
Abstract
Key message
we identified a functional chromogen gene C from wild rice, providing a new insight of anthocyanin biosynthesis pathway in indica and japonica.
Abstract
Accumulation of anthocyanin is a desirable trait to be selected in rice domestication, but the molecular mechanism of anthocyanin biosynthesis in rice remains largely unknown. In this study, a novel allele of chromogen gene C, OrC1, from Oryza rufipongon was cloned and identified as a determinant regulator of anthocyanin biosynthesis. Although OrC1 functions in purple apiculus, leaf sheath and stigma in indica background, it only promotes purple apiculus in japonica. Transcriptome analysis revealed that OrC1 regulates flavonoid biosynthesis pathway and activates a few bHLH and WD40 genes of ternary MYB-bHLH-WD40 complex in indica. Differentially expressed genes and metabolites were found in the indica and japonica backgrounds, indicating that OrC1 activated the anthocyanin biosynthetic genes OsCHI, OsF3H and OsANS and produced six metabolites independently. Artificial selection and domestication of C1 gene in rice occurred on the coding region in the two subspecies independently. Our results reveal the regulatory system and domestication of C1, provide new insights into MYB transcript factor involved in anthocyanin biosynthesis, and show the potential of engineering anthocyanin biosynthesis in rice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Most seed plants have different colors in different parts of seeds mostly due to the anthocyanin pigmentation (Aizza et al. 2011). Anthocyanins and pro-anthocyanins are a class of favonoid, as one of the largest groups of secondary metabolites, and are widely distributed in plants (Xu et al. 2015). Anthocyanins contribute to not only multiple physiological roles in the responses of plant to biotic and abiotic stresses, but also commercial value of plant products. It was reported that various anthocyanin also have a profound impact on food quality beneficial to human health (Vinayagam et al. 2015), which is of significant interest to both crop breeders and consumers.
Biosynthesis and accumulation of anthocyanin depend on the inherent genetic factors and external environmental factors. Inherent factors in anthocyanin biosynthesis include the structural and regulatory genes. Structural genes encode enzymes, include phenylalanine ammonia lyase (PAL), chalcone synthase (CHS), chalcone isomerase (CHI), flavonoid-3-hydroxylase (F3H), flavonoid-3′-hydroxylase (F3′H), dihydroflavanol reductase (DFR), and anthocyanidin synthase (ANS). The expression of these structural genes is controlled by transcription factors (TFs) and has been well characterized in a range of plant species (Honda et al. 2002; Grotewold 2006; Hugueney et al. 2009). A ternary MBW complex, comprising R2R3-MYB TFs, basic-helix-loop-helix (bHLH) TFs and WDR (WD-repeat) proteins, was believed to tightly regulate the common pathway of anthocyanins and pro-anthocyanins biosyntheses (de Vetten et al.1997; Baudry et al. 2004; Hichri et al. 2011; Xu et al. 2015). The regulating network of anthocyanin biosynthesis is well documented in maize and Arabidopsis but much less in rice. Sun et al. (2018) reported that a C-S-A gene system regulated anthocyanin pigmentation in rice hull. In this system, C1 encodes a R2R3-MYB transcription factor and acts as a color-producing gene, and S1 encodes a bHLH protein that functions in a tissue-specific manner. C1 interacts with S1 and activates the expression of A1, which encodes a dihydroflavonol reductase (Sun et al. 2018).
The R2R3-MYB TFs were often identified as determinants of variation in anthocyanin pigmentation and have been identified in many higher plant species (Chagne et al. 2013; Jin et al. 2016; Jian et al. 2019; Tian et al. 2015; Jun et al. 2015; Xu et al. 2020). The R2R3-MYB gene OsC1 was previously isolated from cultivated rice through comparative mapping between rice and maize or according to the nucleotide sequence homology with known maize orthologues (Mikami et al. 2000; Saitoh et al. 2004), and was also cloned from cultivated rice using various methods recently (Liu et al. 2012; Zhao et al. 2016). Several studies determined that OsC1 acted as a chromogen gene and mainly functioned in apiculus and leaf sheath (Saitoh et al. 2004; Gao et al. 2011; Fan et al. 2008; Chin et al. 2016; Hu et al. 2020). The apiculus, as the remnant of awn, maintains its color in some modern varieties and seems not to have undergone artificial selection during domestication (Saitoh et al. 2004). The purple sheath trait was reported to be a morphological marker in rice (Gao et al. 2011), which can be easily observed at the seedling stage, and have often been used in the screening of the authentic hybrids. However, molecular function of OsC1 was not fully understood, the exact genetic determinants for purple apiculus and leaf sheath in rice remain to be unraveled.
Rice ancestor wild rice (Oryza rufipogon) has accumulated anthocyanin in various tissues, most of O. rufipogon appear to have purple or red pigment mainly on awn, apiculus, stigma, pericarp, pulvinus, leaf blade, leaf sheath, internode and palea, but most cultivated rice have lost these pigments due to artificial selection. It had been demonstrated that OsC1 was also a domestication-related gene for the loss of pigments in cultivated rice (Huang et al. 2012). Identification of the C1 allele in wild rice is of great significance for understanding the origin and evolution of rice through investigation of the genes controlling color formation. In this study, a R2R3 MYB transcription factor was fine mapped in the wild rice using a set of chromosome segment substitution lines. The C gene from wild rice, OrC1, was cloned for functional analysis, transcriptom and metabolome profiling and was shown to be a functional allele for anthocyanin biosynthsis. Interestingly, we found that six anthocyanin metabolites were simultaneously regulated by OrC1 in different genetic backgrounds. Our findings emphasize the importance and value of using wild relatives to uncover useful genes that have been lost during crop domestication in order to expand the genetic repertoire for rice domestication study and modern crop breeding.
Materials and methods
Plant materials
A set of CSSLs produced from common wild rice (O. rufipogon) as the donor and an elite indica variety, 9311, as the recurrent parent was developed in our laboratory (Qiao et al. 2016). The CSSLs and 9311 were grown under multiple environmental conditions (Qi et al. 2018). A panel of 180 rice accessions (Supplementary 1) including 89 O. sativa indica, 48 O. sativa japonica, 21 O. rufipogon and 22 O. nivara, was selected from a natural rice germplasm population that is maintained in our laboratory. The purple traits were recorded on multiple environments.
Sequence and phylogenetic analyses
The predicted amino acid sequences of C1 in O. rufipongon and MYB homologous proteins in other species were downloaded from NCBI. Multiple sequence alignments were performed with DNAMAN software. A phylogenic tree was constructed using the Maximum Likelihood method and software MEGA version 5.1 (Tamura et al. 2011). The de novo genomic sequencing of the panel of 180 rice accessions was performed by our laboratory previously. C1 gene genomic sequence was isolated from the genomic sequencing data. The SSR primers used in this study were previously published (Cho et al. 2000), InDel primers were designed in our laboratory (Qi et al. 2018). The other primers used in this study were designed online at the NCBI website based on the 9311 reference genome sequence. Alignments were performed on the Gramene (http://www.gramene.org/) website to ensure the accuracy of the location and the specificity of the primers. Sequences of all primers used in this study are shown in Supplementary 2.
Subcellular localization
To develop the OrC1-GFP fusion protein for subcellular localization, full-length coding sequence of OrC1 was cloned into SpeI and BamHI site of Pan580 plasmid, to generate 35S::OrC1-GFP constructs. Rice protoplast preparations and transfections were performed as previously described (Zhang et al. 2011). The fusion construct (35S::OsC1-GFP) and nucleus marker were co-transformed into rice protoplasts prepared from 2-week-old rice seedlings and using 40% PEG-4000 incubated in the dark at 25 ℃ for 16 to 20 h. In addition, 35S:OrC1-GFP constructs transformed into A.tumefaciens strain GV3101.Then, the strains were injected into tobacco leaves. The 35S::NLS-RFP construct was used as a nuclear positive control. The GFP and RFP fluorescence in rice protoplasts and leaf epidermal cells were observed using a laser scanning confocal microscope (LSM880, Leica).
RNA extraction and real-time PCR
Total RNA was extracted from different rice tissues using the RNA RNeasy Plant Mini Kit (Qiagen, Beijing, China), followed by treatment with RNase-free DNase (TaKaRa, Dalian, China) to remove genomic DNA contamination. qRT-PCR was performed using an ABI 7500 real-time PCR system (Applied Biosystems) following the manufacturer’s instructions. The popular endogenous control gene (Actin) was used as an internal control. Gene-specific primers are listed in Supplementary 2. Relative expression was calculated using the 2−[Δ][Δ]Ct method (Livak et al. 2001). Each sample was amplified in triplicate.
Transcriptome analysis
The leaf sheath of 15-day seedlings of near isogenic line (NIL) NIL-OrC1 and 9311 were collected for RNA extraction. RNA samples were sent to Genedenovo Biotechnology Co., Ltd (Guangzhou, China) for RNA sequencing. The reconstruction of transcripts was carried out with Cufflinks (http://cole-trapnell-lab.github.io/cufflinks/install/) and TopHat2 (Kim et al. 2013). Principal component analysis (PCA) was performed with R package Rmodest (http://www.r-project.org/). Three biological replicates were tested. The PCA of the samples based on the number of fragments per kilobase of exon per million fragments mapped (FPKM) values showed that one replicate of NIL-OrC1 (NIL-3) did not cluster with the other two (Supplementary 3), so only two replicates of NIL-OrC1 were used for further analysis. Differentially expressed genes (DEGs) were identified with a fold change ≥ 2 and a false discovery rate (FDR) using edgeR package (http://www.rproject.org/). DEGs were then subjected to enrichment analysis of Gene Ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway. Data analyses were performed by Genedenovo Biotechnology Co., Ltd.
Rice transformation
Overexpression vectors were constructed by amplifying the OrC1 from cDNA of CSSL52, the PCR products were digested by cloning into the binary vector pBWA(V)HS. The constructed plasmids were introduced into Agrobacterium tumefaciens strain EHA105 for infection. Plasmid construction and rice transformation were performed by Biorun Biosciences Co., Ltd (Wuhan, China) following their standard procedures and protocols.
Anthocyanin metabolite determination
The leaf sheath of 15-day seedlings of NIL, 9311, OrC1 overexpression lines (OE) and Nipponbare, the upper one third of hull at 10 days after flowering of OE and Nipponbare were collected. The sample extracts were analyzed using an LC-ESI-MS/MS system (HPLC, Shim-pack UFLC SHIMADZU CBM30A system, www.shimadzu.com.cn/; MS, Applied Biosystems 4500 Q TRAP, www.appliedbiosystems.com.cn/). Metabolite quantification was performed using a scheduled multiple reaction monitoring (MRM) method, which has been previously described (Chen et al. 2013). The identified metabolites were subjected to orthogonal partial least squares discriminant analysis (OPLS-DA), and metabolites with |Log2 (fold change)|≥ 1, p-value < 0.05, and VIP(variable importance in project) ≥ 1 were considered as differentially accumulated metabolites (DAMs) (Zhang et al. 2020). The sample preparation, extract analysis, metabolite identification and quantification were performed at Wuhan MetWare Biotechnology Co., Ltd. (www.metware.cn) following their standard procedures and were previously described in details by (Chen et al. 2013; Zhu et al. 2013; Kanehisa et al. 2000).
Results
Fine mapping of OrC1 using chromosome segment substitution lines
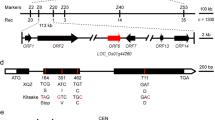
In our previous study, a set of Chinese common wild rice chromosome segment substitution lines (CSSL) was developed (Qiao et al. 2016). The donor parent, O. rufipogon, has a significant purple coloration in apiculus, leaf sheath, and stigma and black hull. The recipient parent, an indica variety 9311, has no purple pigment in the whole plant. In total 150 CSSLs were investigated under multiple environments for purple coloration. One hundred twenty-one SSR and 62 InDel markers, which were polymorphic between the two parents and evenly distributed on all 12 chromosomes, were selected for genotyping. In the CSSL populations, the purple apiculus, leaf sheath and stigma were completely correlated, while the purple or black hull was independently segregated. Using SSR/InDel genotyping, one QTL related to purple apiculus, leaf sheath and stigma (hereafter named purple coloration trait) was identified under multiple environments and located near RM314 on chromosome 6 (Fig. 1a).

Fine mapping of OrC1. (a) Genetic map of chromosome 6 of CSSL52. The red star represents the marker closely linked with the QTL for the purple coloration trait. (b, c) The location of OrC1 was narrowed down to a 131-kb interval between markers RM19552 and RM19561. The number of recombinants obtained is indicated under the marker names, and the number of individuals (n) used in mapping are shown on the left. (d) Seventeen predicted open reading frames (ORF) were identified in the fine mapped region. ORF14 encodes a homolog of C1 gene. The deletions in 9311 and Nipponbare alleles were shown in (d) (color figure online)
One chromosome segment substitution line, CSSL52, which harbors this QTL and purple coloration trait, was selected for further fine mapping of purple genes. CSSL52 carried only one wild rice introgression segment and exhibited purple apiculus, leaf sheath and stigma (Fig. 1a and Fig. 2). We observed no significant difference in other agronomic traits such as plant height and heading date, between 9311 and CSSL52, indicating that the single introgression segment from wild rice was only involved in purple coloration. CSSL52 was back-crossed with 9311 to produce F1 plants, which showed purple apiculus, leaf sheath and stigma (Fig. 2). The color of apiculus, leaf sheath and stigma in F2 population was completely correlated. A nearly 3:1 segregation ratio (1492/472) of purple coloration to achromatic was observed in 1,964 F2 individuals, indicating that the purple coloration trait was controlled by a single dominant gene. Using F2 segregating population, this gene was narrowed down to a 131-kb region between markers RM19552 and RM19561 with recessive class analysis (Fig. 1c), in which there were 17 predicted open reading frames (Supplementary 4). This region contains an expressed gene (LOC_Os06g10350) encoding a R2R3 MYB transcription factor that has been reported as the rice homolog of maize C1 gene (Yuan et al. 2018). Sequence analysis of Os06g10350 revealed that 9311 contained a 10-bp deletion at the start of the third exon, and Nipponbare contains a 3-bp deletion in the second exon (Fig. 1d). Therefore, we focused on Os06g10350 as a candidate for purple coloration and named it as OrC1. Furthermore, a NIL of OrC1 was developed using the positive F2 individual of CSSL52/9311.

Phenotype of CSSL52, 9311 and F1 individual. (a–e) CSSL52 has purple apiculus (c), leaf sheath (d) and stigma (a–c) compared with 9311. (f) The F1 of CSSL52/9311 showed the same purple coloration trait as CSSL52
Characterization of OrC1 from wild rice
OrC1 encodes a 272 amino-acid protein containing two highly conserved SANT domains, similar to the R2 and R3 motifs of R2R3-MYB transcriptional factors in plants. A phylogenetic tree was constructed using the amino acid sequences of OrC1 and other anthocyanin biosynthesis related genes from several plant species. Phylogenetic analysis showed that OrC1 exhibited highest homology to OsC1 in cultivated rice and R2R3 MYB transcription factors in maize (Fig. 3a). To investigate the expression patterns of C1 in wild and cultivated rice, the levels of C1 mRNA were quantified in different tissues including leaves, leaf sheaths, stems, and panicles of NIL-OrC1 and 9311, at the booting stage and 3 days after flowering. No significant difference was detected between NIL and 9311. C1 transcripts were detected at a high level in leaf sheaths, the lowest level of OrC1 transcripts was detected in stems. Three days after flowering, the expression levels of C1 in panicles were significantly increased in both NIL and 9311 (Fig. 3b).

Characterization of OrC1. a Phylogenetic analysis of wild rice OrC1 and other R2R3-MYB proteins. b Expression pattern of OrC1 gene in different tissues at two time points. c Subcellular localization of C1 (35S::OrC1-GFP) in rice protoplasts and tobacco leaf epidermal cells. Scale bar 20 μm
To examine the subcellular localization of OrC1 protein, an OrC1:GFP fusion gene was generated and transformed into rice protoplasts and tobacco leaves under the control of the cauliflower mosaic virus 35S promoter. As shown in Fig. 3c, the control GFP protein was distributed throughout the entire cell, whereas the OrC1-GFP fluorescent signals were exclusively localized in nucleus, consistent with the function of OrC1 as a transcription factor.
