
Abstract
Key message
QTL mapping for fiber-related traits and elucidation of a stable and novel QTL affecting seed lignin content, cellulose content and seed oil content.
Abstract
Dissection of the genetic networks for fiber biosynthesis is important for improving the seed oil content and meal value of Brassica napus. In this study, the genetic basis of seed fiber biosynthesis in B. napus was investigated via quantitative trait locus (QTL) analysis using a doubled haploid population derived from ‘KenC-8’ crossed with ‘N53-2.’ Seed lignin content (LC), cellulose content (CC) and hemicellulose content (HC) were significantly negatively correlated with seed oil content (OC). Co-localization QTLs among LC, CC, HC and OC on A09 were found with contributions ranging from 9.87 to 48.50%. Seven co-localization QTLs involved in the fiber component and OC were further verified by bulked segregant analysis (BSA). The unique QTL uqA9-12 might be a real and new QTL that was commonly identified by QTL mapping and BSA and simultaneously affected LC, CC and OC with opposite additive effects. A potential regulatory network controlling seed fiber biosynthesis was constructed to dissect the complex mechanism of seed fiber and oil accumulation, and numerous candidate genes were identified in the fiber-related QTL regions. These results provided an enrichment of QTLs and potential candidates for fiber biosynthesis, as well as useful new information for understanding the complex genetic mechanism underlying rapeseed seed fiber accumulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brassica napus (AACC, 2n = 38) is widely cultivated globally for the production of vegetable oil and biodiesel, and it provides a high-quality meal for animal feed after oil extraction (Liu et al. 2012). The development of B. napus varieties with high seed oil content, and meal quality is one of the most important tasks for rapeseed breeding. Seed fiber components, including lignin, cellulose and hemicellulose, have a negative effect on the seed oil content and meal value of B. napus (Liu et al. 2012; Wang et al. 2015a). Lignin is a major antinutritional fiber component that leads to a reduction of seed meal value, and the synthesis of cellulose and hemicellulose can redirect photosynthetic assimilates from sugar biosynthesis into seed oil biosynthesis and result in a reduction of oil content (Liang et al. 2002; Liu et al. 2013; Wang et al. 2015a). Understanding the genetic mechanism of seed fiber traits in B. napus is pivotal for improving the seed oil content and meal value of B. napus.
Fibers have complex quantitative traits that are controlled by a number of genes and are highly influenced by environmental conditions (Badani et al. 2006; Liu et al. 2012, 2013; Stein et al. 2013). Quantitative trait loci (QTLs) influencing the fiber content are detected using different populations (Badani et al. 2006; Fu et al. 2007; Snowdon et al. 2010; Wang et al. 2013), especially yellow-seeded B. napus, which is generally considered to contain larger amounts of oil and protein (Fu et al. 2007; Wittkop et al. 2009), a thinner seed coat (Wittkop et al. 2009) and lower content of lignin, cellulose and hemicellulose (Simbaya et al. 1995) compared with black-seeded B. napus.
Seed fiber components are highly correlated with seed phenolic compounds and seed color (Badani et al. 2006; Fu et al. 2007; Snowdon et al. 2010; Wang et al. 2013). Seed phenolic compounds represent important antinutritive fiber components, the intermediate and final metabolites of the phenylpropanoid and flavonoid pathways and affect oil quality, meal value and seed color of B. napus (Liu et al. 2012; Qu et al. 2013; Snowdon et al. 2010). Low fiber content, particularly the content of acid detergent lignin (ADL), was significantly correlated with seed color (Liu et al. 2012, 2013; Snowdon et al. 2010) because lignin and pigment synthesis had common precursors (Badani et al. 2006). Liu et al. (2012) identified markers with tight linkage to a major QTL for seed ADL content in a recombinant inbred line (RIL) population from a cross between yellow-seeded GH06 and black-seeded P174, and a single, dominant and major locus for the reduction of ADL was detected. Liu et al. (2013) detected 11 QTLs for fiber traits and seed color, including a major QTL for seed color and ADL on A09, a minor QTL for ADL on C05, and five QTLs for cellulose variation and three QTLs for hemicellulose variation, in a late-generation RIL population. Snowdon et al. (2010) demonstrated that the broad-sense heritability for three detergent fiber components, especially ADL, was higher than for seed color, suggesting that seed fiber content is a more applicable selection marker in breeding than seed color. Some candidate genes in B. napus were found to be involved in seed fiber biosynthesis (Liu et al. 2012; Snowdon et al. 2010; Stein et al. 2013; Wang et al. 2015a) and the accumulation of seed pigments (Liu et al. 2013; Stein et al. 2013). Additionally, to improve the precision of QTL localization, bulk segregant analysis (BSA) combined with next-genomic sequencing technologies was used to rapidly locate QTLs and identify candidate genes (Chao et al. 2017; Zou et al. 2016). For example, Chao et al. (2017) performed precise QTL mapping of the seed oil and seed protein content of B. napus based on a high-density genetic linkage map and rapid detection of associated genomic regions using BSA. Although numerous QTLs and candidate genes for fiber traits had been discovered in B. napus, QTLs with a prominent effect on fiber content and candidate genes related to these QTLs have not been fully elucidated. Moreover, the mechanism of metabolism in seed fibers and the interaction of genes involved in fiber synthesis have not yet been clarified based on the co-localization of mapped candidate genes with QTLs in B. napus.
