
Abstract
Key message
Identification of marker–trait associations and trait-associated haplotypes in breeding germplasm identifies regions under selection and highlights changes in haplotype diversity over decades of soybean improvement in Canada.
Abstract
Understanding marker–trait associations using genome-wide association in soybean is typically carried out in diverse germplasm groups where identified loci are often not applicable to soybean breeding efforts. To address this challenge, this study focuses on defining marker–trait associations in breeding germplasm and studying the underlying haplotypes in these regions to assess genetic change through decades of selection. Phenotype data were generated for 175 accessions across multiple environments in Ontario, Canada. A set of 76,549 SNPs were used in the association analysis. A total of 23 genomic regions were identified as significantly associated with yield (5), days to maturity (5), seed oil (3), seed protein (5) and 100-seed weight (5), of which 14 are novel. Each significant region was haplotyped to assess haplotype diversity of the underlying genomic region, identifying ten regions with trait-associated haplotypes in the breeding germplasm. The range of genomic length for these regions (7.2 kb to 6.8 Mb) indicates variation in regional LD for the trait-associated regions. Six of these regions showed changes between eras of breeding, from historical to modern and experimental soybean accessions. Continued selection on these regions may necessitate introgression of novel parental genetic diversity as some haplotypes were fixed within the breeding germplasm. This finding highlights the importance of studying associations and haplotype diversity at a breeding program scale to understand breeders’ selections and trends in soybean improvement over time. The haplotypes may also be used as a tool for selection of parental germplasm to inform breeder’s decisions on further soybean improvement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The relationships between genotype and phenotype in soybean using genome-wide association (GWAS) are often studied in diverse germplasm panels; however, the findings from these types of studies are often difficult to apply directly to regional crop improvement efforts. A first step in using data from germplasm studies for crop improvement is thorough characterization of germplasm in the breeding programs where selection is occurring. A second step is to apply the knowledge gained through specific studies to improve traits of high importance using all available resources. As most agronomic traits in soybean are inherited in a quantitative fashion with effects from environment, efforts must be undertaken to identify genomic regions with associations to these traits in a breeding program environment to improve breeder selections for crop improvement (Chaudhary et al. 2015).
Genome-wide association studies have been extensively conducted in soybean using diverse germplasm panels to identify genomic regions with associations to agronomic traits. Sonah et al. (2015) used 139 accessions representing Canadian short-season diversity to characterize major agronomic traits using 17 k SNP markers, identifying a total of 25 genomic regions associated with multiple agronomic traits corresponding to known trait-associated regions from quantitative trait loci (QTL) studies. A comprehensive study of 809 soybean accessions assessed 84 agronomic traits, identifying 245 significant loci including many previously identified genomic regions (Fang et al. 2017). The largest GWAS study in soybean used 12,000 accessions from the USDA germplasm bank to identify SNPs across five chromosomes with significant associations with seed protein and seed oil (Bandillo et al. 2015). Wild soybean has also been characterized using association analysis as demonstrated by Leamy et al. (2017), revealing novel regions associated with seed traits which may be useful in future breeding efforts.
Following up on their GWAS analysis, Bandillo et al. (2015) conducted further haplotyping of the significant genomic regions, identifying regional trends in haplotype frequency which may be attributed to historical culinary preferences. Other haplotyping efforts have been undertaken for pathway-specific genes such as the E genes controlling maturity in soybean (Tardivel et al. 2014; Langewisch et al. 2014), soybean cyst nematode resistance (Liu et al. 2017) and salinity tolerance (Patil et al. 2017).
The comparison of genome-wide haplotypes in domesticated and landrace soybeans and their wild ancestors have shown that, on average, linkage block size has increased in modern soybean cultivars while the number of linkage blocks in a given accession has decreased (Song et al. 2015). Lam et al. (2010) also identified tag SNPs using a linkage disequilibrium (LD)-based haplotyping approach in wild and cultivated soybeans to characterize genomic change related to domestication and selection. Haplotypes have also been used as input for association analysis, as the multi-allelic nature allows for better capture of the underlying alleles in the soybean genome (Contreras-Soto et al. 2017). Genome-wide haplotyping in soybean has recently been demonstrated (GmHapMap), where the entire catalogue of soybean genes has been haplotyped, demonstrating that haplotypes can be used to identify the trait-associated alleles responsible for phenotypic differences in germplasm (Torkamaneh et al. 2019). Understanding haplotype frequencies in breeding germplasm can help breeders identify important genomic regions for future crop improvement.
This study aims to improve the understanding of marker–trait associations in a single breeding program and assess whether selection has altered these associations throughout the breeding process. We hypothesize that marker–trait associations identified in breeding germplasm relate directly to the impacts of breeder selections. To understand the genetic impacts of soybean breeding within a single breeding program, this study has several objectives: (1) test and identify marker–trait associations in soybean breeding germplasm for major soybean traits, (2) haplotype the genomic regions underlying marker–trait associations within a breeding program, (3) assess the changes in haplotype frequency within these haplotyped genomic regions across decades of soybean breeding and (4) define favorable haplotypes for soybean improvement in regional breeding germplasm and identify markers associated with these haplotypes.
Materials and methods
Panel composition and genotypes
The 296 accessions studied capture the pedigree relationships in a breeding program over decades of selection (Table S1). Modern elite cultivars were traced to historical accessions using pedigree records in the University of Guelph soybean breeding program (Bruce et al. 2019a). Additional accessions were collected from several RIL (recombinant inbred line) populations (ten Chinese by Canadian RILs and eight Glycine max by Glycine soja RILs). Genotyping methods and data were described in Bruce et al. (2019b), with 76,549 genome-wide SNPs available for analysis in this study. In brief, the 296 accessions were genotyped using a genotyping-by-sequencing protocol (Elshire et al. 2011; Sonah et al. 2013) using multiple restriction enzymes across the panel (Bruce et al. 2019b), with GBS-derived reads aligned against the soybean Williams 82 reference genome (Gmax_275_Wm82.a2.v1) (Schmutz et al. 2010) and SNPs called using Fast-GBS (Torkamaneh et al. 2017). A minor allele frequency (MAF) filter of 0.05 and heterozygous SNP filter of 0.5 were applied prior to GWAS and haplotyping. No missing data were present in the SNP dataset.
Plant and seed phenotyping
Soybean phenotype data were generated in multi-year trials as described and analyzed by Bruce et al. (2019a) and further described here. In brief, 175 accessions were trialed at two locations (Woodstock, Ontario, and St Pauls, Ontario) in 3 years (2015, 2016 and 2017) where major agronomic and seed traits (yield, oil, protein, days to maturity (DTM), 100-seed weight (SDWT)) were measured in field trials (two replicates per location). Data were first processed per location using a radial smoothing procedure, followed by a combined analysis in PROC GLIMMIX in SAS 9.4 (SAS Institute 2013), both previously described for this data (Bruce et al. 2019a). A best linear unbiased estimator (BLUE) for every trait in each accession was generated for input in the GWAS procedure (Figure S1) with a final total of n = 175 observations for DTM and 100 seed weight, n = 167 observations for yield and n = 166 observations for oil and protein.
Broad-sense heritability (H2) and standard error (se) of H2 estimates for seed and agronomic traits for Guelph germplasm were calculated on a plot-mean basis according to Holland et al. (2003) in SAS 9.4 using the PROC MIXED procedure. No fixed effects were fitted in the model, while the random effects were environment, block within environment, genotype and genotype by environment interaction, with the covariance parameters used for H2 estimation.
Genome-wide association analysis
GWAS analyses were performed using the rMVP package in R (https://github.com/XiaoleiLiuBio/rMVP) using the Fixed and random model Circulating Probability Unification (FarmCPU) model (Liu et al. 2016). Two different matrices (PCA (covariate P) and fastStructure (covariate Q)) were used to capture population structure. Two kinship matrices provided estimates of the relatedness among individuals (covariates K = EMMA and K* = VanRaden) (Kang et al. 2008; VanRaden 2008; Li et al. 2013). Based on the cumulative distribution of p values for different traits, models that took into account kinship and PCA (P + K*) were found to provide the best fit. An adjusted p value (q value) to ensure a false discovery rate (FDR) < 0.05 was used to establish a significance threshold (Wang et al. 2012), with multiple models shown in Figure S2. The population structure and diversity of the panel are previously described (Bruce et al. 2019b), but in brief, the population was generally homogeneous due to the close relationships of the breeding germplasm. Associations were in general at moderate frequency within the germplasm, as shown by the concordance data of significant SNPs to haplotypes (Table S2).
Association analysis for the haplotypes was conducted in Tassel 5 (Glaubitz et al. 2014) using the mixed linear model (MLM) method with kinship (Zhang et al. 2010). Haplotypes were coded as multi-allelic SNPs (A- > AA, B- > TT, C- > CC,…), and significant associations of the haplotypes were tested at p < 0.05 using a FDR multiple testing correction.
Haplotyping the germplasm
Haplotypes were generated across the germplasm using the HaplotypeMiner (https://github.com/malemay/HaplotypeMiner) R package (Tardivel et al. 2019) in R 3.5.3 (R Core Team 2018). The location input used for haplotyping was the significant SNP associated with the trait of interest in each genomic region, defined as the “gene_center” position for the analysis. The analysis included kinship (centered_IBS) as generated in Tassel 5 (Bradbury et al. 2007) using the “cluster_r2_measure = r2v.” A range of parameters in HaplotypeMiner was tested for each input, with final analysis conducted using: “max_marker_to_gene_distance = 4 Mb,” “max_flanking_pair_distance = 8 Mb,” “cluster_threshold = 0.9,” “marker_independence_threshold = 0.7,” “min_allele_count = 2” and no minor allele frequency (MAF) filtering and heterozygote filtering. The SNP dataset was filtered for MAF and heterozygous SNPs prior to the GWAS, and the same dataset was used for the HaplotypeMiner analysis. A large max_marker_to_gene_distance was chosen so that regional LD would define the haplotypes, resulting in various lengths of haplotyped regions (Table S3). The concordance of haplotypes to underlying significant SNPs from the association analysis was checked through comparison of marker classes found within each haplotype (Table S2).
Haplotypes were filtered to remove haplotypes containing heterozygous markers. For the calling of haplotypes, any haplotype with fewer than five observations across the panel was not used. For analyses using trait data, accessions not containing phenotype data (on a per-trait basis) were not used. Haplotype trait data was plotted in the ggplot2 R package (Wickham 2016). Haplotypes were named according to the underlying significant SNP for the region of interest, with a “_hap” after the trait name for differentiation (Table S1).
Haplotype trends over time
Year of cultivar release was used to assess trends in haplotypes within cultivars over time. Accessions were split into four groups: historical (n = 38), Guelph 1985–2005 (n = 32), Guelph 2006–2016 (n = 31) and experimental (n = 74). Historical accessions were those released before 1985, both Guelph groups contained released cultivars based on year of release, and experimental accessions included unreleased experimental accessions and other research germplasm tested at the University of Guelph (Bruce et al. 2019a). Haplotype counts were plotted as frequencies of the total haplotypes within a given group. Significant differences among groups were tested with Fisher’s exact test in R 3.5.3 (R Core Team 2018).
Results
Genotypes and phenotypes for the association analysis
In total, 175 accessions representing historical, modern and current experimental soybean breeding accessions at the University of Guelph were used for the GWAS (Table S1). A singular value (BLUE) for each trait and accession was used as input for the association analysis, with the distributions for all traits plotted (Figure S1). For 100-seed weight, outliers for low seed weight were identified, though these were kept in the analysis as they were representative of the SCN RILs and natto-type soybeans in the field tests. Both oil and protein were normally distributed (Shapiro–Wilk, p < 0.05), while yield and DTM showed skewed distributions as a result of field conditions and panel composition. The SNP markers were evenly distributed genome-wide and at a high density (> 7 SNPs per 100 kb). A plot-mean H2 was calculated for the five studied traits, where estimates ranged from 0.58 for yield to 0.93 for 100 seed weight (Table S4). Overall, the SNP and phenotypic data were of high quality for GWAS.
Marker–trait associations in breeding germplasm
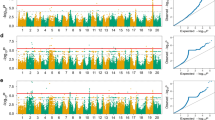
Genome-wide association analyses identified 23 significant marker–trait associations within the Guelph breeding germplasm for five traits: days to maturity (DTM), oil (%), protein (%), 100-seed weight (g) and yield (kg ha−1) (Fig. 1). For DTM, five regions were identified as significant including annotated regions comprising the E1 gene (Xia et al. 2012) with a 3.7-day effect on maturity, E2 (Watanabe et al. 2011) with a 2.9-day effect, E3 (Watanabe et al. 2009) with a 2.1-day effect and a region putatively associated with E8 (Cober et al. 2010) with a 4.5-day effect. A region on chromosome 8 identified as E10 (Samanfar et al. 2017) was identified with a 2.8-day effect on maturity (Table 1). In all cases except the putative E10 locus, the major allele was associated with later maturity.

Multi-trait Manhattan plot of association mapping results for yield (kg ha−1) in green, seed oil (%) in yellow, seed protein (%) in purple, maturity (days) in dark blue and 100-seed weight (g) in pink within Guelph breeding germplasm using 77 k SNPs and FarmCPU in rMVP at FDR = 0.05
For seed oil, three regions were identified as significant on chromosomes 2, 13 and 15 with effect magnitudes ranging from 0.2 to 0.5% of oil (Table 1). Five regions were significantly associated with seed protein, two on chromosome 2 and one each on chromosomes 13, 18 and 20, with effect magnitudes of 0.3–1.1% of seed protein (Table 1). Five regions were associated with 100 seed weight on chromosomes 4, 8, 10, 14 ad 19 with effect magnitudes ranging from 1.1 to 3.4 g per 100 seeds (Table 1). Five regions were associated with yield on chromosomes 3, 6, 8, 17 and 19, with effect magnitudes ranging from 91 to 230 kg ha−1 (Table 1).
Haplotypes in the trait-associated regions
Using a LD-based haplotyping method, haplotypes were generated to overlap the identified marker–trait associations in the breeding germplasm (Table S1). Of the 23 significant loci identified through GWAS analysis, 21 were successfully haplotyped (Table S1). Of these 21 haplotyped regions, two were identified as monomorphic for the defined haplotype within phenotyped germplasm due to removal of haplotypes with heterozygous SNPs, while the maximum number of haplotypes observed at a given region was five. The shortest identified haplotype was 7218 bp for sdwt_hap_c14_32Mb and the longest extended over 6.8 Mb for oil_hap_c2_35Mb, with an average haplotype length of 1.56 Mb (Table S3).
To confirm the association of haplotypes and the traits studied, violin plots were generated to assess trait distribution within haplotype groups for each haplotyped region (Fig. 2). Then each haplotyped region was tested using a multi-allelic MLM association analysis with kinship, where haplotyped regions with significant differences between haplotypes were identified (starred plots, Fig. 2). In total, 10 of these haplotyped regions were found to be significantly associated (p < 0.05) with the trait originally identified through the GWAS.

Haplotype–trait violin plots for the haplotyped regions. Starred plots indicate a significant association was identified between the haplotypes and trait using a mixed linear model with kinship at p < 0.05
To further understand the concordance of the SNP–trait associations and the haplotype–trait associations, the concordance of SNP and haplotypes was assessed (Table S2). Significant SNP associations where the underlying SNP was in concordance with the haplotypes typically showed significance when tested with a haplotype MLM association model including yield_hap_c3_03Mb, yield_hap_c8_47Mb, yield_hap_c17_40Mb, oil_hap_c2_35Mb, prot_hap_c13_31Mb, sdwt_hap_c10_2Mb, sdwt_hap_c14_32Mb, dtm_hap_c8_47Mb, dtm_hap_c10_44Mb, dtm_hap_c19_47Mb. When the defined haplotypes were not consistent with the genotypes of the significant SNP, no haplotype–trait association was identified such as for yield_hap_c6_16Mb and prot_hap_c2_2Mb (Table S3). Several haplotyped regions had only a single haplotype with phenotype data due to lack of phenotype data for alternate haplotypes such as for oil_hap_c15_5Mb, sdwt_hap_c4_40Mb, dtm_hap_c4_30Mb.
Changes in haplotype frequencies through breeding
Haplotypes with significant trait association were plotted by group, with each group defining an era of time in the breeding program at the University of Guelph for a total of 175 accessions to assess changes in haplotype frequency over time. A total of six regions were identified to show significant changes between eras of breeding in the Guelph accessions using Fisher’s exact test (p < 0.05) (Fig. 3).

Haplotype frequencies compared by group (historical (Hist), Guelph 1985–2005, Guelph 2006–2016 and experimental (Exp)) for the ten trait-associated haplotypes. Starred plots indicate significant differences in allele frequency between groups were identified using Fisher’s exact test at p < 0.05
Among the DTM-associated haplotypes, only dtm_hap_c8_47Mb had significant differences between the eras of breeding, where haplotypes C and E have been removed from Guelph 2006–2016 accessions, and haplotype D has been introduced to the experimental accession group (Fig. 3). The remaining DTM regions did not show significant changes in haplotype frequency. No 100-seed weight regions showed significant differences for haplotype frequency between eras of breeding, and in all three regions, the Guelph accessions were fixed for the major (A) haplotype (Fig. 3). The low seed weight haplotype was only found in RILs resulting from a G.Max by G.Soja cross, though the differences were not significant.
Both oil (oil_hap_c2_35Mb) and protein (prot_hap_c13_31Mb) showed significant changes in haplotype frequency over the eras of breeding (Fig. 3). For oil_hap_c2_35Mb, the historical germplasm was nearly fixed for the A haplotype conferring higher seed oil content, with a higher frequency of the minor haplotypes in the Guelph breeding germplasm. For prot_hap_c13_31Mb, the historical accessions had the lowest frequency of the A haplotype conferring higher seed protein, similar frequencies in the two Guelph eras and the highest frequency in the experimental accessions (Fig. 3).
All three yield regions showed significant changes between eras of accessions. The yield_hap_c3_03Mb region has lost the B haplotype conferring lower yields in the Guelph 2006–2016 group (Fig. 3). For the yield_hap_c8_47Mb region, the Guelph 2006–2016 group has only the A and B haplotypes conferring high yield, where haplotype C was present in earlier accession groups. A mid-yielding haplotype E was only found in historical and experimental accessions (Fig. 3). The lower yielding E haplotype is found at low frequencies across the germplasm, although it is lowest in Guelph 2006–2016 accessions (Fig. 3). The yield_hap_c17_40Mb region shows increasing haplotype C frequency compared to historical accessions.
Discussion
Association analysis has been proven to be a robust method for identification of genomic regions associated with phenotypic traits to improve the understanding of the genetic architecture of many traits in crop species. Previously, marker–trait associations within closely related germplasm have been difficult to assess using association techniques due to methodological limitations in implementing GWAS in these types of populations. The FarmCPU method (Liu et al. 2016) has worked well to identify these associations within breeding germplasm and allow for further characterization of these trait-associated regions. As a confirmation for the efficacy of the association analysis, identifying significant SNPs associated with maturity for previously characterized E genes (E1, E2, E3, E10 and a putative region containing E8) shows that this approach is appropriate to study marker–trait associations in closely related breeding accessions. Additionally, other traits studied here such as protein show overlap between the significant region identified here compared to multiple QTL studies such as protein on chromosome 20 (Zhao-ming et al. 2011).
As GWAS has become a routine technique for assessing traits in plant germplasm panels (Bandillo et al. 2015; Fang et al. 2017; Zhang et al. 2018), the value of the individual significant results has decreased as many crop genomes become fully annotated, necessitating the deeper study of these regions (Qian et al. 2017), especially in a well characterized species like soybean. The approach taken here was to define haplotypes surrounding these single-marker associations, as selection within breeding programs is on larger genomic regions rather than at the single gene scale. Studying trends over time for these larger genomic regions allows the understanding of how breeder selections have changed haplotype frequencies as a direct result of selection. The results presented are consistent with Fu et al. (2007) who assessed diversity using SSR markers in Canadian and exotic germplasm finding changes due to breeder selections.
The availability of fast and reproducible haplotyping methods for SNP data (Tardivel et al. 2019) allows for the assessment of large haplotypes within the soybean genome which may contribute to the understanding of genomic changes due to breeding and selection in soybean which typically act in large genomic regions, rather than at the gene level due to LD within the crop. The range of sizes for the described haplotypes shows that there is variability in LD within the group of accessions used in this study, similar to previously observed results for soybean LD (Hyten et al. 2007).
This work also demonstrated a repeatable approach for a study of haplotypes within crop germplasm panels with hundreds or thousands of samples using genome-wide SNP data, rather than single region haplotyping, or looking at major and minor allele frequencies for bi-allelic SNP markers at significant trait-associated regions. The study of multi-allelic haplotypes can reveal patterns of diversity not visible using bi-allelic SNPs (Grainger and Rajcan 2013). From the identified haplotypes, SNP markers can be identified to use in marker-assisted selection of progeny or for informed selection of parents within a breeding program.
A benefit to studying haplotypes defined by significant marker–trait associations in dense SNP data is the ability to narrow the region of interest using marker LD (Qian et al. 2017). The mapping resolution will still be limited by the recombination with the population; however, this is population dependent. Defining haplotypes using SNP markers helps to identify functional alleles for traits of interest as previously demonstrated in wheat (Jiang et al. 2015), maize (Yang et al. 2013) and rice (Yano et al. 2016). It should be noted that while multiple haplotypes can be identified for numerous regions of the genome, this approach does not help to differentiate functional alleles for a given trait as it is impossible to determine if the underlying causative alleles are the same for haplotypes with the same mean trait performance. While novel trait associations were identified in the set of breeding accessions studied, there were previous associations for other traits at these regions which may be correlated with the traits identified here.
Based on the results presented, there is more study to be conducted on the pleiotropic effects of haplotypes within breeding germplasm. This is evidenced by the selection of haplotypes within the breeding program that do not appear to increase the trait value for traits which have been under improvement within the studied germplasm. A possible explanation is that these haplotypes have minor effects on other traits below the threshold of detection. A more complete dissection of these regions could address this question in the future, including fine mapping using the boundaries identified through haplotyping and functional characterization of the underlying causative genes. Some of the haplotypes studied were not associated with the trait of interest as shown in the MLM analyses. Several possibilities exist to explain negative results including a low minor allele frequency of the causative allele, the haplotypes generated do not represent the underlying SNPs very well, or the underlying genomic region is not well characterized with the SNP markers used in this study. Previous usage of the HaplotypeMiner package has shown that not all regions could be properly haplotyped even when using dense, whole genome sequencing SNP data (Tardivel et al. 2019).
Application of a haplotyping approach within breeding germplasm will allow for targeted trait improvement and data-driven parental selection (Fig. 4). Identification of accessions with favorable haplotypes in a target environment will improve crossing outcomes. This approach was described by Qian et al. (2017) to harness haplotype data for crop improvement. Studying and assessing these haplotypes in routine breeding efforts will help breeders make informed decisions about their germplasm and allow for understanding of the trends within their breeding programs. While major-effect haplotypes are easily selected for, this study has shown that these regions are already under selection pressure in a breeding context, and continued selection will require novel sources of diversity for future crop improvement.

A model for parental selection using haplotypes in a breeding program. In this example, the haplotypes are combined to select for high protein, high oil and short days to maturity (preferred haplotypes highlighted in green with unwanted haplotypes shown in red). The two example parental accessions show partial overlap with the ideal haplotypes for early maturity, high protein and high oil. A cross between these two accessions combined with marker-assisted selection could produce the ideal cultivar. This approach for assessing haplotypes in breeding germplasm can be used to select parents with beneficial haplotypes in targeted crosses and progeny screening
An important consideration for the use of these haplotypes in breeding is that the erosion of diversity in these regions may be due to breeders having selected against unfavorable haplotypes. A remaining question about these regions is whether the absolute best haplotype has been selected for, or the best of which was available in the germplasm under selection. Further crossing, testing and study using diverse accessions will help to identify trends in wider germplasm collections, rather than local trends within single breeding programs. This may be exemplified by looking at the SDWT results, where the novel haplotypes identified were within G. soja by G. max RILs, while no haplotypes were identified to have changed within standard breeding germplasm.
Future extension of this research could be to conduct haplotype analyses within the United States Department of Agriculture (USDA) soybean germplasm collection data available through Soybase (Grant et al. 2010). Identification of breeding-related haplotypes would allow for screening of the USDA germplasm collection to identify novel haplotypes for introgression to a breeding program while minimizing the linkage drag of the wide-crosses conducted in germplasm improvement efforts. Additionally, characterizing haplotypes to understand haplotype by environment interactions will inform breeders on the best deployment of specific haplotypes for a given geographical region. The data from haplotype–trait associations combined with regional LD patterns provide valuable information for data-driven parental selection in a breeding program. For example, using a target of high seed protein, a breeder could identify parents containing high protein haplotypes prior to crossing, ensuring that progeny contain desired haplotypes for the trait of interest.
Conclusion
This work has identified genomic regions controlling major agronomic and seed traits within breeding program germplasm at the University of Guelph soybean breeding program. Haplotype analysis revealed significant haplotype–trait associations within this germplasm, and further uncovered changes in haplotype frequencies over time within the breeding germplasm as a result of breeders’ crossing and selection. Continued selection on these haplotypes could erode genetic diversity at major-effect loci, requiring the addition of novel genetic diversity to continue crop improvement. Our work demonstrates the importance of studying these trends in soybean breeding germplasm as it serves as the gene pool from which new cultivars are developed.
References
Bandillo N, Jarquin D, Song Q et al (2015) A population structure and genome-wide association analysis on the USDA soybean germplasm collection. Plant Genome. https://doi.org/10.3835/plantgenome2015.04.0024
Bradbury PJ, Zhang Z, Kroon DE et al (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Bruce RW, Grainger CM, Ficht A et al (2019a) Trends in soybean trait improvement over generations of selective breeding. Crop Sci 59:12. https://doi.org/10.2135/cropsci2018.11.0664
Bruce RW, Torkamaneh D, Grainger C et al (2019b) Genome-wide genetic diversity is maintained through decades of soybean breeding in Canada. Theor Appl Genet. https://doi.org/10.1007/s00122-019-03408-y
Chaudhary J, Patil GB, Sonah H et al (2015) Expanding omics resources for improvement of soybean seed composition traits. Front Plant Sci. https://doi.org/10.3389/fpls.2015.01021
Cober ER, Molnar SJ, Charette M, Voldeng HD (2010) A new locus for early maturity in soybean. Crop Sci 50:524. https://doi.org/10.2135/cropsci2009.04.0174
Contreras-Soto RI, Mora F, de Oliveira MAR et al (2017) A genome-wide association study for agronomic traits in soybean using SNP markers and SNP-based haplotype analysis. PLoS ONE 12:e0171105. https://doi.org/10.1371/journal.pone.0171105
Core Team R (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna Austria
Elshire RJ, Glaubitz JC, Sun Q et al (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. https://doi.org/10.1371/journal.pone.0019379
Fang C, Ma Y, Wu S et al (2017) Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. https://doi.org/10.1186/s13059-017-1289-9
Fu Y-B, Peterson GW, Morrison MJ (2007) Genetic diversity of Canadian soybean cultivars and exotic germplasm revealed by simple sequence repeat markers. Crop Sci 47:1947. https://doi.org/10.2135/cropsci2006.12.0843
Glaubitz JC, Casstevens TM, Lu F et al (2014) TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9:e90346. https://doi.org/10.1371/journal.pone.0090346
Grainger CM, Rajcan I (2013) Characterization of the genetic changes in a multi-generational pedigree of an elite Canadian soybean cultivar. Theor Appl Genet. https://doi.org/10.1007/s00122-013-2211-9
Grant D, Nelson RT, Cannon SB, Shoemaker RC (2010) SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res 38:D843–D846. https://doi.org/10.1093/nar/gkp798
Holland JB, Nyquist WE, Cervantes-Martinez CT (2003) Estimating and interpreting heritability for plant breeding: an update. Plant Breed Rev 22:9–112
Hyten DL, Choi I-Y, Song Q et al (2007) Highly variable patterns of linkage disequilibrium in multiple soybean populations. Genetics 175:1937–1944. https://doi.org/10.1534/genetics.106.069740
Jiang Y, Jiang Q, Hao C et al (2015) A yield-associated gene TaCWI, in wheat: its function, selection and evolution in global breeding revealed by haplotype analysis. Theor Appl Genet 128:131–143. https://doi.org/10.1007/s00122-014-2417-5
Kang HM, Zaitlen NA, Wade CM et al (2008) Efficient control of population structure in model organism association mapping. Genetics 178:1709–1723. https://doi.org/10.1534/genetics.107.080101
Lam H-M, Xu X, Liu X et al (2010) Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42:1053–1059. https://doi.org/10.1038/ng.715
Langewisch T, Zhang H, Vincent R et al (2014) Major soybean maturity gene haplotypes revealed by SNPViz analysis of 72 sequenced soybean genomes. PLoS ONE 9:e94150. https://doi.org/10.1371/journal.pone.0094150
Leamy LJ, Zhang H, Li C et al (2017) A genome-wide association study of seed composition traits in wild soybean (Glycine soja). BMC Genom. https://doi.org/10.1186/s12864-016-3397-4
Li H, Peng Z, Yang X et al (2013) Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat Genet 45:43–50. https://doi.org/10.1038/ng.2484
Liu X, Huang M, Fan B et al (2016) Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet 12:e1005767. https://doi.org/10.1371/journal.pgen.1005767
Liu S, Kandoth PK, Lakhssassi N et al (2017) The soybean GmSNAP18 gene underlies two types of resistance to soybean cyst nematode. Nat Commun. https://doi.org/10.1038/ncomms14822
Patil G, Mian R, Vuong T et al (2017) Molecular mapping and genomics of soybean seed protein: a review and perspective for the future. Theor Appl Genet 130:1975–1991. https://doi.org/10.1007/s00122-017-2955-8
Qian L, Hickey LT, Stahl A et al (2017) Exploring and harnessing haplotype diversity to improve yield stability in crops. Front Plant Sci. https://doi.org/10.3389/fpls.2017.01534
Samanfar B, Molnar SJ, Charette M et al (2017) Mapping and identification of a potential candidate gene for a novel maturity locus, E10, in soybean. Theor Appl Genet 130:377–390. https://doi.org/10.1007/s00122-016-2819-7
SAS Institute (2013) The SAS system for windows. Release 9.4. SAS Inst., Cary, NC
Schmutz J, Cannon SB, Schlueter J et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183. https://doi.org/10.1038/nature08670
Sonah H, Bastien M, Iquira E et al (2013) An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS ONE 8:e54603. https://doi.org/10.1371/journal.pone.0054603
Sonah H, O’Donoughue L, Cober E et al (2015) Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean. Plant Biotechnol J 13:211–221. https://doi.org/10.1111/pbi.12249
Song Q, Hyten DL, Jia G et al (2015) Fingerprinting soybean germplasm and its utility in genomic research. G3 Genes Genomes Genet 5:1999–2006. https://doi.org/10.1534/g3.115.019000
Tardivel A, Sonah H, Belzile F, O’Donoughue LS (2014) Rapid identification of alleles at the soybean maturity gene E3 using genotyping by sequencing and a haplotype-based approach. Plant Genome. https://doi.org/10.3835/plantgenome2013.10.0034
Tardivel A, Torkamaneh D, Lemay M-A et al (2019) A systematic gene-centric approach to define haplotypes and identify alleles based on dense SNP datasets. Plant Genome. https://doi.org/10.3835/plantgenome2018.08.0061
Torkamaneh D, Laroche J, Bastien M et al (2017) Fast-GBS: a new pipeline for the efficient and highly accurate calling of SNPs from genotyping-by-sequencing data. BMC Bioinform. https://doi.org/10.1186/s12859-016-1431-9
Torkamaneh D, Laroche J, Valliyodan B et al (2019) Soybean haplotype map (GmHapMap): a universal resource for soybean translational and functional genomics. bioRxiv. https://doi.org/10.1101/534578
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423. https://doi.org/10.3168/jds.2007-0980
Wang M, Yan J, Zhao J et al (2012) Genome-wide association study (GWAS) of resistance to head smut in maize. Plant Sci 196:125–131. https://doi.org/10.1016/j.plantsci.2012.08.004
Watanabe S, Hideshima R, Xia Z et al (2009) Map-based cloning of the gene associated with the soybean maturity locus E3. Genetics 182:1251–1262. https://doi.org/10.1534/genetics.108.098772
Watanabe S, Xia Z, Hideshima R et al (2011) A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genetics 188:395–407. https://doi.org/10.1534/genetics.110.125062
Wickham H (2016) ggplot2: elegant graphics for data analysis. Springer, New York
Xia Z, Watanabe S, Yamada T et al (2012) Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc Natl Acad Sci 109:E2155–E2164. https://doi.org/10.1073/pnas.1117982109
Yan L, Hofmann N, Li S et al (2017) Identification of QTL with large effect on seed weight in a selective population of soybean with genome-wide association and fixation index analyses. BMC Genom. https://doi.org/10.1186/s12864-017-3922-0
Yang Q, Li Z, Li W et al (2013) CACTA-like transposable element in ZmCCT attenuated photoperiod sensitivity and accelerated the postdomestication spread of maize. Proc Natl Acad Sci 110:16969–16974. https://doi.org/10.1073/pnas.1310949110
Yano K, Yamamoto E, Aya K et al (2016) Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat Genet 48:927–934. https://doi.org/10.1038/ng.3596
Zhang Z, Ersoz E, Lai C-Q et al (2010) Mixed linear model approach adapted for genome-wide association studies. Nat Genet 42:355–360. https://doi.org/10.1038/ng.546
Zhang J, Wang X, Lu Y et al (2018) Genome-wide scan for seed composition provides insights into soybean quality improvement and the impacts of domestication and breeding. Mol Plant 11:460–472. https://doi.org/10.1016/j.molp.2017.12.016
Zhao-ming Q, Ya-nan S, Qiong W et al (2011) A meta-analysis of seed protein concentration QTL in soybean. Can J Plant Sci 91:221–230. https://doi.org/10.4141/cjps09193
Acknowledgements
We would like to acknowledge funding support from the Natural Sciences and Engineering Research Council of Canada Collaborative Research and Development Grant (NSERC CRD) program and our industry partners including Grain Farmers of Ontario, Secan and Huron Commodities Inc. All of the mentioned funding is part of the NSERC CRD Grant No. CRDPJ 447948 – 13. We would also like to acknowledge Marc-André Lemay at Université Laval for his help with the HaplotypeMiner R package, the staff in Dr. Rajcan’s research program in Guelph, ON, Colbey Templeman, Yesenia Salazar, Martha Jimenez, Mei Wang and Lin Liao, as well as Bryan Stirling in Dr. Eskandari’s laboratory at Ridgetown, ON.
Author information
Authors and Affiliations
Contributions
RB, CG, IR and ME performed project planning. RB, DT and FB analyzed the data. RB, DT and IR prepared the manuscript. All authors have reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by Albrecht E. Melchinger.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bruce, R.W., Torkamaneh, D., Grainger, C.M. et al. Haplotype diversity underlying quantitative traits in Canadian soybean breeding germplasm. Theor Appl Genet 133, 1967–1976 (2020). https://doi.org/10.1007/s00122-020-03569-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-020-03569-1