Transcriptome analysis revealed differentially expressed genes between NIL and 9311
To further investigate the molecular mechanism of OrC1-mediated anthocyanin synthesis pathway, we performed a transcriptome analysis with the leaf sheath of 9311 and NIL at the seedling stage, because the purple color phenotype was most obvious at this stage. In total, 2388 DEGs were identified with the stringent criteria (|log2(FC)|> 1, and FDR < 0.05). Of these DEGs, 1,225 were up-regulated and 1,163 were down-regulated in NIL compared with 9311. Furthermore, all DEGs were assigned to 98 KEGG. KEGG pathway enrichment analysis revealed that OrC1 mostly affected the expression of genes involved in phenylpropanoid biosynthesis, diterpenoid biosynthesis, plant hormone signal transduction, and flavonoid biosynthesis (Fig. 4a).

OrC1 regulates gene expressions. a Top 20 significant enrichment KEGG pathway; b Relative expression levels of seven structure genes involved in anthocyanin synthesis determined by qRT- PCR. The values represent the mean ± s.d. *P < 0.05 and **P < 0.01 indicate significant differences in two-tailed Student’s t-tests. Three biological replicates. The actin gene was used for normalization
Seven structural genes involved in the anthocyanin metabolic pathway were used for the validation of the sequencing results. The relative gene expression levels and FPKM (RNA-Seq) of the seven genes showed similar regulatory patterns in the NIL compared with 9311. A linear regression coefficient (R2) of 0.9711 was obtained between the results of qRT-PCR and RNA-Seq. These consistencies validated the results of RNA-Seq analysis. All seven anthocyanin biosynthetic genes were consistently up-regulated in the NIL compared with those in 9311, among them, expression levels of OsPAL, OsDFR, OsANS, OsF3′H, and OsF3H, were significantly increased in the NIL (Fig. 4b).
Anthocyanin biosynthesis are largely influenced by some key pathways and MBW complex. According to transcriptome results, 116, 84, and 19 DEGs were detected in phenylpropanoid biosynthesis, MAPK signaling and flavonoid biosynthesis pathways, respectively (Fig. 5a). At the same time, five R2R3-MYB transcription factors, six bHLH proteins and four OsWD40 proteins were also found in DEGs (Fig. 5b, Supplementary 5), including the cloned gene Rc (Os07g0211500) which was involved in proanthocyanin synthesis (Furukawa et al. 2007).

Heat map diagram of expression levels for anthocyanin biosynthesis related genes in a three KEGG pathways and b MBW complex. The heat map was drawn according to FPKM values. Columns and rows in the heat map represent samples and genes, respectively. The KEGG pathway or gene ID were displayed on the left. Color scale indicates fold changes in gene expression (color figure online)
Overexpression of OrC1 results in purple apiculus in transgenic Nipponbare
To explore the biological function of OrC1, its ORF was transformed into japonica cultivar Nipponbare with the 35S promoter. The transgenic individuals were confirmed by PCR analysis with gene-specific primers. Ten positive transgenic lines were analyzed by qRT-PCR to determine the transgene expression level and two independent lines, OE1 and OE2, which differed in transcription levels were selected for additional analysis (Supplementary 6). Under natural conditions, only the apiculus of T1 plants showed a purple pigmentation phenotype, the sheath was as the same as wild type Nipponbare. Purple apiculus of transgenic plants could be observed when panicles were exposed to sunlight at the initial heading stage. It turned into brown color at the wax ripeness stage. T2 transgenic plants showed a perfect segregation ratio 3:1 of purple or brown apiculus to normal. Both purple and brown apiculus could be easily distinguished at the fully ripen stage. There is no difference between the two OrC1 overexpression lines, OE1 and OE2, and no other phenotypes were different between OE and Nipponbare except apiculus color (Fig. 6a). Seven above-mentioned structural genes were also detected by qRT-PCR, only OsCHI, OsF3H and OsANS showed higher expression level in OE lines than Nipponbare (Fig. 6b).

Overexpression of OrC1 results in purple apiculus of Nipponbare. a Phenotypes of transgenic OrC1 overexpression line (OE) and wild type Nipponbare (NIP) plants. The purple coloration was only found in the apiculus of transgenic plant. From left to right: whole plant, young spikelet, mature spikelet, mature and young seeds of NIP and OE. b The relative expression levels of seven structure genes determined by qRT-PCR in NIP and two OE. Three biological replicates. The values represent the mean ± s.d. *P < 0.05 and **P < 0.01 indicate significant differences in two-tailed Student’s t-tests. The actin gene was used for normalization
Identification of differential metabolites in the anthocyanin biosynthesis pathways
Liquid chromatography-electrospray ionization-tandem mass spectrometry analysis was used to quantify the anthocyanin content in leaf sheath of both NIL and 9311. A total of 28 metabolites of anthocyanins and pro-anthocyanidins were detected, and the relative contents for each metabolite were normalized before being subjected to downstream data analyses. Among them, 11 metabolites in NIL were significantly higher than that in 9311, two metabolites were higher in 9311 than that in NIL. For Nipponbare and transgenic plants, given the OE lines only have purple apiculus and no color in other organs, the leaf sheath and upper one third of the hull of two OE lines and Nipponbare were used for metabolite analysis. A total of 13 metabolites of anthocyanins were detected, and among them six metabolites in apiculus of OE were significantly higher than that in Nipponbare (Fig. 7). In sheath, there is no significant difference between OE and Nipponbare. No difference was found in apiculus or sheath between the two OE lines.