In our previous study, the KN doubled haploid (DH) population with a wide range of variations in fatty acid concentration was derived from a cross between black-seeded ‘KenC-8’ and yellow-seeded ‘N53-2’ (Wang et al. 2013). This population also showed high phenotypic variation for fiber traits. In this study, using the KN DH population, the QTLs for seed fiber traits (lignin content, LC; cellulose content, CC; hemicellulose content, HC) associated with the seed oil content (OC) in multiple environments were detected and further verified through BSA, and a regulatory network for fiber biosynthesis was also proposed.
Materials and methods
Plant materials
A total of 300 lines in the KN DH population derived from female ‘N53-2’ and male ‘KenC-8’ (Wang et al. 2013; Chao et al. 2017) were used to analyze the phenotype and to detect QTLs for fiber traits in this study. ‘KenC-8’ is the parental line of the variety ‘Zayou59’ released in 1996 in China, which was selected from a multiway hybridization combination: ‘1721-1B’ ‘start’ × ‘955’ ‘ChunShan2B’ (Wang et al. 2013). ‘KenC-8’ is a spring B. napus with low oil (approximately 40%), low erucic acid and high glucosinolate content. ‘N53-2’ is a DH line developed from the Canadian canola cultivar ‘Midas’ and a Chinese inbred line ‘SE8’ (Wang et al. 2013). ‘N53-2’ is a winter-type B. napus with high oil (> 50%), high erucic acid and low glucosinolate content.
Field experiments
The KN DH lines and their parents were planted in two independent environments in China, including a semi-winter-type rapeseed planting area, Wuhan in Hubei Province (coded WH, E113.68°, N30.58°), for two successive years (October–May of 2012–2013 and 2013–2014), and a winter-type rapeseed planting area, Dali in Shaanxi Province (coded DL, E109.93°, N34.80°), for four successive years (September–June of 2009–2010, 2010–2011, 2011–2012, 2012–2013 and 2013–2014). The location was treated as a macroenvironment, and the year-location combination was treated as a microenvironment. For example, WH indicated that the experiment was planted in a semi-winter ecological condition, and 12WH indicated that the experiment was performed in 2012 in Wuhan of Hubei province. The field experiments were performed in a completely randomized block design with three replications in DL and two replications in WH. Each line was grown in two rows per plot in both locations, every row contained approximately 12 plants, and the distance between rows and between plants was 0.4 and 0.2 m, respectively. Open pollinated seeds were collected from five randomly chosen plants in each line at maturity for analysis. The climatic conditions during the growing season for experiments have been reported by Wang et al. (2015b).
Measurements of fiber components and trait analysis
Seed fiber components were measured by near-infrared reflectance spectroscopy (NIRS) using an NIR System 6500 with WinISI II software (FOSS GmbH, Rellingen, Germany) according to previous reports (Liu et al. 2013; Wang et al. 2015a). Phenotype values for acid detergent lignin (ADL; % seed dry weight), acid detergent fiber (ADF; % seed dry weight) and neutral detergent fiber (NDF; % seed dry weight) were speculated from NIRS spectra using NIRS calibrations for these traits as described by Wang et al. (2015). In the external validation, the R-squared value (RSQ) for NDF, ADF and ADL was 0.847, 0.938 and 0.979, the standard error of prediction (SEP) was 0.624, 0.479 and 0.172, and the standard error of prediction corrected for bias (SEP(c)) was 0.687, 0.467 and 0.172, respectively. LC was represented by the ADL content. CC was determined by subtracting ADL from ADF, and HC was determined by subtracting ADF from NDF. The data for OC in the KN DH population were based on our previous report (Chao et al. 2017). All phenotype data were averaged over three technical replicates.
QTL analysis and meta-analysis for fiber traits and clarification of unique QTLs by BSA
A high-density genetic linkage map of the KN population has been constructed based on phenotyping using the Brassica 60 K SNP Array (developed by the international Brassica SNP consortium in cooperation with Illumina Inc. San Diego, CA, USA) together with simple sequence repeat (SSR) and sequence tagged site (STS) markers, the total length of which reached 3072.7 cM, with an average distance of only 0.96 cM between adjacent markers (Chao et al. 2017). QTL analysis for LC, CC and HC was performed by composite interval mapping (CIM) using the QTL Cartographer 2.5 software (Wang et al. 2012). The walking speed was set to 2 cM and a window size of 10 cM with five background cofactors. The LOD threshold was determined by the 1000-permutation test based on a 5% error rate, and an LOD of 3.4 was used to identify the existence of QTLs in each environment. These QTLs were termed ‘identified QTLs.’ The method for QTL nomenclature was performed according to Wang et al. (2013).
Then, the identified QTLs that were repeatedly detected in different trials and with overlapping confidence intervals (CIs) were integrated into a consensus QTL by meta-analysis using BioMercator 4.2 software with default parameters (Arcade et al. 2004). They were then named with the initial letters ‘cq,’ for instance, cqLC-A9-1. Consensus QTLs occurring in at least one microenvironment with phenotypic variation (PV) > 20%, or in at least two microenvironments with PV > 10%, were considered major QTLs (Shi et al. 2009). The consensus QTLs with overlapping CIs for different traits were then further integrated into one unique QTL and designated with the initial letters ‘uq’ (for example, uq-A5-1). If a consensus QTL had no overlapping CI with others, it was also regarded as a unique QTL.
Bulked segregant analysis (BSA) based on high-throughput sequencing for OC in the KN DH population has been reported by Chao et al. (2017). Two extreme pools for OC corresponded to the two opposite extreme pools of LC, CC and HC. Thus, BSA for OC was used to investigate the genetic correlation of fiber components and OC herein. Because reads were mapped to the reference genome ‘Darmor-bzh’ (http://www.genoscope.cns.fr/brassicanapus/data/) to calculate the SNP index in the BSA analysis, the closely linked SNP markers of unique QTLs were aligned to the reference genome ‘Darmor-bzh’ through BLASTn to identify whether the QTLs were consistent with the significantly associated genomic region (AGR) detected by BSA.
Alignment of QTLs for fibers and OC to the ‘ZS11’ reference genome
Since the ‘ZS11’ reference genome was more comprehensive than the ‘Darmor-bzh’ genome (Sun et al. 2017), OC-QTLs and fiber-related QTLs from previous reports (Snowdon et al. 2010; Stein et al. 2013; Liu et al. 2012, 2013; Delourme et al. 2006; Chen et al. 2013; Jiang et al. 2014; Wang et al. 2015a; Cheng et al. 2016) and this present research were aligned herein into the physical map of the ‘ZS11’ reference genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000686985.2/). The closely linked markers were obtained from the linkage map, and the primer sequences of those markers were acquired from related published papers. The genomic position of the closely linked markers was identified using e-PCR (Rotmistrovsky et al. 2004), and then, the QTLs were projected onto the physical map of the ‘ZS11’ reference genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000686985.2/) for comparison. This procedure was performed as described by Raboanatahiry et al. (2017). Finally, if one or more QTLs were identified on a chromosome in this study but not in previous reports, these were considered novel QTLs.
Identification of fiber-related candidate genes and potential regulatory networks
The genetic linkage map can be aligned to the ‘ZS11’ reference genome by BLAST, as described by Alschul et al. (1997) and Chao et al. (2017). Based on the collinearity relationship between the genetic linkage map and the ‘ZS11’ reference genome, genes within QTL regions, which had SNP or InDel variation in introns, exons or within 1 kb up- and down-stream between two parents according to the genome resequencing of the two parents from BSA (Chao et al. 2017), were regarded as potential candidate genes.
The orthologous of potential candidate genes in QTL regions and their annotations were obtained by BLASTn based on the A. thaliana database (http://www.arabidopsis.org/). The same method was used for the whole genome search of fiber-related orthologs in B. napus. Candidate genes were clustered and analyzed using Mapman software (Thimm et al. 2004) according to their orthologs and annotation. The interaction networks were analyzed and constructed using String software (http://string-db.org/) and visualized by Cytoscape_V3.6.0 (Shannon et al. 2003). The degree and betweenness centrality were calculated by network analyzer and mapped as the node size and node color, respectively. All interactions, clusters and analysis were based on A. thaliana orthologs.
qRT-PCR validation of fiber-related candidate genes
The siliques at 5, 15, 25 days after flowering and the seeds at 35, 45 days after flowering from the parent materials were, respectively, collected for total RNA extraction with TRIzol® reagent (Thermo Fisher Scientific). One microgram of total RNA was reverse-transcribed using SuperScript III Reverse Transcriptase (Invitrogen) and oligo (dT)18 according to the manufacturer’s protocol. qRT-PCR validation for 6 potential fiber-related candidate genes from different associate loci and a reference B. napus ACT 7 gene were performed with the SYBR premix EX TaqTM kit (TaKaRa, Japan) using an ABI 7500 Real-Time PCR System (ABI). For each reaction, three technical replicates were assessed. The primers of candidate genes and the ACT 7 gene are listed in Online Resource 1.
Results
Variation and correlation analysis of fiber components
Three fiber traits of the KN DH population in seven environments were measured (Online Resource 2). The LC (6.95%) and CC (6.17%) of ‘KenC-8’ were more than ‘N53-2’ (LC and CC was 3.05% and 4.76%, respectively), whereas no significant difference in HC was observed. Moreover, three fiber traits and OC showed a wide range of phenotypic variation and strong transgressive segregations in the KN DH population, and their mean values were close to the mid-parent values (Fig. 1). LC showed a partial normal distribution in the population, and CC and HC exhibited a normal distribution. The distribution patterns of LC, CC and HC indicated that they were genetically stable but also affected by the environment. The correlation among the three fiber components and OC was calculated (Table 1), revealing that LC was significantly positively correlated with CC (coefficient of 0.621) and CC was significantly positively correlated with HC (0.419), but LC was moderately negatively correlated with HC (− 0.156). Moreover, LC, CC and HC all showed a negative correlation with OC (− 0.512 to − 0.156), suggesting that seed OC might be increased by reducing the three fiber components in B. napus.

Phenotypic variation of seed fiber components and oil content in the KN DH population. The units of the x-axis are the phenotypic values, and the units of the y-axis are the number of lines
QTL mapping for LC, CC and HC
A total of 85 identified QTLs for LC, CC and HC were obtained in 17 linkage groups (except for A01 and C07) (Fig. 2), and a single QTL could explain the phenotypic variation (PV) from 2.57 to 54.76% (Online Resource 3). All 85 identified QTLs were then integrated into 60 consensus QTLs by meta-analysis (Table 2; Online Resource 3), and 52 environment-specific QTLs were further detected. Some environment-stable QTLs were identified, of which, 6, 3, 1 and 2 consensus QTLs were identified in two, three, four and six microenvironments, respectively.

Genetic linkage map and locations of QTLs for fiber traits and oil content in the KN map. The QTLs distributed in 19 linkage groups in different natural environments are indicated by different backgrounds on the cycle (backgrounds of light green, the winter macroenvironment; backgrounds of pink, the semi-winter macroenvironment). Different environments and years are indicated by ‘years’ + ‘location.’ Blue bar, LC (lignin content); green bar, CC (cellulose content); red bar, HC (hemicellulose content); purple, OC (oil content). The gray traits indicate that QTLs detected in different traits are co-localized
For LC, 29 identified QTLs were detected on eight chromosomes, had an additive effect of − 1.18 to 0.27 and explained 2.57–54.77% of PV (Table 2; Online Resource 3). The 29 identified QTLs were integrated into 16 consensus QTLs, and 3 and 2 consensus QTLs were detected in two and six microenvironments, respectively. Three major QTLs (cqLC-A9-1, cqLC-A9-3 and cqLC-C5-1) were repeatedly detected in at least two trials and displayed a large effect (PV > 10%). cqLC-A9-1 and cqLC-C5-1 were stably detected in six microenvironments, and cqLC-A9-1 explained the highest PV (48.50%). Additionally, 6 and 5 consensus QTLs were detected as semi-winter and winter environment-specific QTLs, respectively, among which cqLC-A9-2 with a PV of 40.97% was a major and winter environment-specific QTL.
For CC, 35 identified QTLs with an additive effect ranging from − 0.41 to 0.22 were detected across nine chromosomes and explained 4.24–23.85% of PV (Table 2; Online Resource 3). The 35 identified QTLs were integrated into 23 consensus QTLs, and 3, 3 and 1 consensus QTLs were detected in two, three and four microenvironments, respectively. Three QTLs (cqCC-A9-1, cqCC-A9-2, cqCC-A9-5) were repeatedly detected in at least three trials and showed a large effect (PV > 10%), and cqCC-A9-2 was stably detected in four microenvironments and had the highest PV of 23.85%. A total of 7 and 13 consensus QTLs were detected as semi-winter and winter environment-specific QTLs, respectively.
For HC, 21 identified QTLs with an additive effect ranging from − 0.41 to 0.47 were detected on seven chromosomes and explained 5.68–13.10% of PV, which were all environment-specific QTLs, including 12 semi-winter environment-specific QTLs and 9 winter environment-specific QTLs (Table 2; Online Resource 3).
Co-localization of fiber components and OC
The genetic relationship between three fiber components and OC of B. napus could be confirmed by the mapping of colocated loci. A trait-by-trait meta-analysis revealed that 94 consensus QTLs for the four traits were integrated into 77 unique QTLs (Online Resource 4). Among them, 13 unique co-localized QTLs had pleiotropic effects and controlled at least two traits (Table 3). These co-localized QTLs were generally classified into two kinds: unique QTLs integrated from QTLs with the same additive effect direction, and unique QTLs with opposite additive effect directions. For instance, uq.C5-9 and uq.C5-10 obtained from QTLs controlling LC and CC showed the same additive effect, in accordance with the significant positive correlations between LC and CC. However, uq.A9-11 showed an opposite additive effect, and the additive effect of cqCC-A9-4 (− 0.19) was negative, while cqOC-A9-4 (0.73) was positive, which might explain the negative phenotype correlation between CC and OC. Interestingly, uq.A9-9 integrated from QTLs controlling LC, CC, HC and OC exhibited not only the same but also opposite additive effect directions (Fig. 3a; Table 3; Online Resource 4). The additive effect of cqLC-A9-1 (− 1.16) and cqCC-A9-2 (-0.32), cqOC-A9-3 (0.89) and cqHC-A9-6 (0.36) was the same, while the additive effects of cqLC-A9-1 and cqOC-A9-3, cqCC-A9-2 and cqOC-A9-3, cqLC-A9-1 and cqHC-A9-6, cqCC-A9-2 and cqHC-A9-6 were all opposite, suggesting a complexity of the competitive mechanism of these four traits.

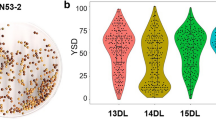
Co-localization of fiber components and oil content on A09. a. Unique QTLs with the same and opposite additive effects on A09. The identified QTLs for fiber traits and OC in different experiments are shown by curves above the line of the linkage group. The short-term localization above the horizontal axis represents QTLs detected at the corresponding position. Additive effects are shown by curves of the same color below the line of the linkage group. b. The unique QTLs uq.A9-9 and uq.A9-12 corresponding to AGRs detected by BSA for OC. The Δ (SNP index) plot with statistically significantly associated regions (two green significant threshold lines, P < 0.01 and P < 0.05) is drawn at the bottom. In the two coordinate axis, the X-axis represents the position of linkage group A9 and chromosome A09, and the Y-axis represents the LOD value and Δ (SNP index), respectively. uq.A9-9 and uq.A9-12 are linked by black lines with their corresponding AGRs. The linked SNPs and their positions in the reference genome ‘Darmor-bzh’ drawn in the middle. c. OC, LC, CC and HC of two extreme pools for OC-BSA analysis. H and L on the abscissa represent high oil and low oil pools, respectively. Boxes represent the variation in the phenotype average of the 24 individuals used to construct the two extreme pools (24 individuals each pool) in seven environments
Two extreme pools for OC from the KN DH population also corresponded to the two opposite extremes of LC, CC and HC in the bulked segregant construction for BSA analysis (Fig. 3c). In this study, of eight unique QTLs involved in OC, seven were detected in the BSA analysis (Online Resource 5). An uq.A9-9 simultaneously controlling OC, LC, CC and HC was linked to Bn-A09-p32954471, which was aligned in the significantly associated region AGR_A9-8 (Fig. 3b), while uq.A9-11 (controlling OC and CC) and uq.A9-12 (controlling OC, LC and CC) were detected in positions inside AGR_A9-12 based on the linked SNP markers Bn-A09-p35389045 and Bn-A09-p35661872 (Fig. 3b). It is noteworthy that all seven unique QTLs that were commonly detected by QTL mapping and BSA affected OC and CC with opposite additive effects, indicating a comprehensive genetic basis for the synthesis and metabolism of OC and CC. In addition, uq.A9-9 and uq.A9-12 were integrated from two major QTLs of LC with the highest PV (cqLC-A9-1, 48.50% of PV and cqLC-A9-3, 31.68%), which demonstrated that the extreme phenotype pool for OC corresponded to the opposite trend for LC.
To analyze whether the fiber-related QTLs in the present research were novel, these QTLs and other reported QTLs from six populations were projected on the physical map of the ‘ZS11’ reference genome to identify overlapping regions (Online Resource 6). Six of eight major fiber-related QTLs were overlapped with previously reported QTLs, including cqLC-A9-1, cqLC-A9-2, cqCC-A9-1, cqCC-A9-2, cqLC-C5-1 and cqCC-C5-2. Among them, cqLC-C5-1 and cqCC-C5-2 were co-localized with qADLC05 reported by Liu et al. (2013) and the significant SNPs rs37338, rs37369 and rs37408 reported by Wang et al. (2015a, b). Through integration analysis with previously reported OC-QTLs, a QTL hotspot related to OC and fiber content was found in the region ranging from 37 to 45 Mb in A09 (Fig. 4). The uq.A9-9, integrated from cqLC-A9-1, cqCC-A9-2, cqHC-A9-6 and cqOC-A9-3, was identical to the significant SNP rs21223 associated with seed ADL content (Wang et al. 2015a). However, uq.A9-12, integrated from the three major QTLs cqLC-A9-3, cqCC-A9-5 and cqOC-A9-5, showed no overlap with other reports and might be a potential novel QTL.

Locations of QTLs for OC and fiber that have been previously reported and detected in this study on the ‘ZS11’ reference genome. The A09 chromosome, markers linked to fiber-related QTL and their genome location in the reference genome are drawn on the left. Different colors of QTLs correspond to OC and fiber. The color code for the QTLs is shown on the top right of the figure. The QTL name is suffixed according to the population in which it is detected: YB (‘Express 617’ × ‘1012-98,’ Snowdon et al. 2010), GP (‘GH06’ × ‘P174,’ Liu et al. 2012 and 2013), YE2 and ExpV8 (‘Express 617’ × ‘1012-98’ and ‘Express 617’ × ‘V8,’ Stein et al. 2013), RNSL (‘Rapid’ × ‘NSL96/25,’ Delourme et al. 2006), SG (‘Sollux’ × ‘Gaoyou,’ Chen et al. 2013), TN (‘Tapidor’ × ‘Ningyou7,’ Jiang et al. 2014), M201 × M202 (Cheng et al. 2016), and GWAS represent significant SNP signals from the GWA mapping using B. napus germplasm resources (Wang et al. 2015a). The two dashed lines represent the positions of two unique QTLs, uq.A9-9 and uq.A9-12
Identification of fiber-related candidate genes and potential regulatory networks
Based on the collinearity relationship between the genetic linkage map and the B. napus ‘ZS11’ reference genome, 80 potential candidate genes in B. napus were identified to correspond to 74 homologous A. thaliana genes involved in the fiber biosynthesis (Online Resource 7). Some noteworthy potential fiber-related candidates were identified in overlapping regions of LC-, CC-, HC- and OC-QTLs. For instance, BnA09g0383130 corresponding to AtMYB52, a transcription factor that regulates the biosynthesis of lignin, xylem, and cellulose (Dubos et al. 2010), was located in the overlapping CI region of three major QTLs (cqLC-A9-1, cqCC-A9-2, cqOC-A9-3). BnA09g0382840 corresponding to AtMYB51, a transcription factor involved in indole glucosinolate biosynthesis (Gigolashvili et al. 2007), was also located in the overlapping region of cqLC-A9-1, cqCC-A9-2 and cqOC-A9-3. BnC05g0730820, which is homologous to DHQD (3-dehydroquinate dehydratase), an enzyme involved in phenylalanine biosynthesis, was mapped to the overlapping region of two major QTLs cqLC-C5-1 and cqCC-C5-2.
To investigate the genetic basis of fiber accumulation in B. napus seeds, a potential metabolic pathway was constructed with phosphoenolpyruvate and β-d-fructose-6P as the junction based on potential candidates underlying LC-, CC- and HC-QTLs (Fig. 5; Online Resource 7a). The pathway was involved in glycolysis/gluconeogenesis, starch and source metabolism, phenylpropanoid biosynthesis, and lignin synthesis. GAPC1 (glyceraldehyde-3-phosphate dehydrogenase) was one key enzyme in the glycolytic pathway (Rius et al. 2008) and involved in membrane lipid-based signaling, energy metabolism and growth control in the plant response to ROS (Guo et al. 2012). BnC08g0881270 corresponding to GAPC1 was identified in the region of cqHC-C8-2. COMT1 (caffeic acid-O-methyltransferase) was a key enzyme involved in the synthesis of the flavonolignin unit derived from tricin (Fornalé et al. 2016), and BnA03g0105070 corresponding to COMT1 located in the region of cqLC-A3-3. AtCSLC08 (cellulose synthase-like C8) was a cellulose synthase-like (CSL) protein that functions in the biosynthesis of noncellulosic polysaccharides such as xylans, xyloglucans, galactans and mannans (Richmond and Somerville 2000), and BnA09g0379870, which is homologous to AtCSLC08, was mapped to the overlapping region of cqCC-A9-1 and cqHC-A9-4 and might be an important candidate gene for cellulose and hemicellulose biosynthesis.

Potential regulatory pathways controlling seed fiber synthesis in B. napus. The pathway is constructed based on potential candidate genes within QTL regions of LC, CC and HC. The red, green and blue characters indicate A. thaliana orthologs of underlying LC-, CC- and HC-QTL regions, respectively. The broken arrows indicate multiple steps between the two compounds
To further dissect the accumulation mechanism of lignin, cellulose, hemicellulose and oil in seeds, an interaction network of the potential candidate genes in QTL regions of LC, CC, HC and their co-localized OC-QTLs was constructed based on A. thaliana orthologs (Fig. 6; Online Resource 7b). The whole network, including 32 enzymes and 16 transcription factors, was incorporated into 48 nodes and 95 edges. A number of important genes and regulatory factors in the accumulation of lignin, cellulose, hemicellulose and oil in seeds were observed, such as GAPC1, DHQD, PFK, HCT, MYB51 and WRKY33 (Fig. 6). GAPC1 might directly interact with PFK7 and then SPS1F and SUS1 functioning in the ‘starch and sucrose metabolism’ pathway to flow into cellulose/hemicellulose biosynthesis. GAPC1 might also interact with DHQD or PFK7 to DHQD to function in the ‘phenylalanine biosynthesis’ pathway involving a methyl donor. AT5G55070 in the methyl donor might be linked to ACX2, LACS6, FATB and GPAT5 to flow into fatty acid synthesis and oil body formation. MYB51, which influenced the connection of the network, interacted with two transcription factors MYB97 and WRKY33 to regulate phenylpropanoid biosynthesis, and MYB51 also interacted with CESA3 to participate in cellulose biosynthesis. Moreover, four transcription factors, MYB83, MYB85, MYB58 and MYB46, which function as regulators of phenylpropanoid metabolism in plants (Liu et al. 2015), were also associated with HCT and CCR2 to participate in the regulation of lignin biosynthesis.

Interaction network of potential candidate genes for fiber traits and oil content. Network visualization of the interaction of the 52 candidate genes and regulatory factors constructed using String software and observed using Cytoscape_V3.6.0 software. Genes are presented as nodes, and gene interactions are presented as edges
Furthermore, 6 candidate genes were selected for qRT-PCR validation (Fig. 7). Characterization of the expression of these candidate genes was basically in agreement with LC and CC of the two parents, e.g., the expression levels of BnC08g0881270 and BnC01g0466620 in black-seeded ‘KenC-8’ with high LC and CC were upregulated in comparison with yellow-seeded ‘N53-2’ with low LC and CC.

qRT-PCR analysis of fiber synthesis-related candidate genes in the two parents. The x-axis indicates five different developmental stages of seed, and the y-axis indicates the mRNA expression level of each candidate gene. Green and purple represent ‘N53-2’ and ‘KenC-8,’ respectively. The 5 DAF, 15 DAF, 25 DAF, 35 DAF and 45 DAF indicate that the materials were collected at 5, 15, 25, 35 and 45 days after flowering, respectively. The expression of the gene for ‘N53-2’ at 5 days after flowering was used as the reference, and the expression of each gene was calculated based on three biological replicates. The bar represents the standard deviation calculated by the ΔΔCt method
Discussion
Seed fiber components cause a considerable reduction of the seed oil content and meal value of B. napus, and thus a reduction of seed fiber compounds, including lignin, cellulose and hemicellulose, which can improve the oil content and meal value (Liang et al. 2002; Liu et al. 2013). Thus, they are new targets for B. napus with high oil content and meal quality (Wang et al. 2015a). The dissection of QTLs with significant effects on seed fiber components enabled the identification of positional and functional candidate genes for these traits in B. napus.
In our KN DH population derived from ‘N53-2’ and ‘KenC-8,’ LC, CC and HC showed high phenotypic variation, and CC and HC were negatively correlated with OC, which was identical to the previous report that seed CC and HC have negative correlations with OC in the yellow-seeded GH06 × black-seeded P174 RIL population (Liu et al. 2013). It is noteworthy that LC in the present study showed a high negative correlation with the level of OC. In addition, the co-localized QTLs identified for CC and OC showed greater consistency with the significantly associated loci from BSA based on OC, but with negative effects. These results suggested that these traits shared some underlying genetic bases, providing a clue for increasing OC by reducing two fiber components.
Most of the major QTLs with a large effect on seed fiber content and seed color have been mapped to A09 of B. napus in different genetic backgrounds (Badani et al. 2006; Fu et al. 2007; Liu et al. 2012, 2013; Stein et al. 2013; Snowdon et al. 2010). Snowdon et al. (2010) mapped a major QTL influencing seed color, fiber content and phenolic compounds on A09 in biparental populations from two different yellow-seeded × black-seeded B. napus crosses. Liu et al. (2012) detected a major QTL for seed ADL on A09 in a yellow-seeded GH06 × black-seeded P174 RIL population. Stein et al. (2013) dissected a major QTL influencing seed ADL, ADF, NDF and seed color on A09 in a black-seeded ‘Express 617’ × yellow-seeded ‘1012–98’ DH population. In addition to A09, QTLs for seed fiber traits have also been reported on other chromosomes. Wang et al. (2015) detected three significant association signals for seed ADL not only on A09 but also on A05 and C05. Behnke et al. (2018) identified a major QTL on C05 that resulted in a significant reduction of ADL content and increase in seed oil and protein content in B. napus.
In the present results from the KN DH population, single QTL mapping of each fiber component showed that QTLs on A09 were highly correlated to seed fiber traits. QTLs uqA9-9 and uqA9-10 could explain the PV of individual fiber content ranging from 7.27% to 48.50%, integrated from two major QTLs cqLC-A9-1 and cqLC-A9-2, respectively (Online Resource 4), consistent with previous studies (Online Resource 6) (Liu et al. 2012, 2013; Snowdon et al. 2010; Stein et al. 2013; Wang et al. 2015a). The uqA9-9 could control as many as four traits, including the QTLs for fiber components (cqLC-A9-1, cqCC-A9-2 and cqHC-A9-6) and the QTL (cqOC-A9-3) for oil content (Chao et al. 2017). In addition, uq.A9-9 was found to overlap with the significant SNP rs21223 associated with seed ADL content (Wang et al. 2015a). Thus, as a stable QTL, uq.A9-9 is valuable for further molecular-assisted breeding. The unique QTL uq.A9-12, integrated from two major QTLs cqLC-A9-3 and cqCC-A9-5 (Online Resource 4), could explain the PV of individual fiber content ranging from 12.59% to 31.68% and was a novel QTL and not reported in previous studies (Fig. 4). In the present study, uq.A9-12 was identified by both linkage-based QTL mapping and the BSA method for simultaneously controlling LC, CC and OC but with opposite additive effect (Fig. 3b and Online Resource 5), which indicated that it increased OC while reducing fiber content. As potential novel and major locus for OC and fiber content simultaneously, uq.A9-12 was more valuable for MAS and further map-based cloning. Additionally, uq.C5-9, integrated from cqLC-C5-1 and cqCC-C5-2, was identical to qADLC05 reported by Liu et al. (2013), the significant SNPs rs37338, rs37369 and rs37408 reported by Wang et al. (2015), and one major QTL for ADLm reported by Behnke et al. (2018); however, uq.C5-9 had no influence on seed oil content.
A number of potential genes involved in seed fiber biosynthesis in B. napus have been reported. Wang et al. (2015) identified 123 seed filtered candidate genes, 68% of which were associated with seed coat development and fiber biosynthesis and 17% to the phenylpropanoid pathway of flavonoid biosynthetic processes and anthocyanin accumulation. Some candidate genes for major QTLs with effects on seed fiber content and color have been located on A09 in B. napus (Liu et al. 2012, 2013; Snowdon et al. 2010; Stein et al. 2013). Two candidate genes encoding cinnamoyl-CoA reductase 1 (CCR1) and cinnamyl alcohol dehydrogenase (CAD2/CAD3) have been detected on A09 for a major QTL for low seed ADL in B. napus, and the Bna.CCR1 homolog might interact with neighboring homologues of a CAD family member to regulate seed phenylpropanoid biosynthesis (Liu et al. 2012; Stein et al. 2013; Wang et al. 2015a). Additionally, Wang et al. (2015) identified four key genes (BnaA.SEC8, BnaA.PAL4, BnaA.CESA3, BnaA.GPAT5) that are associated with the phenylpropanoid-lignin pathways and seed coat development in the R-QTL containing rs11682 and rs11683 on A05. Snowdon et al. (2010) found two potential candidate genes corresponding to At3g17390 encoding an S-adenosyl-l-methionine synthetase 3 (SAMS3) and At3g17310 encoding a similar domain-rearranged methyltransferase (DRM) involved in lignin biosynthesis.
In the present study, a total of 74 A. thaliana orthologs of candidate genes underlying fiber components QTL were used to construct a potential regulatory network controlling seed fiber synthesis, and a number of important genes and regulatory factors were involved, such as CCR2, HCT, MYB51 and WRKY33 (Fig. 5, 6). Among these candidate genes, MYB51 (BnA09g0382840) located in the overlapping regions of cqLC-A9-1, cqCC-A9-2 and cqOC-A9-3 was focused on. MYB51 strongly regulated CESA3 in the interaction network of the candidate genes, and CESA3 with CESA1 and CESA6 are considered the core components of the primary wall cellulose synthesis complex (Desprez et al. 2007; Persson et al. 2007). MYB51 strongly interacted with WRKY33 to regulate MYB77 and then with F3H to participate in flavonoid biosynthesis and lignin biosynthesis. These findings suggested that MYB51 might play a potential role in seed cellulose, lignin and flavonoid biosynthesis in B. napus.
Additionally, 25 and 27 fiber-related QTLs were identified as semi-winter and winter environment-specific QTLs, respectively, with cqLC-A9-2 as a representative major and winter environment-specific QTL. Some fiber-related genes were detected within environment-specific QTL regions. For example, the gene BnA09g0389350 corresponding to MYB61, an activator regulating monolignol biosynthesis (Newman et al. 2004), was located in the winter environment-specific QTL cqLC-A9-2. The gene BnC05g0727720 corresponding to CESA3, one of the core components of the cellulose synthase complex, was located in the semi-winter environment-specific QTL cqCC-C5-2. These environment-specific QTLs and fiber-related genes within these specific QTLs will be beneficial for breeding new varieties of rapeseed with broad adaptability.
References
Alschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, Joets J (2004) BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics 20:2324–2326
Badani AG, Snowdon RJ, Wittkop B, Lipsa FD, Baetzel R, Horn R, De Haro A, Font R, Lühs W, Friedt W (2006) Colocalization of a partially dominant gene for yellow seed colour with a major QTL influencing acid detergent fibre (ADF) content in different crosses of oilseed rape (Brassica napus). Genome 49:1499–1509
Behnke N, Suprianto E, Möllers C (2018) A major QTL on chromosome C05 significantly reduces acid detergent lignin (ADL) content and increases seed oil and protein content in oilseed rape (Brassica napus L.). Theor Appl Genet 131:2477–2492
Chao H, Wang H, Wang X, Guo L, Gu J, Zhao W, Li B et al (2017) Genetic dissection of seed oil and protein content and identification of networks associated with oil content in Brassica napus. Sci Rep 7:46295
Chen Y, Qi L, Zhang X, Huang J, Wang J, Chen H, Ni X et al (2013) Characterization of the quantitative trait locus OilA1 for oil content in Brassica napus. Theor Appl Genet 26:2499–2509
Cheng X, Xia MS, Zeng MX, Gu MJ, Yang MY, Xu J, Liu L et al (2016) Identification of quantitative trait loci associated with oil content and development of near isogenic lines for stable qOC-A10 in Brasscia napus L. Can J Plant Sci 96:423–432
Delourme R, Falentin C, Huteau V, Clouet V, