg body weight during the second hour of the clamp divided by the mean of the insulin levels at 60 and 120 min of the clamp (i.e. nmol glucose kg−1 min−1/pmol insulin l−1), in the subjects divided according to quartiles of insulin sensitivity. Means±bi-directional SEM
Relationship between changes in glucagon secretion and insulin secretion
Two adaptive processes in glucagon secretion were thus evident in response to changes in insulin sensitivity: an increased glucagon response to arginine, and an increased suppression of baseline glucagon secretion in response to glucose and in line with reduced insulin sensitivity. To examine whether these two processes are dependent on changes in insulin secretion, multivariate analysis was undertaken. The results were similar when introducing the islet hormone responses from either the first, second or third arginine challenge; to avoid repeating data only the results from the second arginine bolus are reported here. In a first analysis, the AGR2 was used as the dependent variable and insulin sensitivity and AI2 (i.e. the insulin response to arginine at 15 mmol/l glucose, which is a parameter also increased by insulin resistance) were used as independent variables. It was found that both these variables independently contributed to the AGR2, with a combined r value of 0.48 (p<0.001). Similarly, when suppression of baseline glucagon by glucose at 13–15 mmol/l (baseline suppression1) was used as the dependent variable and insulin sensitivity and AIR2 were used as independent variables, it was found that both these variables independently contributed to the suppression of glucagon by glucose with a combined r value of 0.45 (p<0.001). In contrast, adding also fasting (or 2-h) glucose to the models did not change these conclusions.
Relationship between changes in glucagon secretion and glucose, BMI and age
The 2-h glucose value correlated with AGR (correlation with AGR2; r=0.36; p<0.001) as well as with suppression of glucagon by glucose at 13–15 mmol/l (baseline suppression; r=0.21; p=0.012). Also BMI correlated with AGR (with AGR2; r=0.21; p=0.028), but not with glucagon suppression. However, neither 2-h glucose nor BMI correlated with AGR or glucagon suppression in a model where also insulin sensitivity was introduced. It should be emphasised, however, that all subjects had NGT and therefore that the variation in 2-h glucose was small. Age did not correlate with glucagon secretion.
Discussion
It has been documented that the body adapts to insulin resistance by a compensatory increase in insulin secretion [3, 18–21]. This concept is important for understanding the key role of the islet beta cells for the development of IGT and type 2 diabetes: if beta cell compensation to insulin resistance fails, glucose homeostasis deranges, which will result in IGT and type 2 diabetes [3]. However, we, and others, have demonstrated that glucagon secretion is also important for the development of IGT and type 2 diabetes, in view of the hyperglucagonaemia and increased glucagon secretion which accompany these conditions [4–12]. This raises the issue of the relationship between insulin resistance and glucagon secretion under normal conditions. Is there, as for insulin secretion, a compensatory response in glucagon secretion in persons with insulin resistance, and may failure of the alpha cells to adapt to insulin resistance be a mechanism of glucose intolerance?
In this study we have examined glucagon secretion, as judged from the glucose-dependent arginine-stimulation test, in relation to insulin sensitivity, as judged from the euglycaemic–hyperinsulinaemic clamp technique, in a large number of healthy women with NGT. The glucose-dependent arginine-stimulation test was initially developed for the careful analysis of beta cell function, since it displays both basal and maximal insulin secretion as well as glucose sensitivity of the beta cells [15, 16]. However, since arginine stimulates glucagon secretion as well, and since glucose suppresses alpha cell secretion, the test is also useful for characterising alpha cell function. Indeed, we have previously used this test to show that increased glucagon secretion contributes to future worsening of glucose intolerance and that this is independent of changes in insulin secretion [22]. A potential limitation is that changes in glucagon secretion are inferred from changes in glucagon levels. However, it is unlikely that the large and rapid changes in glucagon levels seen after challenging with glucose and arginine would be secondary to other processes than glucagon secretion, such as glucagon disposal.
We show here that glucose suppresses glucagon levels and that arginine markedly stimulates glucagon secretion by an effect which is suppressed by raising the glucose level; both the absolute increase in glucagon levels by arginine and the rapidity with which arginine stimulated glucagon secretion were reduced by glucose. The main findings of this study were, however, that two processes in alpha cell function are different when comparing subjects with insulin resistance than in those with high insulin sensitivity: first there is an increased glucagon response to arginine and second there is an increased suppression of baseline glucagon by glucose in insulin resistance. The finding that insulin resistance is associated with increased glucagon response to arginine confirms a previous study [10]. That study showed an inverse relationship between the glucagon response to arginine in relation to insulin sensitivity with an r value of 0.5. In the present study, it was also demonstrated that insulin resistance is associated with augmented glucagon suppression by glucose, i.e. the direct action of glucose, not only arginine, on glucagon secretion seems to be perturbed in insulin resistance.
It should be emphasised that the women studied here all had NGT, and therefore that the changes in glucagon secretion reported here should not be regarded as secondary to changes in glycaemia. Instead, the changes should be regarded as being adaptations to the development of insulin resistance. Hence, islet compensation in response to insulin resistance does not involve insulin secretion only, but also changes in glucagon secretion. The mechanisms by which the alpha cell adapts to insulin resistance remain to be established. It is possible that several adaptive responses to insulin resistance might mediate the adaptation. These potential mediators include NEFA, as well as adipokines released from adipocytes in relation to insulin sensitivity, as well as neural reflexes activated by insulin resistance and signalling to the islets, in analogy to discussions on mechanisms of changes in insulin secretion during insulin resistance [23]. One intriguing aspect is also whether the changes in glucagon secretion are secondary to insulin resistance in the glucagon-producing alpha cells. It is known that insulin inhibits glucagon secretion [24, 25] and, therefore, resistance in the alpha cells to insulin would augment glucagon secretion [26], which would explain the higher glucagon response to arginine in the arginine test. On the other hand, insulin secretion is increased in insulin resistance, which would counterbalance a defective insulin action also in the alpha cells. Also, the augmented suppression by glucose would not be explained by such a mechanism. Interestingly, subjects with IGT and type 2 diabetes have reduced suppression by glucose of glucagon secretion [4–12]. It remains to be established whether failure of the augmenting action of glucose to suppress glucagon levels in insulin resistance represents the mechanism that initiates IGT.
In conclusion, this study has demonstrated that in healthy subjects with NGT, insulin resistance is associated with increased fasting glucagon levels, augmented glucagon response to arginine and an augmented suppression by glucose of glucagon levels. This suggests that islet adaptation to insulin resistance involves not only increased insulin secretion but also changes in glucagon secretion.
Abbreviations
- AGR:
-
acute glucagon response
- AIR:
-
acute insulin response
References
Reaven GM, Hollenbeck CB, Chen YDI (1989) Relationship between glucose tolerance, insulin secretion, and insulin action in non-obese individuals with varying degrees of glucose tolerance. Diabetologia 32:52–55
De Fronzo RA, Bonadonna RC, Ferrannini E (1992) Pathogenesis of NIDDM. A balanced overview. Diabetes Care 15:318–368
Ahrén B, Pacini G (2004) Importance of quantifying insulin secretion in relation to insulin sensitivity to accurately assess beta cell function in clinical studies. Eur J Endocrinol 150:97–104
Muller WA, Faloona GR, Aguilar-Parada E, Unger RH (1970) Abnormal α-cell function in diabetes. N Engl J Med 283:109–115
Reaven GM, Chen YD, Golay A et al. (1987) Documentation of hyperglucagonaemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 64:106–110
Butler PC, Rizza RA (1991) Contribution to postprandial hyperglycaemia and effect on initial splanchnic glucose clearance of hepatic glucose cycling in glucose-intolerant or NIDDM patients. Diabetes 40:73–81
Unger RH, Orci L (1981) Glucagon and the A cell: physiology and pathophysiology, part 1. N Engl J Med 304:1518–1524
Unger RH, Orci L (1981) Glucagon and the A cell: physiology and pathophysiology, part 2. N Engl J Med 304:1575–1580
Gerich JE (1997) Metabolic abnormalities in impaired glucose tolerance. Metabolism 46:40–43
Ahrén B, Larsson H (2000) Islet dysfunction in insulin resistance involves impaired insulin secretion and increased glucagon secretion in postmenopausal women with impaired glucose tolerance. Diabetes Care 23:650–657
Ahrén B, Larsson H (2001) Impaired glucose tolerance (IGT) is associated with reduced insulin-induced suppression of glucagon concentrations. Diabetologia 44:1998–2003
Shah P, Vella A, Basu A et al. (2000) Lack of suppression of glucagon contributes to postprandial hyperglycaemia in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab 85:4053–4059
Shah P, Basu A, Basu R, Rizza R (1999) Impact of lack of suppression of glucagon on glucose tolerance in humans. Am J Physiol 277:E283–E290
Larsson H, Ahrén B, Lindgärde F, Berglund G (1995) Fasting blood glucose in determining the prevalence of diabetes in a large, homogeneous population of Caucasian middle-aged women. J Intern Med 237:537–541
Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D Jr (1984) Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest 74:1318–1328
Larsson H, Ahrén B (1998) Glucose-dependent arginine stimulation test for characterization of islet function: studies on reproducibility and priming effect of arginine. Diabetologia 41:772–777
DeFronzo RA, Tobin JD, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237:E214–E223
Bergman RN, Phillips LS, Cobelli C (1981) Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and β-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest 68:1456–1467
Kahn SE, Prigeon RL, McCulloch DK et al. (1993) Quantification of the relationship between insulin sensitivity and β-cell function in human subjects: evidence for a hyperbolic function. Diabetes 42:1663–1672
Van Haeften TW, Pimenta W, Mitrakou A et al. (2000) Relative contributions of β-cell function and tissue insulin sensitivity to fasting and postload glycaemia. Metabolism 49:1318–1325
Ahrén B, Larsson H (2002) Quantification of insulin secretion in relation to insulin sensitivity in nondiabetic postmenopausal women. Diabetes 52 [Suppl 1]:202–211
Larsson H, Ahrén B (2000) Glucose intolerance is predicted by low insulin secretion and high glucagon secretion: outcome of a prospective study in postmenopausal Caucasian women. Diabetologia 43:194–202
Ahrén B, Pacini G (2003) Signals adapting the beta cells to changes in insulin sensitivity. Int Congr Ser 1253:105–113
Maruyama H, Hisatomi A, Orci L et al. (1984) Insulin within islets is a physiologic glucagon release inhibitor. J Clin Invest 74:2296–2299
Samols E, Stagner JI (1988) Intra-islet regulation. Am J Med 85:31–35
Ipp E (2000) Impaired glucose tolerance. The irrepressible α-cell? Diabetes Care 23:569–570
Acknowledgements
The study was supported by the Swedish Research Council (Grant No. 6834), the Swedish Diabetes Association, Region Skåne and the Faculty of Medicine, Lund University. The author is grateful to L. Bengtsson and M. Persson for expert assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ahrén, B. Glucagon secretion in relation to insulin sensitivity in healthy subjects. Diabetologia 49, 117–122 (2006). https://doi.org/10.1007/s00125-005-0056-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-0056-8