Identification of the differentially accumulated metabolites between NIL and 9311 (above), and between OE and Nipponbare (below). Red * indicates a significant increase at fold change ≥ 2, and blue * indicates a significant decrease at fold change ≤ 0.5 (color figure online)
The DAMs between pairs of samples were determined. Comparative analysis of the four groups of DAMs revealed that six common anthocyanin metabolites were differentially accumulated in NIL-Sheath vs 9311-Sheath, OE- apiculus vs Nip-apiculus and OE-apiculus vs OE-Sheath, including rosinidin, delphinidin and four cyanidins (Supplementary 7). And all of them are up regulated in NIL compared with 9311, and in OE lines compared with Nipponbare.
Three genes were involved in delphinidin and cyanidin biosynthesis pathway in the flavonoid biosynthesis KEGG pathway, map 00,941 (https://www.genome.jp/kegg-bin/show_pathway?map00941) (Supplementary 8). According to our transcriptome profile results, two genes, Os06g0626700 (OsINS) and Os01g0372500 (OsANS), were upregulated and one gene Os04g0630800 (OsANR) was down regulated in NIL compared with 9311 (Supplementary 8, blue rectangle). OsANS was also significantly upregulated in OE compared with Nipponbare (Fig. 6b). We tested OsINS and OsANR in OE lines and Nipponbare, the OsINS was upregulated and the OsANR was downregulated in OE lines but not significantly (data not shown), indicated that OrC1 regulated some of the same genes in indica and japonica background.
Haplotype analysis
The promoter and coding regions of C1 alleles from 180 rice accessions were isolated and analyzed. The haplotype analysis of promoter region showed that the variation in this region was not associate with coloration traits (Supplementary 9). For coding region, 12 haplotypes (H_1 to H_12) were identified. Among them, H_2, 9, 10, 12 were functional, and H_1, 3, 4–8, 11 were non-functional with different deletions (Fig. 8a). Only two O. rufipogon and seven O. nivara have non-functional allele. The combination of the phenotypes and genotypes of the association panel showed that 24 cultivated rice accessions with non-functional allele showed at least one coloration trait (purple hull or stigma), indicating the presence of other determinant factors besides C1. All indica and wild rice in H_2 displayed purple apiculus, leaf sheath and stigma, almost all japonica in H_2 only displayed purple apiculus (Supplementary 1). Haplotype network analysis showed that the most frequent non-functional alleles in indica and japonica are different, which were H_1 and H_7 in indica and japonica, respectively (Fig. 8b), indicating multiple origins of C1 in two subspecies.

Genotype analysis of coding region of C1. a Sequence polymorphism of different haplotypes of C1 in 180 accessions. No. of Acc.: number of accessions, I, indica; J, japonica; R, O. rufipongon; N, O. nivara; F, functional; Non-F, non-functional. b Haplotype network of C1
Discussion
Although the spatiotemporal regulation of anthocyanin biosynthesis has been well documented and the chromogen C gene, a major coloration gene, has also been fine mapped in cultivated rice (Saitoh et al. 2004; Liu et al. 2012; Zhao et al. 2016), the allele in wild rice or full functional allele of C gene in rice has not been cloned. Given the high heterogeneity in the genome of O. rufipogon, it is difficult to clone novel genes which have been lost or weakened in cultivated rice during domestication. The CSSLs in this study has been testified to be an excellent platform for large-scale gene discovery in wild rice (Qi et al. 2018; Li et al. 2018; Zhang et al. 2020). Our study identified a stablely expressed QTL associated with purple coloration which harbor OrC1, emphasizing the importance of CSSLs to uncover useful genes of wild relatives. Although the other organs of NIL such as stem and leaf have no purple coloration, the C1 gene constitutively expressed in all tissues, and has consistent expression patterns in both NIL and 9311 (Fig. 3b). We also sequenced the promoter region of C1 gene in 180 rice accessions, no haplotype was associated with purple traits (Supplementary 1 and 9), indicating that the anthocyanin biosynthesis in rice was mostly controlled by C1 protein function but not at gene expression level. As domestication related gene, we deduced that the artificial selection occurs in its coding region but not the promoter region.
Anthocyanin biosynthesis in different rice tissues is primarily controlled by R2R3-MYB TFs and the ternary MBW complexes (Sakamoto et al. 2001; Zheng et al. 2019). In this study, the NIL-OrC1 in indica background shown a co-segregated purple leaf sheath, stigmas and apiculus while the OE lines of OrC1 in japonica background only had a purple apiculus. We compared other bHLH and WDR genes in NIL, 9311 and Nipponbare. All the gene sequences in NIL is exactly the same as 9311, which indicated that only one coloration gene OrC1 in NIL was from wild rice. Comparison between 9311 and Nipponbare showed that six bHLH genes and three WDR genes have non-synonymous SNP (Supplementary 10). Therefore, we predicted that the different purple traits in NIL and OE were due to their different MBW complex. Moreover, both 9311 and Nipponbare have a non-functional allele of Os04g0557500 (Supplementary 10), which was reported as a tissue regulator and controls pigmentation in hull (Sun et al. 2018). Hence, both NIL and OE accumulated pigments only in apiculus but not the hull. Previously studies demonstrated that OsC1 is the determinant factor of anthocyanin biosynthesis in leaf sheath and apiculus (Fan et al. 2008; Chin et al. 2016), but has not been reported to be involved in pigmentation in stigmas. It is likely that an MBW complex consisting of OrC1 and a tissue-specific regulator regulated the anthocyanin biosynthesis in stigmas of NIL. Further work is needed to determine the exact bHLH TF combined with OrC1. Moreover, haplotype analysis showed that the functional C1 allele led to anthocyanin accumulation in three tissues in indica and only in apiculus in japonica (Fig. 8 and Supplementary 1), consistent with the results of OrC1 function in 9311 and Nipponbare (Figs. 2 and 6). Besides C1, other redundant genes must be involved in the regulation of anthocyanin biosynthesis in rice stigma.
