
Abstract
Key message
A total of 449 barley accessions were phenotyped for Pyrenophora teres f. teres resistance at three locations and in greenhouse trials. Genome-wide association studies identified 254 marker–trait associations corresponding to 15 QTLs.
Abstract
Net form of net blotch is one of the most important diseases of barley and is present in all barley growing regions. Under optimal conditions, it causes high yield losses of 10–40% and reduces grain quality. The most cost-effective and environmentally friendly way to prevent losses is growing resistant cultivars, and markers linked to effective resistance factors can accelerate the breeding process. Here, 449 barley accessions expressing different levels of resistance comprising landraces and commercial cultivars from the centres of diversity were selected. The set was phenotyped for seedling resistance to three isolates in controlled-environment tests and for adult plant resistance at three field locations (Belarus, Germany and Australia) and genotyped with the 50 k iSelect chip. Genome-wide association studies using 33,818 markers and a compressed mixed linear model to account for population structure and kinship revealed 254 significant marker–trait associations corresponding to 15 distinct QTL regions. Four of these regions were new QTL that were not described in previous studies, while a total of seven regions influenced resistance in both seedlings and adult plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Net blotch caused by Pyrenophora teres Drechsler is one of the most important, damaging and widely distributed diseases of barley (Mathre 1997). Pyrenophora teres exists in two forms: Pyrenophora teres f. teres and P. teres f. maculata, causing ‘net form’ net blotch (NFNB) and ‘spot form’ net blotch (SFNB), respectively. These two forms are similar in morphology during the sexual and asexual stages and only differ in the symptoms they cause (Lightfoot and Able 2010; Smedegård-Petersen 1971). NFNB produces brown netted lesions containing longitudinal and transverse striations, while SFNB produces brown spotted lesions (Smedegård-Petersen 1976). Under favourable conditions, NFNB causes significant reductions in both yield (Brandl and Hoffmann 1991; Kangas et al. 2005; Mathre 1997) and quality (Burleigh et al. 1988). Yield losses caused by NFNB on susceptible barley cultivars can reach up to 40% under favourable epidemic conditions (Steffenson et al. 1996). In the Northern Caucasus, the North-West and Central regions of the Non-Chernozem region, the South Ural, in the far east of Russia and in Belarus P. teres f. teres is the most important disease in barley. In these regions, an epidemic appears every 4–5 years. Yield losses due to epidemics are estimated to be at 36–45% (Afonin et al. 2008). In Australia, P. teres f. teres is considered a major disease in barley and is estimated to cause average annual losses of $AUD 19 million (Murray and Brennan 2009). Conservation tillage is standard practice in Australian farming systems resulting in a plentiful bank of over-seasoning inoculum. Losses of up to 70% with severe lodging were recorded in 2009 in South Australia on barley cultivar ‘Maritime’ (Wallwork et al. 2016). Resistance to P. teres f. teres is a major priority of all barley breeding programs in Australia.
Pyrenophora teres f. teres is a highly variable pathogen (Khan 1982; Liu et al. 2011; Serenius 2006; Steffenson and Webster 1992; Tekauz 1990). As a result of global virulence studies, 153 pathotypes among 1162 isolates were identified in different geographic populations of P. teres f. teres originating from Europe, Syria and Canada on a set of 9 barley differential lines (Anisimova et al. 2017). One of the reasons for the high diversity of P. teres f. teres populations is the ability to reproduce both sexually and asexually (Mathre 1997). This high heterogeneity concerning virulence of the pathogen implies high genetic diversity in host resistance. In several studies, the complexity of the P. teres f. teres–barley interaction was shown to be controlled by major qualitative genes (Afanasenko et al. 1999; Cakir et al. 2003; Friesen et al. 2006; Grewal et al. 2012; Ma et al. 2004; Manninen et al. 2006) and quantitative trait loci (QTL) (Douglas and Gordon 1985; König et al. 2013, 2014; Robinson and Jalli 1997; Steffenson et al. 1996; Vatter et al. 2017).
Resistance genes and QTL against NFNB were identified on all seven barley chromosomes in bi-parental mapping populations and by association genetics studies (Afanasenko et al. 2015; Berger et al. 2013; Cakir et al. 2011; Cakir et al. 2003; Graner et al. 1996; Grewal et al. 2008, 2012; Gupta et al. 2004; Koladia et al. 2017; König et al. 2013, 2014; Ma et al. 2004; Manninen et al. 2006; O’Boyle et al. 2014; Raman et al. 2003; Richards et al. 2017; Richter et al. 1998; Steffenson et al. 1996; Wonneberger et al. 2017a, b). Several studies report that resistance genes identified in adult plants are often different from genes conferring NFNB resistance at the seedling stage (Cakir et al. 2003; Grewal et al. 2012; Steffenson et al. 1996; Wonneberger et al. 2017a). Pathotype-specific resistance QTL were found when different P. teres f. teres isolates were used for phenotyping different mapping populations (Afanasenko et al. 2015; Grewal et al. 2012; Koladia et al. 2017; Richards et al. 2017).
In recent years, genome-wide association studies (GWAS) have become popular for mapping QTL and major genes for several reasons: (1) segregating populations and the construction of own genetic linkage maps are not needed for GWAS, (2) It enables discovery of useful genetic variation in a broader portion of the genetic diversity present in a species than bi-parental mapping approaches, and (3) it exploits historic recombination events, and by using populations including breeding lines and commercial cultivars, there is a higher probability that markers are directly transferable into current breeding programmes. Nevertheless, a limiting factor in GWAS can be population size. As shown by Wang et al. (2012), population size in GWAS approaches should be at least around 380 individuals to ensure statistically sound and consistently detectable marker–trait associations (MTAs). Another limitation is the presence of extensive linkage disequilibrium (LD) associated with natural or artificial selection, particularly in crop species, which have been subject to intense breeding. LD is the non-random association between two alleles at different loci and is affected by population size and mutation rate, but mostly by recombination rate (Flint-Garcia et al. 2003; Rafalski and Morgante 2004). In outcrossing species, such as maize, recombination rates are high and LD therefore decays within hundreds to a few thousands of base pairs (Remington et al. 2001; Yan et al. 2009). In contrast, in selfing species, which are usually homozygous, recombination is less effective (Flint-Garcia et al. 2003).The highly inbreeding model species Arabidopsis thaliana, for example, displays an average genome-wide LD decay of around 250 kilobases, corresponding to about 1 cM (Nordborg et al. 2002). Thus, compared to its very small genome size of roughly 130 Mb, LD extends over large blocks. In barley GWAS panels, LD was reported to be between 18 and 1.3 cM, depending on the diversity of the evaluated materials (Bellucci et al. 2017; Bengtsson et al. 2017; Burlakoti et al. 2017; Gyawali et al. 2017; Massman et al. 2010; Mitterbauer et al. 2017; Tamang et al. 2015; Vatter et al. 2017; Wehner et al. 2015; Wonneberger et al. 2017a). It was shown that LD decays faster in global populations. In other words, genetically and geographically diverse GWAS sets, which include inter alia landraces, have a comparatively low LD (Mohammadi et al. 2015; Nordborg et al. 2002). Low LD has the advantage of higher mapping resolution, narrowing the intervals of interesting QTL. However, this also means that a higher marker density is required (Zhu et al. 2008). Recently developed genotyping methods for barley, like the 50 k Barley iSelect SNP Chip (Bayer et al. 2017) or genotyping-by-sequencing (GBS) (Poland et al. 2012), reduce genotyping costs and ensure high marker density and coverage of the barley genome and overcome some of the limitations outlined. Based on these considerations, the main objectives of this study were (1) to screen a diverse set of barley accessions for resistance against Pyrenophora teres f. teres under greenhouse and field conditions, (2) to genotype this set with the 50 k iSelect chip, (3) to identify QTL for resistance against NFNB and (4) to compare these with previously known QTL in order to identify potentially new resistance loci along with associated markers for resistance breeding.