Transcriptome profiles revealed that almost all the structural genes in the flavonoid biosynthesis pathway were induced by OrC1 in NIL, but only CHI, F3H and ANS were induced in OE (Figs. 4 and 6). In rice, structure genes are activated by MYB and MBW with redundancy (Zheng et al. 2019), we believe that some bHLH TFs of 9311 is functional and some of Nipponbare is non-functional, the coloration of NIL was due to both OrC1 and MBW complex, but the purple apiculus of OE lines was only caused by OrC1 protein. Furthermore, OrC1 is involved not only in flavonoid and phenylpropanoid biosynthesis, but also in singling pathways like MAPK, and regulated bHLH and WD40 genes (Fig. 5). This indicates that C1 gene is involved in other physiological processes besides anthocyanin biosynthesis. The exact metabolites of anthocyanin that C1 gene produced is still not clear. Shin et al. (2006) transformed maize C1 and R-S regulatory genes into rice using endosperm specific promoters, the produced flavonoids mostly are anthocyanins including dihydroquercetin (taxifolin), dihydroisorhamnetin (3′-O-methyl taxifolin) and 3′-O-methyl quercetin. We have detected six DAMs accumulated in both NIL and OE line (Supplementary 7). Because we only tested the anthocyanin metabolites, so other flavonoids metabolites are not excluded. Take together the seven structural gene expression profiles, we deduced that OrC1 regulated OsCHI, OsF3H, OsANS and produce six anthocyanin metabolites independently from the MBW complexes. These results set a foundation to understand the regulatory mechanisms of C1 gene in the anthocyanin biosynthesis pathway.
The C1 involved anthocyanin biosynthesis pathway pre-exists in wild rice but is absent in most cultivated rice, indicating a strong negative human selection of this trait. Haplotype network analysis showed that the functional mutations of C1 had multiple origins and been selected independently in two subspecies (Fig. 8). In Zheng et al. (2019)’s study, all C1 alleles of the wild rice accessions had no null mutations. In this study, we found that non-function allele in both O. rufipogon and O. nivara (Fig. 8; Supplementary 1). There are two main hypotheses for the cultivated rice domestication: single origin and multiple origin (Choi et al. 2017). Some well-documented domestication genes of rice, such as sh4 for seed shattering (Li et al. 2006), and prog1 for erect growth (Tan et al. 2008), supporting the single-origin hypothesis. Our haplotype analysis of C1 supports multiple origin. More interestingly, the non-functional C1 allele is much closer to O. nivara than O. rufipongon, which supports the theory that the origin of cultivated rice is O. nivara, an annual wild rice species that is generally considered an intermediate between O. rufipongon and cultivated rice; however, the direct ancestor of cultivated rice remains controversial.
References
Aizza LCB, Dornelas MC (2011) A genomic approach to study anthocyanin synthesis and flower pigmentation in passionflowers. J Nucleic Acids 37:15–17
Baudry A, Heim MA, Dubreucq B, Caboche M, Weisshaar B, Lepiniec L (2004) TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J 39:366–380
Chagne D, Lin-Wang K, Espley RV, Volz RK, How NM, Rouse S, Brendolise C, Carlisle CM, Kumar S, De Silva N, Micheletti D, McGhie T, Crowhurst RN, Storey RD, Velasco R, Hellens RP, Gardiner SE, Allan AC (2013) An ancient duplication of apple MYB transcription factors is responsible for novel red fruit-flesh phenotypes. Plant Physiol 161:225–239
Chen W, Gong L, Guo Z, Wang WS, Zhang HY, Liu XQ, Yu SB (2013) A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: application in the study of rice metabolomics. Mol Plant 6:1769–1780
Cho YG, Ishii T, Temnykh S, Chen X, Lipovich L, McCouch SR, Park WD, Ayres N, Cartinhour S (2000) Diversity of microsatellites derived from genomic libraries and GenBank sequences in rice (Oryza sativa L.). Theor Appl Genet 100:713–722
Choi J, Platts A, Fuller D, Hsing Y, Wing R, Purugganan M (2017) The rice paradox: multiple origins but single domestication in Asian rice. Mol Biol Evol 34:969–979
Chin H, Wu Y, Hour A, Hong C, Lin Y (2016) Genetic and evolutionary analysis of purple leaf sheath in rice. Rice 9:8
de Vetten N, Quattrocchio F, Mol J, Koes, (1997) The an11 locus controlling flower pigmentation in petunia encodes a novel WD-repeat protein conserved in yeast, plants, and animals. Genes Dev 11:1422–1434
Fan FJ, Fan YY, Du JH (2008) Fine mapping of C (chromogen for anthocyanin) gene in rice. Rice Sci 15:1–6
Furukawa T, Maekawa M, Oki T, Suda I, Iida S, Shimada H, Takamure I, Kadowaki K (2007) The Rc and Rd genes are involved in pro-anthocyanidin synthesis in rice pericarp. Plant J 49:91–102
Gao DY, He B, Zhou YH, Sun LH (2011) Genetic and molecular analysis of a purple sheath somaclonal mutant in japonica rice. Plant Cell Rep 30:901–911
Grotewold E (2006) The genetics and biochemistry of floral pigments. Ann Rev Plant Biol 57:761–780
Hugueney P, Provenzano S, Verriès C, Ferrandino A, Meudec E, Batelli G, Merdinoglu D, Cheynier V, Schubert A, Ageorges A (2009) A novel cation-dependent O-methyltransferase involved in anthocyanin methylation in grapevine. Plant Physiol 150:2057–2070
Honda C, Kotoda N, Wada M, Kondo S, Kobayashi S, Soejima J, Zhang Z, Tsuda T, Moriguchi T (2002) Anthocyanin biosynthetic genes are coordinately expressed during red coloration in apple skin. Plant Physiol Biochem 40:955–962
Hichri I, Barrieu F, Bogs J, Kappel C, Delrot S, Lauvergeat V (2011) Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J Exp Bot 62:2465–2483
Hu W, Zhou T, Han Z, Tan C, Xing Y (2020) Dominant complementary interaction between OsC1 and two tightly linked genes, Rb1 and Rb2, controls the purple leaf sheath in rice. Theor Appl Genet 133:2555–2566
Huang X, Kurata N, Wei X, Wang Z, Wang A, Zhao Q, Zhao Y, Liu K, Lu H, Li W, Guo Y, Lu Y, Zhou C, Fan D, Weng Q, Zhu C, Huang T, Zhang L, Wang Y, Feng L, Furuumi H, Kubo T, Miyabayashi T, Yuan X, Xu Q, Dong G, Zhan Q, Li C, Fujiyama A, Toyoda A, Lu T, Feng Q, Qian Q, Li J, Han B (2012) A map of rice genome variation reveals the origin of cultivated rice. Nature 490:497–501
Jian W, Cao H, Yuan S, Liu Y, Lu J, Lu W, Li N, Wang J, Zou J, Tang N, Xu C, Cheng Y, Gao Y, Xi W, Bouzayen M, Li Z (2019) SlMYB75, an MYB-type transcription factor, promotes anthocyanin accumulation and enhances volatile aroma production in tomato fruits. Hortic Res 6:22
Jin W, Wang H, Li M, Wang J, Yang Y, Zhang X (2016) The R2R3 MYB transcription factor PavMYB10.1 involves in anthocyanin biosynthesis and determines fruit skin colour in sweet cherry (Prunus avium L.). Plant Biotechnol J 14:2120–2133
Jun JH, Liu C, Xiao X, Dixon RA (2015) The transcriptional repressor MYB2 regulates both spatial and temporal patterns of proanthocyandin and anthocyanin pigmentation in Medicago truncatula. Plant Cell 27:2860–2879
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Kim D, Pertea G, Trapnell C, Pimentel H, Ryan K, Salzberg S (2013) TopHat2: accurat e alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:36
Li CB, Zhou A, Sang T (2006) Rice domestication by reducing shattering. Science 311:1936–1939
Liu X, Sun X, Wang W, Ding H, Liu W, Li G, Jiang M, Zhu C, Yao F (2012) Fine mapping of Pa-6 gene for purple apiculus in rice. Plant Biotechnol J 55:218–225
Li J, Xu R, Wang C, Qi L, Zheng XM, Wang WS, Ding YB, Zhang LZ, Wang YY, Cheng YL, Zhang LF, Qiao WH, Yang QW (2018) A heading date QTL, qHD7.2, from wild rice (Oryza rufipogon) delays flowering and shortens panicle length under long-day conditions. Sci Rep 8:2928
Livak K, Schmittgen T (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-[Δ][Δ]Ct method. Methods 25:402–408
Mikami I, Takahashi A, Khin-Thidar SY (2000) A candidate for C (chromogen for anthocyanin) gene. Rice Genet Newsl 17:54–56
Qi L, Ding YB, Zheng XM, Xu R, Zhang LZ, Wang YY, Wang XN, Zhang LF, Cheng YL, Qiao WH, Yang QW (2018) Fine mapping and identification of a novel locus qGL12.2 controls grain length in wild rice (Oryza rufipogon Griff.). Theor Appl Genet 131:1497–1508
Qiao WH, Qi L, Cheng ZJ, Su L, Li J, Sun Y, Ren JF, Zheng XM, Yang QW (2016) Development and characterization of chromosome segment substitution lines derived from Oryza rufipogon in the genetic background of O. sativa spp. indica cultivar 9311. BMC Genomics 17:580
Saitoh K, Onishi K, Mikami I, Thidar K, Sano Y (2004) Allelic diversification at the C (OsC1) locus of wild and cultivated rice: nucleotide changes associated with phenotypes. Genetics 168:997–1007
Sakamoto W, Ohmori T, Kageyama K, Miyazaki C, Saito A, Murata M, Noda K, Maekawa M (2001) The purple leaf (Pl) locus of rice: the Pl(W) allele has a complex organization and includes two genes encoding basic helix-loop-helix proteins involved in anthocyanin biosynthesis. Plant Cell Physiol 42:982–991
Sun X, Zhang Z, Chen C, Wu W, Ren N, Jiang C, Yu J, Zhao Y, Zheng X, Yang Q, Zhang H, Li J, Li Z (2018) The C-S-A gene system regulates hull pigmentation and reveals evolution of anthocyanin biosynthesis pathway in rice. J Exp Bot 69:1485–1498
Shin Y, Park H, Yim S, Baek N, Lee C, An G, Woo Y (2006) Transgenic rice lines expressing maize C1 and R-S regulatory genes produce various flavonoids in the endosperm. Plant Biotechnol J 4:303–315
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tan L, Li X, Liu F, Sun X, Li C, Zhu Z, Fu Y, Cai H, Wang X, Xie D, Sun C (2008) Control of a key transition from prostrate to erect growth in rice domestication. Nat Genet 40:1360–1364
Tian J, Peng Z, Zhang J, Song T, Wan H, Zhang M, Yao Y (2015) McMYB10 regulates coloration via activating McF3’H and later structural genes in ever-red leaf crabapple. Plant Biotechnol J 13:948–961
Vinayagam R, Xu B (2015) Antidiabetic properties of dietary flavonoids: a cellular mechanism review. Nutr Metab 12:60
Xu W, Dubos C, Lepiniec L (2015) Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci 20:176–185
Xu Z, Yang Q, Feng K, Yu X, Xiong A (2020) DcMYB113, a root-specific R2R3-MYB, conditions anthocyanin biosynthesis and modification in carrot. Plant Biotechnol J 18:1585–1597
Yuan H, Zeng X, Shi J, Xu Q, Wang Y, Jabu D, Sang Z, Nyima T (2018) Time-course comparative metabolite profiling under osmotic stress in tolerant and sensitive Tibetan hulless barley. BioMed Res Int 9415409:1–12
Zhang LZ, Huang JF, Wang YY, Xu R, Yang ZY, Zhao ZG, Liu SJ, Tian Y, Zheng XM, Li F, Wang JR, Song Y, Li JQ, Cui YX, Zhang LF, Cheng YL, Lan JH, Qiao WH, Yang QW (2020) Identification and genetic analysis of qCL1.2, a novel allele of the “green revolution” gene SD1 from wild rice (Oryza rufipogon) that enhances plant height. BMC Genet 21:62
Zhang Q, Wang L, Liu Z, Zhao Z, Zhao J, Wang Z, Zhou G, Liu P, Liu M (2020) Transcriptome and metabolome profiling unveil the mechanisms of Ziziphus jujuba Mill. peel coloration. Food Chem 312:125903
Zhang Y, Su JB, Duan S, Ao DJR, Liu J, Wang P, Li YG, Liu B, Feng DR, Wang JF, Wang HB (2011) A highly efficient rice green tissue protoplast system for transient gene expression and studying light/chloroplast-related processes. Plant Methods 7:30
Zhao SS, Wang CH, Ma J, Wang S, Tian P, Wang JL, Cheng ZJ, Zhang X, Guo XP, Lei CL (2016) Map-based cloning and functional analysis of the chromogen gene C in rice (Oryza sativa L.). J Plant Biol 59:496–505
Zheng J, Wu H, Zhu HB, Huang C, Liu C, Chang YS, Kong ZC, Zou ZH, Wang GW, Lin YJ, Chen H (2019) Determining factors, regulation system, and domestication of anthocyanin biosynthesis in rice leaves. New Phytologis 223:705–721
Zhu Z, Schultz AW, Wang J, Johnson CH, Yannone SM, Patti GJ, Siuzdak G (2013) Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat Protoc 8:451–456
Acknowledgements
We especially thank Dr. Peng Zhang (The University of Sydney, Australia) and Dr. Hao Chen (Huazhong Agricultural University, China) for critical reading of the manuscript. This work was supported by the National Key R&D Program of China (2016YFD0100101) and the National Natural Science Foundation of China (31471471).
Author information
Authors and Affiliations
Contributions
WH Qiao, JH Lan and QW Yang conceived and designed the experiments. WH Qiao and YY Wang performed the experiments and wrote the paper. R Xu, ZY Yang, JR Wang and JF Huang analyzed the data. Y Sun, L Su and LZ Zhang performed fine mapping. SJ Liu, YL Tian, LM Chen and X Liu contributed to field investigation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Jessica Rutkoski.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
122_2021_3787_MOESM9_ESM.tif
Genotype analysis of promoter region of C1. (a) Sequence polymorphism of different haplotypes of C1 promoter from 180 accessions. No. of Acc.: number of accessions, I: O. sativa. indica, J: O. sativa. japonica, R: O. rufipongon, N: O. nivara. (b) Haplotype network of C1 promoter.
Supplementary file 8 (TIF 8402 kb)
122_2021_3787_MOESM10_ESM.tif
Genotype analysis of promoter region of C1. (a) Sequence polymorphism of different haplotypes of C1 promoter from 180 accessions. No. of Acc.: number of accessions, I: O. sativa. indica, J: O. sativa. japonica, R: O. rufipongon, N: O. nivara. (b) Haplotype network of C1 promoter.
Supplementary file 8 (TIF 2267 kb)
Rights and permissions
About this article

Cite this article
Qiao, W., Wang, Y., Xu, R. et al. A functional chromogen gene C from wild rice is involved in a different anthocyanin biosynthesis pathway in indica and japonica. Theor Appl Genet 134, 1531–1543 (2021). https://doi.org/10.1007/s00122-021-03787-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-021-03787-1