Materials and methods
Germplasm set
For GWAS, a set of 277 barley landraces and 172 commercial cultivars (total 449 accessions) from the N. I. Vavilov Research Institute of Plant Genetic Resources (VIR) collection were studied (Online Resource 1). This set was the result of a long-term joint research project between the All-Russian Institute of Plant Protection (VIZR), the VIR and the Institute of Resistance Research and Stress Tolerance of the Federal Research Centre for Cultivated Plants (JKI). A total of 12,000 barley accessions from different centres of barley diversity and commercial cultivars were screened for resistance to Pyrenophora teres f. teres and Cochliobolus sativus under greenhouse conditions and in detached leaf assays at the JKI and VIZR (Afanasenko 1995; Silvar et al. 2010; Trofimovskaya et al. 1983). Out of these, about 300 and 150 accessions with different levels of resistance to P. teres f. teres and C. sativus were identified, respectively, and investigated in the present study. With respect to the landraces, the set comprises 31 accessions from Ethiopia and Sudan, 56 from the Middle East (Turkey, Syria, Israel, Palestine), 20 from the Mediterranean Region (Cyprus, Italy, Spain, Crete, Greece), 59 from Central Asia (Tajikistan, Turkmenistan, Kyrgyzstan, Uzbekistan, Kazakhstan), 33 from China, Japan and Mongolia, 37 from South America (Peru, Ecuador, Bolivia), 4 from India, Korea, and Pakistan, 2 from Tunisia and Egypt, 8 from the Caucasus region (Georgia, Armenia, Dagestan) and 6 from the USA. With regard to commercial cultivars, the set includes one cultivar each from Austria, Denmark, Finland, Italy, Kyrgyzstan, Manchuria, Sardinia, Tunisia, Turkey, Turkmenistan, Uruguay and Yugoslavia. Mexico, the Netherlands, Poland, Portugal and Sweden are represented with two cultivars each, India with three and Germany and Ethiopia with four cultivars each. Six cultivars each are from China, France and Kazakhstan, eight and nine from the Czech Republic and Ukraine, respectively. Canada, and the USA and Japan are represented with 11, 12 and 18 cultivars, respectively. Thirty-one cultivars are from Australia and 33 from Russia.
Some of the landrace accessions date back to Nikolai I. Vavilov, who collected them on different expeditions. Accession VIR CI 3175 dates back to his first expedition in Pamir during 1916. Accessions VIR CI 7687–8378 were collected in Syria, Tunis and Cyprus during 1926, and accessions VIR CI 8515–8877 were collected in Italy, Spain and Ethiopia in 1927.
The set represents 31 morphological forms of Hordeum vulgare (Online Resource 1). It includes 178 two-rowed and 271 six-rowed accessions. Out of 449 accessions, 20 are winter types, 28 have black kernels, and 51 have naked kernels.
Single plant selections were made for each accession, and these selections were self-pollinated by bagging in 2013 and 2014 under field conditions at VIR (Pushkin, Russia) and in Zhodino (Belarus).
Fungal isolates
Five single-spore-derived P. teres f. teres isolates were used in this study. They were selected based on their origin, virulence and sporulation ability. Isolate No 13 was collected in 2014 near Volosovo in the Leningrad Region, Russia, and isolate Hoehnstedt was collected in 2016 on infected fields close to the village of Hoehnstedt in Saxony-Anhalt (Germany). The three isolates NFNB 50, NFNB 73 and NFNB 85 are Australian isolates from Queensland, each with different virulence profiles. NFNB 50 and NFNB 85 were from the Gatton area, while NFNB 73 was collected from Tansey in the South Burnett region. NFNB 50 was used in both greenhouse and field trials, but NFNB 73 and NFNB 85 were used in field trials only.
Pyrenophora teres isolates No 13 and Hoehnstedt were grown on V8-agar medium containing 150 ml V8 juice, 10.0 g Difco PDA, 3.0 g CaCO3, 10.0 g agar and 850 ml distilled water. Petri dishes were placed in a dark chamber at room temperature for 5 to 7 days, exposed to light for 24 h and placed again in a dark chamber at 13 °C for 24 h. Conidia were then harvested by adding sterile water to the Petri dish and scraping conidia off with a sterile spatula. Conidia were counted with a haemocytometer, and the concentration was adjusted to 5000 conidia/ ml. Australian isolates were grown as described by Martin et al. (2018).
In the Australian field trials, single isolates were used for inoculation. For this, isolated blocks of highly susceptible varieties were sown in early to mid-April. Inoculum for these blocks was multiplied in the laboratory and applied to the blocks at the 4–5 leaf stage. Epidemics in these blocks were promoted by sprinkler irrigation at least twice a week when conditions for infection were favourable. These blocks provided the inoculum for the subsequent field screening (Martin et al. 2018).
Greenhouse trials
Greenhouse trials were performed at the Julius Kuehn-Institute in Quedlinburg, Germany, in 2015 and 2017 with isolates No 13 and Hoehnstedt, respectively. Three seeds per accession were grown in plastic pots (8 × 8 × 8 cm) for 2–3 weeks at 16–18 °C with alternating 12 h periods of light/darkness (exposure min 5000 lx). The experiment was set up in four replications in a complete randomized block design. The NFNB differential set proposed by Afanasenko et al. (2009) was included in each replication as a standard. When the second leaf was fully developed (BBCH 12-13), plants were spray-inoculated with the spore suspension until the inoculum was at the point of running off (approximately 0.35 mL/plant). Plants were then covered with plastic foil for 48 h to ensure 100% humidity and grown for another 10–14 days at 20–22 °C and 70% humidity until symptoms were clearly visible.
Isolate NFNB 50 was tested at the Hermitage Research Facility in Warwick, Queensland, Australia, in 2017. Plants were grown in commercial potting mix (Searles Premium Potting Mix) in plastic maxipots (10 cm in diameter, 17 cm tall). Four to five seeds were sown at 0°, 120° and 240° around the circumference of each pot. The experiment was set up in two replications in an incomplete block design, where pots corresponded to blocks and there were three lines per block. Pots were maintained in the greenhouse at 15/27 °C for 2 weeks until the second leaf was fully expanded. Field-collected conidia were suspended in water at 3000 conidia/mL and applied from four directions using a WallWick® commercial spray gun delivering an average 3 mL/pot. Immediately after inoculation, pots were placed into a fogging chamber for 20 h at 19 °C with 14 h in the dark. After incubation, pots were returned to the greenhouse, double-spaced and bottom watered, and grown for another 8 days. Pots were fertilized twice weekly after emergence with a soluble complete fertilizer (Grow Force EX7) until notes were taken.
Infection response type was assessed on the second leaf of each plant following the scale of Tekauz (1985).
Field trials
Field trials were conducted at three locations, Belarus, Germany and Australia, during the years 2015, 2016 and 2017.
Trials in Belarus were conducted at the Research and Practical Centre of the National Academy of Sciences of Belarus for Arable Farming (Zhodino, Belarus) in 2016 and 2017. Accessions were sown in rows of 1 m with 15–20 seeds per row and a spacing of 0.3 m between rows. The trial was set up in a complete randomized block design with two replications. The cultivar ‘Thorgal’, susceptible to net blotch and resistant to powdery mildew, was sown around the trial as a border and after every 10th accession to support net blotch and reduce powdery mildew development in the nurseries. Additionally, ‘Thorgal’ was used as a spreader for net blotch. To increase infection, infected barley straw was spread in the rows of ‘Thorgal’ after sowing. The infected straw was harvested in the previous year at the same location. The percentage of leaf area infected was assessed at early-dough to mid-dough stages (BBCH 83–85) on the three upper leaves of the plants.
In Germany, the field trials were conducted at the Julius Kuehn-Institute (Quedlinburg, Germany) in 2015 and 2016 using the Summer Hill design developed by König et al. (2013). Accessions were sown at the beginning of August in hills with 25 seeds/hill and a spacing of 0.5 m between hills. The susceptible varieties ‘Candesse’ and ‘Stamm 4046’ were used as spreader rows and sown in rows between the hills, spacing 1.0 m between rows as described in Vatter et al. (2017). The trials were set up in two replications in a complete randomized block design. Infected barley straw was used as inoculum and was incorporated into the soil prior to sowing. Phenotyping started when symptoms were clearly visible on the susceptible standards. The percentage of leaf area infected (Moll et al. 2010) and the infection response type (Tekauz 1985) were assessed at three different time points with a period of 2 weeks between scoring dates. The area under disease progress curve (AUDPC) was calculated and used to calculate the average ordinate (AO) as described by Vatter et al. (2017).
Field trials in Australia were conducted at the Hermitage Research Facility in Warwick, Queensland in 2017 with three distinct isolates. Trials were sown as hill plots with an in-row spacing of 0.5 m and between-row spacing of 0.76 m. Two rows of datum plots were sown between five rows of spreader (0.19 m spacing). Spreaders were sown 11–19 days before the plots and were inoculated by spreading infected green plant material cut from inoculum increase blocks when the spreaders were at about BBCH 30. Epidemics were promoted with overhead sprinkler irrigation applied in the late afternoon and/or early evening so that the nurseries remained wet overnight. In the absence of rainfall, irrigation was applied two or more nights per week when conditions were favourable for infection. Infection responses were taken on a whole plot basis using a 0 to 9 scale at BBCH stages 70–73. The scale is a variant of the scale by Saari and Prescott (1975). It takes into account the plant response (infection type; IT) and the amount of disease in a plot and therefore correlates very well with the host response and the leaf area diseased (Martin et al. 2018).
Statistical analysis
Statistical analyses and analysis of variance (ANOVA) were performed using the software package SAS 9.4 (SAS Institute Inc. Cary, NC, USA) using proc mixed and proc glimmix for greenhouse trials and field trials in Australia. For field trials, the least square means (lsmeans) and for greenhouse trials the means of the infection response type were calculated and used for genome-wide association studies (GWAS). Broad sense heritability across years was calculated using the formula h2 = VG/(VG + VGY/y + VR/year) as described by Vatter et al. (2017), where VG is genotypic variance, VGY is genotype x year variance, VR is residual variance, and y and r are the number of years and replicates, respectively.
Genotyping, population structure, kinship and linkage disequilibrium
Genomic DNA was extracted from 14-day-old plants according to Stein et al. (2001). The accessions were genotyped on the Illumina iSelect 50 k Barley SNP Chip (Illumina) at Trait Genetics GmbH (Gatersleben, Germany). Physical positions of markers were taken from Bayer et al. (2017), which is based on the barley pseudo-molecule assembly by Mascher et al. (2017). SNPs having failure rates > 10%, heterozygous calls > 12.5% and a minor allele frequency (MAF) < 5% were excluded from the analyses, as well as unmapped SNPs. Thus, 33,818 SNPs were left for subsequent GWAS. In order to calculate the kinship matrix and the population structure, the markers were further filtered with the software PLINK 1.9 (www.cog-genomics.org/plink/1.9/) (Chang et al. 2015). The tool LD prune was used with the following parameters: indep pairwise window size 50, step 5 and an r2 threshold 0.5 (Campoy et al. 2016). This resulted in 8533 markers for calculating the kinship and population structure. Kinship was calculated with the web-based platform Galaxy (Afgan et al. 2016) using the tool Kinship and the modified Roger’s distance (Reif et al. 2005). Population structure was determined with the software STRUCTURE v2.3.4 (Pritchard et al. 2000). In order to identify the optimal subpopulations, an admixture model was used with a burn-in of 50,000, followed by 50,000 Monte Carlo Markov chain (MCMC) replications for k = 1 to k = 10 with 10 iterations. STRUCTURE HARVESTER (Earl and vonHoldt 2012) was used to identify the optimal k. Following this, a new STRUCTURE analysis was performed with a burn-in of 100,000 and 100,000 MCMC iterations at the optimal k value. Accessions were considered as admixed, when their membership probabilities were < 80% (Richards et al. 2017). Linkage disequilibrium (LD) was calculated as squared allele frequency correlations (R2) between all intra-chromosomal marker pairs using the tool linkage disequilibrium in the web-based platform Galaxy. Genome-wide LD decay was plotted as R2 of a marker against the corresponding genetic distance, and a Loess regression was computed. For R2, the default settings were used (Sannemann et al. 2015).
Genome-wide association studies
Genome-wide association studies (GWAS) were performed using the Galaxy implemented tool GAPIT, which uses the R package GAPIT (Lipka et al. 2012). The model used was a compressed mixed linear model (CMLM)(Zhang et al. 2010) including the population structure (Q) and kinship (K). In order to detect significant marker–trait associations, a Bonferroni correction was employed. For this, the reduced marker set of 8533 markers, which was used for calculating population structure and kinship, and a significance level of P = 0.2 was used (Muqaddasi et al. 2017; Storey and Tibshirani 2003). This resulted in a threshold of − log10 (P) ≥ 4.63. GWAS for greenhouse trials, and field trials in Australia were conducted for each isolate separately. For field trials in Zhodino and Quedlinburg, GWAS were conducted across years for each location. Manhattan plots were generated with the R v.3.4.4 package qqman.
In order to compare previously described QTL with QTL identified in the present study, the databases GrainGenes (https://wheat.pw.usda.gov/GG3/) and BARLEX (https://apex.ipk-gatersleben.de/apex/f?p=284:10) were used to obtain marker information and identify physical positions of previously published QTL studies. Where previously described QTL were identified based on iSelect markers, the physical positions were obtained from Bayer et al. (2017).
Results
Phenotypic evaluation of greenhouse trials
Phenotyping in the greenhouse with three different isolates showed a wide range of variability in the infection response type (1–10 scale) for all three isolates tested. The average infection response type (IRT) for isolate Hoehnstedt ranged from 1 to 9 (mean 3.96), for No 13 from 1 to 10 (mean 5.28) and for NFNB 50 from 1 to 10 (mean 3.6) (Fig. 1). Analysis of variance showed significant differences among the barley accessions for seedling resistance to NFNB for all isolates (Table 1). In trials with isolates No 13 and Hoehnstedt, the proposed differential set by Afanasenko et al. (2009) was used as a reference. The infection scores for the differential lines can be seen in Table 2. For isolate No 13, the infection scores ranged from 0.75 (CI 5791) to 8 (Harrington). For Hoehnstedt, the lines showed less variance and the scores ranged from 2.63 (Harbin) to 4.89 (CLS 25282). For isolate NFNB 50, the lines used as references and their respective infection scores can be obtained from Table 3. The scores ranged from 0.8 (Beecher) to 9.1 (Grimmett).

Frequency distribution for Pyrenophora teres f. teres reaction after inoculation with isolates Hoehnstedt, No 13 and NFNB 50
Phenotypic evaluation of field trials
A wide range of disease severity was observed for all three locations (Fig. 2; Table 1).

Frequency distribution of resistance to Pyrenophora teres f. teres for field trials in Quedlinburg (Germany), Zhodino (Belarus) and Warwick (Australia) with isolates NFNB 50, NFNB 73 and NFNB 85
In Germany in both years, infection pressure for NFNB was high in Summer Hill trials. Disease severity scores ranged on average between 4.1 and 31.5% (mean 11.5%). The frequency distribution was slightly right skewed with 184 accessions showing disease severity of < 10% and 7 accessions showing scores of > 25%. The heritability for this location was estimated at h2 = 0.73.
In Belarus in 2017, conditions were unfavourable for NFNB yet favourable for powdery mildew, which did not allow any more than two assessments of net blotch. Hence, for this location the disease score based on the respective last scoring date was used to calculate the mean disease severity across years. Disease severity scores ranged on average between 0.1 and 60% (mean 7.7%). The frequency distribution for this location is right-skewed with 201 accessions showing a disease severity of < 5% and 5 accessions showing scores of 40% and higher. Heritability for field trials in Belarus was h2 = 0.79. In field trials in Germany and Belarus again the differential set by Afanasenko et al. (2009) was scored as a reference (Table 2). In Germany, the disease severity among the differentials ranged from 5.52 (Prior) to 14.51% (CI 5791). In Belarus, the disease severity ranged from 0.75 (CI 5791, CLS 25282) to 19.75% (Harrington).
In Australia, three individual isolates were tested in the field. For all three isolates, disease scores from 1 to 9 were observed. Frequency distributions for isolates NFNB 50 and NFNB 73 were right-skewed towards resistance with 316 and 292 accessions showing disease scores ≤ 3, respectively. For isolate NFNB 85 only 149 accessions showed disease scores ≤ 3. The reference lines used in field trials in Australia are shown in Table 3. The infection scores ranged from 1.5 (WPG8412-9-2-1) to 8.8 (BS89-4-3). Line WPG8412-9-2-1 was resistant against all isolates used. So far no isolate found in Australia has been virulent on this line.
Population structure and linkage disequilibrium
STRUCTURE analysis identified an optimal k value of 3, with 58, 139 and 91 individuals belonging to subpopulation one, two and three, respectively (Online Resource 2; Online Resource 3). Out of the 449 accessions, 161 showed membership probabilities of less than 0.8 and were considered as admixed. Accessions belonging to subpopulations one and two were mainly 6-rowed types, population three comprised mainly 2-rowed accessions. Genome-wide linkage disequilibrium (LD) decay was estimated at 167 kb.
Genome-wide association mapping
Seedling resistance
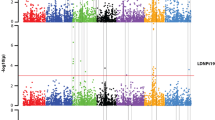
For isolate Hoehnstedt, eight significant marker–trait associations (MTAs) were detected on chromosome 6H (Fig. 3). Their − log10 (p) ranged from 4.63 to 6.3 (Table 4, Online Resource 4). The MTAs corresponded to two regions. The first region spanned from 128 to 165 Mbp (53.52 cM), including seven markers, with the peak marker at 140 Mbp with a − log10 (p) = 6.3 (JHI-Hv50 k-2016-391848), which explained 4.9% of the phenotypic variance. In addition, a second region comprising one significant marker (JHI-Hv50 k-2016-399702) was identified at 370 Mbp with a − log10 (p) = 5.2 and an R2 of 3.9%.

Genome-wide association analyses of resistance to Pyrenophora teres f. teres isolates Hoehnstedt, No 13 and NFNB 50 tested under greenhouse conditions. The x-axis shows the seven barley chromosomes, positions are based on the physical map, and the − log10(p) value is displayed on the y-axis. The green horizontal line represents the significance threshold of − log10(p) = 4.63
GWAS for isolate No 13 revealed 12 significant MTAs with − log10 (p) from 4.71 to 6.92 (Table 4; Fig. 3). Five markers associated with resistance were located on chromosome 4H between 64 and 70 Mbp with a peak marker at 70 Mbp with a − log10 (p) = 6.92 (JHI-Hv50 k-2016-237924), explaining 2.8% of the phenotypic variance. Additional MTAs were detected on chromosomes 4H (JHI-Hv50 k-2016-241935) and 6H (SCRI_RS_176650) at 352 Mbp and 373 Mbp, respectively. In addition, five MTAs were detected on chromosome 7H at 645 Mbp explaining 1.8–2.2% of the phenotypic variance.
For isolate NFNB 50 32, significant MTAs were detected (Fig. 3). The − log10 (p) ranged from 4.64 to 9.24 (Table 4). On chromosome 3H, 15 MTAs were detected, corresponding to three regions. One marker was located at 73 Mbp (46.29 cM, R2 = 2.0%) (JHI-Hv50 k-2016-165152), and three were located between 119 and 138 Mbp (46.68 cM), explaining 2.2–2.6% of the phenotypic variance (Online Resource 4). The third region spanned from 490 to 492 Mbp (48.44–48.63 cM) and included 11 markers, with the peak marker (JHI-Hv50 k-2016-183207) located at 490 Mbp (– log10 (p) = 9.24), which explained 4.4% of the phenotypic variance. On chromosome 6H, MTAs were detected in three regions, i.e. eight MTAs between 64 and 72 Mbp (52.73 cM, R2 = 2.0 to 2.6%), six MTAs between 133 and 140 Mbp (53.52 cM, R2 = 2.1% per marker) and two MTAs at 373 Mpb (SCRI_RS_188243 and SCRI_RS_195914), each explaining 2.1% of the phenotypic variance. One single MTA was detected on chromosome 7H (JHI-Hv50 k-2016-440870) at 5 Mbp with an R2 of 2.4%.
Adult plant resistance
GWAS based on the data of the field trials in Quedlinburg revealed nine MTAs on chromosome 6H with − log10 (p) between 4.78 and 6.62 (Table 4; Fig. 4, Online Resource 5). Five markers were located between 44 and 47 Mbp with three peak markers at 47 Mbp (– log10 (p) = 4.95), explaining 2.8% of the phenotypic variance, each. The remaining four markers were located at 406–410 Mbp with the peak marker (JHI-Hv50 k-2016-403424) at 406 Mbp explaining 3.9% of the phenotypic variance.

Genome-wide association analyses of resistance to Pyrenophora teres f. teres under field conditions in Quedlinburg (Germany) and Zhodino (Belarus). The x-axis shows the seven barley chromosomes, positions are based on the physical map, and the − log10(p) value is displayed on the y-axis. The green horizontal line represents the significance threshold of − log10(p) = 4.63
For the location Zhodino, a total of 61 significant MTAs were detected; 58 of these were located on chromosome 3H (Fig. 4, Online Resource 5). The 58 markers can be divided into five regions based on the physical map, and genetically these regions are less than 1 cM apart (Table 4). The first region comprised two markers located at 58 (46.29 cM) (JHI-Hv50 k-2016-164734) and 101 Mbp (46.29 cM) (JHI-Hv50 k-2016-166000) with − log10 (p) = 4.81 and 4.74, respectively. The second region comprised two markers located at 119 (46.68 cM) (JHI-Hv50 k-2016-166356) and 130 Mbp (46.68 cM) (JHI-Hv50 k-2016-166392) with − log10 (p) = 5.84. The third region spanned from 233 to 350 Mbp and included 50 markers with − log10 (p) = 4.83 to 5.24. The explained phenotypic variance ranged between 2.3 and 2.6% per marker. One marker mapped at 428 Mbp (47.07 cM), and the remaining three markers mapped to 621 Mbp. On chromosomes 4H, 5H and 6H one MTA was identified on each, located at 33, 634 and 140 Mbp, respectively.
For field trials conducted with isolate NFNB 50, eleven MTAs were detected (Fig. 5), four of which were located on chromosome 3H at 446 to 490 Mbp (47.46–48.44 cM) with the peak marker located at 490 Mbp (– log10 (p) = 6.08; JHI-Hv50 k-2016-183351) explaining 3.4% of the phenotypic variance. The remaining MTAs were located on chromosome 6H in the interval 133–135 Mbp (53.52 cM), 368 and 373 Mbp, R2 values ranged between 2.5 and 4% (Table 4, Online Resource 6).

Genome-wide association analyses of resistance to Pyrenophora teres f. teres isolates NFNB 50, NFNB 73 and NFNB 85 tested under field conditions in Warwick (Australia). The x-axis shows the seven barley chromosomes, positions are based on the physical map, and the –log10(p) value is displayed on the y-axis. The green horizontal line represents the significance threshold of − log10(p) = 4.63
GWAS for isolate NFNB 73 revealed 65 significant MTAs (Fig. 5, Online Resource 6). One marker was located on chromosome 5H at 579 Mbp (– log10 (p) = 4.65; JHI-Hv50 k-2016-326506) and one on chromosome 6H at 72 Mbp (JHI-Hv50 k-2016-388677). 51 MTAs mapped to chromosome 6H between 123 and 344 Mbp (53.52–53.91 cM) with − log10 (p) between 4.8 and 8.18, explaining 2.3 to 4% of the phenotypic variance (Table 4). The remaining 12 MTAs were also located on chromosome 6H in an interval from 356 to 373 Mbp with the peak marker at 373 Mbp with a − log10 (p) = 20.07 and a R2 value of 11.1% (SCRI_RS_188243).
A total of 56 markers, all located on chromosome 6H, were detected for isolate NFNB 85, corresponding to two regions (Fig. 5, Online Resource 6). The first region spanned from 37 to 76 Mbp with three peak markers at 47 Mbp (–log10 (p) = 10.22), explaining 6.8% per marker (JHI-Hv50 k-2016-385826, JHI-Hv50 k-2016-385857, JHI-Hv50 k-2016-385944) (Table 4). The second region was located at 355 to 379 Mbp with the peak marker at 373 Mbp and a − log10 (p) = 8.71 (JHI-Hv50 k-2016-399838), which explained 5.7% of the phenotypic variance.
Discussion
The net form of net blotch (NFNB) is a threat to barley producing regions all over the world. The high variability of this pathogen (Khan 1982; Liu et al. 2011; Serenius 2006; Steffenson and Webster 1992; Tekauz 1990) makes it a difficult task for breeders to identify and successfully introduce new resistance genes and QTL into current breeding material. Via bi-parental mapping approaches, several QTL for resistance on all seven barley chromosomes have been identified (König et al. 2014; Liu et al. 2011; Martin et al. 2018; Vatter et al. 2017). In recent years, GWAS has become a prominent approach to identify QTL and major genes for agronomically important traits, e.g. yield, abiotic and biotic stress (Gurung et al. 2011; Lex et al. 2014; Rode et al. 2011; Simmonds et al. 2014; Wehner et al. 2015). So far, only three studies were published showing the use of GWAS to identify NFNB resistance in barley (Amezrou et al. 2018; Richards et al. 2017; Wonneberger et al. 2017a) along with one study using nested association mapping (NAM)(Vatter et al. 2017). All of these previous studies used the low-density 9 k iSelect SNP Chip as a genotyping platform and applied genetic linkage maps for approximation of QTL positions. To our knowledge, the present study represents the first study on NFNB in barley to employ the 50 k iSelect barley SNP chip in association with physical marker positions (Bayer et al. 2017) based on the pseudo-molecule genome assembly of Mascher et al. (2017).
Phenotypic results of all experiments showed a high variability of the accessions towards the pathogen. Heritability ranged from h2 = 0.67 to 0.79 and was in good accordance with previously published studies, reporting on h2 for this trait ranging between 0.62 and 0.99 (Grewal et al. 2012; König et al. 2013; Richards et al. 2017; Vatter et al. 2017; Wonneberger et al. 2017a).
Overall, 254 significant marker–trait associations (MTAs) were detected, which corresponded to 15 distinct regions. Three of them were identified conferring seedling resistance, five conferring adult plant resistance and seven were active at both growth stages. The regions were located on barley chromosomes 3H, 4H, 5H, 6H and 7H.
On chromosome 3H, five regions were identified associated with NFNB resistance (Fig. 6). The first two regions identified based on phenotypic data from the field trials at Zhodino and greenhouse trials using isolate NFNB 50, were located at 58–101 Mbp and 119–130 Mbp corresponding genetically to 46.29 and 46.68 cM, respectively. These MTAs are assumed to contribute resistance at seedling and adult plant stages. König et al. (2014) identified several regions on the short arm of chromosome 3H (QTLuhs-3H, QTLuhs-3H-1 and QTLuhs-3H-2), but with the data available it is not possible to determine whether it corresponds to our regions. The third region on chromosome 3H was mapped to 233–350 Mbp for data based on field trials in Zhodino. Physically, this appears to be a large interval, and Bayer et al. (2017) were not able to anchor markers located in this interval into a genetic map of their RIL population. Physically, there is a gap between the two markers JHI-Hv50 k-2016-169770 (222.344564 Mbp) and JHI-Hv50 k-2016-175163 (352.146586 Mbp); however, genetically both markers are located at 47.07 cM (Bayer et al. 2017). The region is located near or in the centromere, where little recombination occurs, which explains the large physical interval. Graner et al. (1996) identified a gene on the long arm of chromosome 3H conferring resistance to NFNB designated Pt,a. In our study, two regions on chromosome 3HL were identified. The first spanned from 428 to 492 Mbp and was based on data from the field trials in Zhodino and the field and greenhouse trials with isolate NFNB 50, hence contributing to seedling and adult plant resistance. One of the significant associated markers in our studies was SCRI_RS_152172, which was also significantly associated with NFNB resistance in the study by Wonneberger et al. (2017a). Koladia et al. (2017) evaluated nine NFNB isolates and detected a QTL, which was significant for all these isolates, located at 490 Mbp with the peak marker being SCRI_RS_221644. Burlakoti et al. (2017) also found a QTL, which corresponds to our region. Interestingly, they conducted GWAS on resistance for the SFNB, indicating that even though both forms are genetically distinct, this region harbours resistance against both forms and is not isolate specific. The same holds true for the second region identified on chromosome 3HL located at 621 Mbp and identified from field data from Zhodino. Burlakoti et al. (2017) and Tamang et al. (2015), who both worked with SFNB, identified QTL on chromosome 3HL at 625 Mbp and 616–619 Mbp, respectively. Martin et al. (2018), also detected a QTL for NFNB resistance located at 622 Mbp by mapping a DH-population (UVC8 x SABBI Erica) and developed a PCR-based KASP marker (USQ3_1329) for use in breeding programmes.

QTLs for P. teres f. teres resistance in barley on chromosomes 3H and 6H. QTLs detected in the present study are represented by vertical bars. Previous studies reporting overlapping QTLs are shown next to the respective region
On chromosome 4H, two regions harbouring NFNB resistance were identified. The first was located on the short arm of chromosome 4H at 33–70 Mbp and was detected from field trials at Zhodino and greenhouse trials with isolate No 13. This region corresponded to a QTL found by Islamovic et al. (2017), which was flanked by markers 4544-461 (46 Mbp) and 1944-1901 (76 Mbp) and was significantly associated with resistance for all four isolates tested. One of the isolates they used was NFNB 50, which was also tested in our study, but did not reveal any marker–trait association at this chromosomal region. The second region on chromosome 4H was located near the centromere at 352 Mbp. This region was found for seedling tests using isolate No 13. Wonneberger et al. (2017a) detected a QTL at 350 Mbp for seedling resistance and Steffenson et al. (1996) located a major QTL on chromosome 4H near the centromere for seedling resistance, which explained 31% of the phenotypic variance.
Two regions for NFNB resistance at the adult plant stage were detected on chromosome 5HL. The first region was detected using isolate NFNB 73 and was located at 579 Mb. For this region, no overlap with previously published QTL was found. Based on the data from field trials in Zhodino, an association located at 634 Mbp was detected. The resistance locus AL_QRptt5-2, identified in a study using a DH-population of the cross Arve x Lavrans, and in a GWA study (Wonneberger et al. 2017a, b), is located in the same region (648–652 Mbp). In addition, Amezrou et al. (2018) detected a MTA at 624–629 Mbp and Grewal et al. (2012) mapped a QTL to the telomeric region of chromosome 5HL (199.4–206.3 cM).
Chromosome 6H is widely known to harbour several QTL for resistance/susceptibility to NFNB. In this study, four regions associated with resistance were detected (Fig. 6). The first region is located on chromosome 6HS between 37 and 76 Mbp and was detected for field trials in Quedlinburg, field trials using isolates NFNB 73 and NFNB 85 and for greenhouse trials using isolate NFNB 50. This region mapped to the sensitivity locus SPN1, which is flanked by the markers 4191-268 (47.261684 Mbp) and ABC08769-1-1-205 (91.140417 Mbp) (Liu et al. 2015). In that study, Liu et al. screened a RIL-population of Hector x NDB 112. The latter is a highly resistant, North American landrace, which was also included in our study and showed low infection responses for all experiments. Vatter et al. (2017) and Wonneberger et al. (2017a) both found associations at 46 Mbp, while Richards et al. (2017) identified MTAs for all isolates tested at between 42 and 66 Mbp. Interestingly, five of the markers identified in their study (SCRI_RS_162581, SCRI_RS_119674, SCRI_RS_142506, SCRI_RS_168111 and 12_30658), also revealed a significant MTA in our study. Finally, Amezrou et al. (2018) also identified a MTA at 66 Mbp. Furthermore, a large region spanning from 123 to 344 Mbp was detected based on the data for field trials in Zhodino, field trials using isolates NFNB 50 and NFNB 73, and greenhouse trials using isolates NFNB 50 and Hoehnstedt. Genetically, this region spanned from 53.52 to 53.91 cM (Bayer et al. 2017). Wonneberger et al. (2017a) and Islamovic et al. (2017) detected significant QTL at 120–164 Mbp and 340 Mbp (marker 5497-661), respectively. Martin et al. (2018) developed several PCR-based KASP markers for the identification of NFNB resistance QTL. Their marker USQ2_0799 is located at 335.741625 Mbp (Anke Martin, Centre for Crop Health, University of Southern Queensland, Toowoomba, QLD 4350, Australia, unpublished data) and co-located with the microsatellite marker Bmag0173 (67.4 cM). This microsatellite marker has long been associated with NFNB resistance and was used in several studies in the past (Abu Qamar et al. 2008; Cakir et al. 2003; Friesen et al. 2006; Gupta et al. 2010; Gupta et al. 2011; Liu et al. 2015; Manninen et al. 2006; O’Boyle et al. 2014; Shjerve et al. 2014). The third region on chromosome 6H is located near the centromere at 355–379 Mbp and turned out to be significantly associated with NFNB resistance in all experiments, except field trials in Germany and Zhodino. Markers located in this interval could not be anchored genetically in the RIL population by Bayer et al. (2017). Similar to chromosome 3H, there is a gap on chromosome 6H in the region between markers JHI-Hv50 k-2016-397733 (344.799609 Mbp) and JHI-Hv50 k-2016-401495 (387.092035 Mbp). Genetically, they are located at 53.91 and 54.30 cM, respectively (Bayer et al. 2017). Nevertheless, this region corresponds to the major susceptibility locus Spt1, formerly named rpt.r/ rpt.k (Abu Qamar et al. 2008), and was fine-mapped by Richards et al. (2016). In their study, the markers rpt-M12, rpt-M13 and rpt-M20 co-segregated with the Spt1 locus and corresponded to the Morex WGS contigs morex_contig_43862, morex_contig_64570 and morex_contig_37494, respectively. The contig morex_contig_64570 includes the iSelect marker SCRI_RS_176650, which is positioned at 373.424916 Mbp and significantly associated with resistance to isolates No 13, NFNB 73 and NFNB 85. The gene is flanked by the markers rpt-M8 and SCRI_RS_165041 (384.412678 Mbp). Rpt-M8 corresponds to morex_contig_1573477, which includes the SNP markers SCRI_RS_171997 (370.428440 Mbp) and SCRI_RS_186193 (370.429069 Mbp). Hence, the locus is positioned between 370 and 384 Mbp. According to Richards et al. (2016) Spt1 is delimited to 0.24 cM. This region contains 49 genes including 39 high-confidence genes with 6 genes encoding immunity receptor-like proteins. Moreover, this region was also identified via GWAS by Richards et al. (2017), Wonneberger et al. (2017a), Vatter et al. (2017) and Martin et al. (2018). In the latter study, PCR-based KASP markers were developed, namely USQ1_1140 (352 Mbp) and USQ3_0144 (384 Mb). The marker USQ1_1140 co-located with the microsatellite marker HVM 74 (68.0 cM), which was reported in several previous studies to be associated with resistance to NFNB (Cakir et al. 2003; Grewal et al. 2008; Gupta et al. 2011). In a study with SFNB, Tamang et al. (2015) identified significant MTAs at 370–373 Mbp for all isolates tested. One of the significant markers was the above-mentioned iSelect marker SCRI_RS_176650 (373.424916 Mbp). This suggests that Spt1 does not only confer susceptibility to NFNB but also to SFNB and that this SNP (SCRI_RS_176650) can be very valuable for identifying resistant or susceptible genotypes. The last region on 6H is located at 406–410 Mbp and was identified from field trials in Quedlinburg. Amezrou et al. (2018) and Koladia et al. (2017) both reported a QTL at 390 Mbp and in both studies the marker SCRI_RS_140091 was one of the peak markers. Using the genetic map of Bayer et al. (2017), our region is located at 54.69 cM, while the region reported in the other two studies is located at 54.3 cM. Based on the available data, it is not possible to determine whether the regions belong to one QTL or if they have to be considered as individual QTLs.
On chromosome 7H, two regions contributing to seedling resistance were identified. The first region was identified from greenhouse trials using isolate NFNB 50 and was located on chromosome 7HS at 5 Mbp and consisted of one MTA (marker JHI_Hv50 k-2016-440870). Vatter et al. (2017) found significant associations at 2 Mbp. The second region was located on the long arm of chromosome 7H, consisted of five MTAs at 645 Mbp, and was based on greenhouse trials with isolate No 13. Richards et al. (2017) reported a QTL at 643 Mbp against all isolates tested. In a study by Martin et al. (2018), a QTL at 655 Mbp was reported from field trials using isolates NFNB 50 and NFNB 85. These isolates were used in the present study as well, yet we did not detect any significant associations from our trials using these two isolates.
The set of barley accessions used in this study showed a genome-wide LD decay of 167 kb. Notably, for an inbreeding crop such as barley, this can be considered comparably low. LD decay in previous GWA studies in barley were estimated at 18 to 1.3 cM (Bellucci et al. 2017; Bengtsson et al. 2017; Burlakoti et al. 2017; Gyawali et al. 2017; Massman et al. 2010; Mitterbauer et al. 2017; Tamang et al. 2015; Vatter et al. 2017; Wehner et al. 2015; Wonneberger et al. 2017a). Burlakoti et al. (2017) evaluated a barley set of 376 advanced breeding lines from four breeding programmes from the Upper Midwest and reported LD decays between 10 and 18 cM. Wonneberger et al. (2017a) used a set comprising landraces and breeding lines predominantly originated from Norway, Sweden, Denmark and Finland and reported LD decay to be 13 cM. Vatter et al. (2017) used a nested association mapping (NAM) population (Maurer et al. 2015), where the spring barley cultivar Barke was crossed with 25 wild barley accessions originated from the Fertile Crescent. This population showed a LD decay of 8 cM. LD decays between 4 and 8 cM were also reported by Massman et al. (2010), Bellucci et al. (2017) and Tamang et al. (2015), who evaluated 768 advanced breeding lines from the Upper Midwest, 112 cultivated winter barleys from eleven European countries and 2062 geographically diverse barley cultivars, breeding lines and landraces, respectively. Mitterbauer et al. (2017) evaluated a set of 98 winter barleys comprising gene bank accessions and European cultivars released in different years and estimated LD decay at 1.3 cM. This shows that LD varies greatly between studies and is highly dependent on the material investigated. However, it gives an insight into the diversity of the genotypes involved. The barley accessions in the present study originated from almost 50 different countries from all over the world. The high diversity of the set was observed in the phenotypic reactions towards infection with NFNB, but is also reflected in the low LD. A low LD enables a high mapping resolution, which requires a high marker saturation (Zhu et al. 2008). With the 50 k iSelect chip (Bayer et al. 2017), this hurdle can be overcome. The chip comprises 44,040 SNP markers, of which 33,818 SNP markers were informative for the barley set investigated in the present study. This means, with a genome size of 5.1 Gb in barley, there was on average a marker every 150 kb. In combination with the physical map, it will enable researchers to define QTL more accurately. The centromeric region of chromosome 6H is a good example for this. This region has long been known to harbour several resistance/susceptibility QTL and has been described in many studies (Abu Qamar et al. 2008; Friesen et al. 2006; Gupta et al. 2010; Gupta et al. 2011; Liu et al. 2015; Martin et al. 2018; O’Boyle et al. 2014; Richards et al. 2016, 2017; Shjerve et al. 2014; Vatter et al. 2017; Wonneberger et al. 2017a). Two markers located in this region are the SSR markers Bmag0173 and HVM 74. These two markers were repeatedly reported to map to the same region. Martin et al. (2018) mapped them only 0.6 cM apart (Bmag0173: 67.4 cM, HVM 74: 68 cM). However, in the present study it was shown that they can be assigned to two different regions, located at 123–344 Mbp and 356–379 Mbp.
The aim of this study was to identify QTL for resistance against NFNB in a diverse barley set. The MTAs detected corresponded to 15 distinct regions and were located on barley chromosomes 3H, 4H, 5H, 6H and 7H. Eleven of these regions corresponded to QTL already described in previous studies, and seven regions were identified at the seedling and adult plant stage. The four putatively new QTL were located on chromosomes 3H at 233 to 350 Mbp, 5H at 579 Mbp, 6H at 406 to 410 Mbp and on chromosome 7H at 5 Mbp. Most regions were identified across several isolates and/or locations tested, which makes them interesting for breeding purposes, since they do not seem to be race-specific.
Author contribution statement
OA and FO planned and managed the project. OK provided and characterized all accessions. AZ identified varieties and was involved in the resistance screening in field trials in Belarus. AA conducted the field screening in Russia. GJP was in charge of the screenings conducted in Australia (field and greenhouse). OA, FO, GJP and RS contributed to the interpretation and discussion of the results. FN conducted the field and greenhouse screenings in Germany, analysed the data and wrote the manuscript. OA contributed to the introduction and the material and methods parts of the manuscript.
References
Abu Qamar M et al (2008) A region of barley chromosome 6H harbors multiple major genes associated with net type net blotch resistance. Theor Appl Genet 117:1261–1270. https://doi.org/10.1007/s00122-008-0860-x
Afanasenko O (1995) Characteristics of resistance of barley accessions to different Pyrenophora teres populations. Mycol Phytopathol 29:27–32
Afanasenko O, Makarova I, Zubkovich A (1999) The number of genes controlling resistance to Pyrenophora teres Drechs. strains in barley. Russ J Genet C/C Genet 35:274–283
Afanasenko O, Jalli M, Pinnschmidt H, Filatova O, Platz G (2009) Development of an international standard set of barley differential genotypes for Pyrenophora teres f. teres. Plant Pathol 58:665–676
Afanasenko O et al (2015) Mapping of the loci controlling the resistance to Pyrenophora teres f. teres and Cochliobolus sativus in two double haploid barley populations. Russ J Genet Appl Res 5:242–253
Afgan E et al (2016) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res 44:W3–W10. https://doi.org/10.1093/nar/gkw343
Afonin A, Greene S, Dzyubenko N, Frolov A (2008) Interactive Agricultural ecological Atlas of Russia and Neighboring Countries. Economic Plants and their Diseases, Pests and Weeds. http://www.agroatlas.ru/. Accessed 26 Mar 2019
Amezrou R et al (2018) Genome-wide association studies of net form of net blotch resistance at seedling and adult plant stages in spring barley collection. Mol Breed 38:1–14. https://doi.org/10.1007/s11032-018-0813-2
Anisimova A, Novikova L, Novakazi F, Kopahnke D, Zubkovich A, Afanasenko O (2017) Polymorphism on virulence and specificity of microevolution processes in populations of causal agent of barley net blotch Pyrenophora teres f teres. Mikol I Fitopatol 51:229–240
Bayer MM et al (2017) Development and evaluation of a barley 50 k iSelect SNP. Array Front Plant Sci 8:1792. https://doi.org/10.3389/fpls.2017.01792
Bellucci A et al (2017) Genome-wide association mapping in winter barley for grain yield and culm cell wall polymer content using the high-throughput CoMPP technique. PLoS ONE 12:e0173313. https://doi.org/10.1371/journal.pone.0173313
Bengtsson T, Manninen O, Jahoor A, Orabi J (2017) Genetic diversity, population structure and linkage disequilibrium in Nordic spring barley (Hordeum vulgare L. subsp. vulgare). Genet Resour Crop Evolut 64:2021–2033. https://doi.org/10.1007/s10722-017-0493-5
Berger GL et al (2013) Marker-trait associations in Virginia Tech winter barley identified using genome-wide mapping. Theor Appl Genet 126:693–710
Brandl F, Hoffmann G (1991) Differentiation of physiological races of Drechslera teres (Sacc.) Shoem., pathogen net blotch of barley. Zeitschrift fuer Pflanzenkrankheiten und Pflanzenschutz
Burlakoti RR et al (2017) Genome-Wide Association Study of spot form of net blotch resistance in the upper midwest barley breeding programs. Phytopathology 107:100–108. https://doi.org/10.1094/PHYTO-03-16-0136-R
Burleigh J, Tajani M, Seck M (1988) Effects of Pyrenophora teres and weeds on barley yield and yield components. Phytopathology 78:295–299
Cakir M et al (2003) Mapping and validation of the genes for resistance to Pyrenophora teres f. teres in barley (Hordeum vulgare L.). Aust J Agric Res 54:1369–1377
Cakir M et al (2011) Genetic mapping and QTL analysis of disease resistance traits in the barley population Baudin × AC Metcalfe. Crop Pasture Sci 62:152–161
Campoy JA et al (2016) Genetic diversity, linkage disequilibrium, population structure and construction of a core collection of Prunus avium L. landraces and bred cultivars. BMC Plant Biol 16:49. https://doi.org/10.1186/s12870-016-0712-9
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. https://doi.org/10.1186/s13742-015-0047-8
Douglas G, Gordon I (1985) Quantitative genetics of net blotch resistance in barley. N Z J Agric Res 28:157–164
Earl DA, vonHoldt BM (2012) Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Flint-Garcia SA, Thornsberry JM, Buckler ESt (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374. https://doi.org/10.1146/annurev.arplant.54.031902.134907
Friesen TL, Faris JD, Lai Z, Steffenson BJ (2006) Identification and chromosomal location of major genes for resistance to Pyrenophora teres in a doubled-haploid barley population. Genome 49:855–859. https://doi.org/10.1139/g06-024
Graner A, Foroughi-Wehr B, Tekauz A (1996) RFLP mapping of a gene in barley conferring resistance to net blotch (Pyrenophora teres). Euphytica 91:229–234
Grewal TS, Rossnagel BG, Pozniak CJ, Scoles GJ (2008) Mapping quantitative trait loci associated with barley net blotch resistance. Theor Appl Genet 116:529–539. https://doi.org/10.1007/s00122-007-0688-9
Grewal TS, Rossnagel BG, Scoles GJ (2012) Mapping quantitative trait loci associated with spot blotch and net blotch resistance in a doubled-haploid barley population. Mol Breed 30:267–279
Gupta S et al (2004) Gene distribution and SSR markers linked with net type net blotch resistance in barley. In: 9th International Barley Genetics Symposium, 20–26 June. Czech Republic, Brno, pp 668–673
Gupta S et al (2010) Quantitative trait loci and epistatic interactions in barley conferring resistance to net type net blotch (Pyrenophora teres f teres) isolates. Plant Breed 129:362–368
Gupta S, Li C, Loughman R, Cakir M, Westcott S, Lance R (2011) Identifying genetic complexity of 6H locus in barley conferring resistance to Pyrenophora teres f teres. Plant Breed 130:423–429. https://doi.org/10.1111/j.1439-0523.2011.01854.x
Gurung S et al (2011) Identification of novel genomic regions associated with resistance to Pyrenophora tritici-repentis races 1 and 5 in spring wheat landraces using association analysis. Theor Appl Genet 123:1029–1041. https://doi.org/10.1007/s00122-011-1645-1
Gyawali S, Otte ML, Chao S, Jilal A, Jacob DL, Amezrou R, Verma RPS (2017) Genome wide association studies (GWAS) of element contents in grain with a special focus on zinc and iron in a world collection of barley (Hordeum vulgare L.). J Cereal Sci 77:266–274. https://doi.org/10.1016/j.jcs.2017.08.019
Islamovic E, Bregitzer P, Friesen TL (2017) Barley 4H QTL confers NFNB resistance to a global set of P. teres f. teres isolates. Mol Breed 37:29. https://doi.org/10.1007/s11032-017-0621-0
Kangas A, Jalli M, Kedonperä A, Laine A, Niskanen M, Salo Y, Vuorinen M, Jauhiainen L, Ramstadius E 2005 Viljalajikkeiden herkkyys tautitartunnoille virallisissa lajikekokeissa 1998-2005. Disease susceptibility of cereals in Finnish official variety trials. [English summary and titles] Agrifood Res Rep, MTT:n selvityksiä 96: 33
Khan T (1982) Occurence and pathogenicity of Drechslera teres isolates causing spot-type symptoms on barley in Western Australia. Plant Dis 66:423–425
Koladia V, Faris J, Richards J, Brueggeman R, Chao S, Friesen T (2017) Genetic analysis of net form net blotch resistance in barley lines CIho 5791 and Tifang against a global collection of P. teres f teres isolates. Theor Appl Genet 130:163–173
König J, Perovic D, Kopahnke D, Ordon F (2013) Development of an efficient method for assessing resistance to the net type of net blotch (Pyrenophora teres f teres) in winter barley and mapping of quantitative trait loci for resistance. Mol Breed 32:641–650. https://doi.org/10.1007/s11032-013-9897-x
König J, Perovic D, Kopahnke D, Ordon F, Léon J (2014) Mapping seedling resistance to net form of net blotch (Pyrenophora teres f teres) in barley using detached leaf assay. Plant Breed 133:356–365. https://doi.org/10.1111/pbr.12147
Lex J, Ahlemeyer J, Friedt W, Ordon FJ (2014) Genome-wide association studies of agronomic and quality traits in a set of German winter barley (Hordeum vulgare L.) cultivars using Diversity Arrays Technology (DArT). J Appl Genet 55:295–305
Lightfoot DJ, Able AJ (2010) Growth of Pyrenophora teres in planta during barley net blotch disease. Australas Plant Pathol 39:499–507
Lipka AE et al (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28:2397–2399
Liu Z, Ellwood SR, Oliver RP, Friesen TL (2011) Pyrenophora teres: profile of an increasingly damaging barley pathogen. Mol Plant Pathol 12:1–19. https://doi.org/10.1111/j.1364-3703.2010.00649.x
Liu Z, Holmes DJ, Faris JD, Chao S, Brueggeman RS, Edwards MC, Friesen TL (2015) Necrotrophic effector-triggered susceptibility (NETS) underlies the barley-Pyrenophora teres f. teres interaction specific to chromosome 6H. Mol Plant Pathol 16:188–200. https://doi.org/10.1111/mpp.12172
Ma Z, Lapitan NL, Steffenson B (2004) QTL mapping of net blotch resistance genes in a doubled-haploid population of six-rowed barley. Euphytica 137:291–296
Manninen OM, Jalli M, Kalendar R, Schulman A, Afanasenko O, Robinson J (2006) Mapping of major spot-type and net-type net-blotch resistance genes in the Ethiopian barley line CI 9819. Genome 49:1564–1571. https://doi.org/10.1139/g06-119
Martin A, Platz GJ, de Klerk D, Fowler RA, Smit F, Potgieter FG, Prins R (2018) Identification and mapping of net form of net blotch resistance in South African barley. Mol Breed 38:53. https://doi.org/10.1007/s11032-018-0814-1
Mascher M et al (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544:427–433. https://doi.org/10.1038/nature22043
Massman J et al (2010) Genome-wide association mapping of Fusarium head blight resistance in contemporary barley breeding germplasm. Mol Breed 27:439–454. https://doi.org/10.1007/s11032-010-9442-0
Mathre D (1997) Compendium of Barley Diseases. The American Phytopathological Society, St. Paul, MN
Maurer A et al (2015) Modelling the genetic architecture of flowering time control in barley through nested association mapping. BMC Genom 16:290
Mitterbauer E et al (2017) Growth response of 98 barley (Hordeum vulgare L.) genotypes to elevated CO2 and identification of related quantitative trait loci using genome-wide association studies. Plant Breed 136:483–497. https://doi.org/10.1111/pbr.12501
Mohammadi M et al (2015) A genome-wide association study of malting quality across eight US barley breeding programs. Theor Appl Genet 128:705–721. https://doi.org/10.1007/s00122-015-2465-5
Moll E, Flath K, Tessenow I (2010) Assessment of resistance in cereal cultivars Design and analysis of experiments using the SAS-application RESI 2 Berichte aus dem Julius Kühn-Institut: 154
Muqaddasi QH, Reif JC, Li Z, Basnet BR, Dreisigacker S, Röder MS (2017) Genome-wide association mapping and genome-wide prediction of anther extrusion in CIMMYT spring wheat. Euphytica 213:73. https://doi.org/10.1007/s10681-017-1863-y
Murray GM, Brennan JP (2009) The current and potential costs from diseases of barley in Australia. Grains Research and Development Corporation, Barton
Nordborg M et al (2002) The extent of linkage disequilibrium in Arabidopsis thaliana. Nat Genet 30:190–193. https://doi.org/10.1038/ng813
O’Boyle P et al (2014) Mapping net blotch resistance in ‘Nomini’and CIho 2291 barley. Crop Sci 54:2596–2602
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7:e32253. https://doi.org/10.1371/journal.pone.0032253
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rafalski A, Morgante M (2004) Corn and humans: recombination and linkage disequilibrium in two genomes of similar size. Trends Genetics 20:103–111
Raman H, Platz G, Chalmers K, Raman R, Read B, Barr A, Moody D (2003) Mapping of genomic regions associated with net form of netblotch resistance in barley. Aust J Agric Res 54:1359–1367
Reif JC, Melchinger AE, Frisch M (2005) Genetical and mathematical properties of similarity and dissimilarity coefficients applied in plant breeding and seed bank management. Crop Sci 45:1–7
Remington DL et al (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci 98:11479–11484
Richards J, Chao S, Friesen T, Brueggeman R (2016) Fine mapping of the barley chromosome 6H net form net blotch susceptibility locus. G3 (Bethesda) 6:1809–1818. https://doi.org/10.1534/g3.116.028902
Richards JK, Friesen TL, Brueggeman RS (2017) Association mapping utilizing diverse barley lines reveals net form net blotch seedling resistance/susceptibility loci. Theor Appl Genet 130:915–927. https://doi.org/10.1007/s00122-017-2860-1
Richter K, Schondelmaier J, Jung C (1998) Mapping of quantitative trait loci affecting Drechslera teres resistance in barley with molecular markers. Theor Appl Genet 97:1225–1234
Robinson J, Jalli M (1997) Quantitative resistance to Pyrenophora teres in six Nordic spring barley accessions. Euphytica 94:201–208
Rode J, Ahlemeyer J, Friedt W, Ordon F (2011) Identification of marker-trait associations in the German winter barley breeding gene pool (Hordeum vulgare L.). Mol Breed 30:831–843. https://doi.org/10.1007/s11032-011-9667-6
Saari E, Prescott J (1975) Scale for appraising the foliar intensity of wheat diseases. Plant Disease Reporter 59:377–380
Sannemann W, Huang BE, Mathew B, Léon J (2015) Multi-parent advanced generation inter-cross in barley: high-resolution quantitative trait locus mapping for flowering time as a proof of concept. Mol Breed 35:86
Serenius M (2006) Population structure of Pyrenophora teres, the causal agent of net blotch of barley (Doctoral Dissertation). Agrifood Research Reports 78:60
Shjerve RA, Faris JD, Brueggeman RS, Yan C, Zhu Y, Koladia V, Friesen TL (2014) Evaluation of a Pyrenophora teres f teres mapping population reveals multiple independent interactions with a region of barley chromosome 6H. Fungal Genet Biol 70:104–112. https://doi.org/10.1016/j.fgb.2014.07.012
Silvar C et al (2010) Screening the Spanish barley core collection for disease resistance. Plant Breed 129:45–52
Simmonds J et al (2014) Identification and independent validation of a stable yield and thousand grain weight QTL on chromosome 6A of hexaploid wheat (Triticum aestivum L.). BMC Plant Biol 14:191
Smedegård-Petersen V (1971) Pyrenophora teres f. maculata f. nov. and Pyrenophora teres f. teres on barley in Denmark. Yearb R Vet Agric Univ (Copenhagen) 1971:124–144
Smedegård-Petersen V (1976) Pathogenesis and genetics of net-spot blotch and leaf stripe of barley caused by Pyrenophora teres and Pyrenophora graminea (Doctoral Dissertation) Copenhagen, p 176
Steffenson BJ, Webster R (1992) Quantitative resistance to Pyrenophora teres f. teres in barley. Phytopathology 82:407–411
Steffenson B, Hayes P, Kleinhofs A (1996) Genetics of seedling and adult plant resistance to net blotch (Pyrenophora teres f. teres) and spot blotch (Cochliobolus sativus) in barley. Theor Appl Genet 92:552–558
Stein N, Herren G, Keller B (2001) A new DNA extraction method for high-throughput marker analysis in a large-genome species such as Triticum aestivum. Plant Breed 120:354–356
Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci 100:9440–9445
Tamang P, Neupane A, Mamidi S, Friesen T, Brueggeman R (2015) Association mapping of seedling resistance to spot form net blotch in a worldwide collection of barley. Phytopathology 105:500–508. https://doi.org/10.1094/PHYTO-04-14-0106-R
Tekauz A (1985) A numerical scale to classify reactions of barley to Pyrenophora teres. Can J Plant Path 7:181–183
Tekauz A (1990) Characterization and distribution of pathogenic variation in Pyrenophora teres f. teres and P. teres f. maculata from western Canada. Can J Plant Pathol 12:141–148
Trofimovskaya AY, Afanasenko O, Levitin MM (1983) Sources of barley resistance to the causal agent of net blotch (Drechslera teres). Rep Acad Agricu Sci 3:19–21
Vatter T, Maurer A, Kopahnke D, Perovic D, Ordon F, Pillen K (2017) A nested association mapping population identifies multiple small effect QTL conferring resistance against net blotch (Pyrenophora teres f. teres) in wild barley. PLoS ONE 12:e0186803. https://doi.org/10.1371/journal.pone.0186803
Wallwork H, Butt M, Capio E (2016) Pathogen diversity and screening for minor gene resistance to Pyrenophora teres f. teres in barley and its use for plant breeding. Australas Plant Pathol 45:527–531
Wang H, Smith KP, Combs E, Blake T, Horsley RD, Muehlbauer GJ (2012) Effect of population size and unbalanced data sets on QTL detection using genome-wide association mapping in barley breeding germplasm. Theor Appl Genet 124:111–124. https://doi.org/10.1007/s00122-011-1691-8
Wehner GG, Balko CC, Enders MM, Humbeck KK, Ordon FF (2015) Identification of genomic regions involved in tolerance to drought stress and drought stress induced leaf senescence in juvenile barley. BMC Plant Biol 15:125
Wonneberger R, Ficke A, Lillemo M (2017a) Identification of quantitative trait loci associated with resistance to net form net blotch in a collection of Nordic barley germplasm. Theor Appl Genet 130:2025–2043. https://doi.org/10.1007/s00122-017-2940-2
Wonneberger R, Ficke A, Lillemo M (2017b) Mapping of quantitative trait loci associated with resistance to net form net blotch (Pyrenophora teres f teres) in a doubled haploid Norwegian barley population. PLoS ONE 12:e0175773. https://doi.org/10.1371/journal.pone.0175773
Yan J, Shah T, Warburton ML, Buckler ES, McMullen MD, Crouch JJ (2009) Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS ONE 4:e8451
Zhang Z et al (2010) Mixed linear model approach adapted for genome-wide association studies. Nat Genet 42:355
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome J 1:5–20. https://doi.org/10.3835/plantgenome2008.02.0089
Acknowledgements
This research was supported by the German Research Society (DFG) (OR 72/11-1) and the Russian Foundation for Basic Research (RFBR) (No 15-54-12365 NNIO_a).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Communicated by Kevin Smith.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Novakazi, F., Afanasenko, O., Anisimova, A. et al. Genetic analysis of a worldwide barley collection for resistance to net form of net blotch disease (Pyrenophora teres f. teres). Theor Appl Genet 132, 2633–2650 (2019). https://doi.org/10.1007/s00122-019-03378-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03378-1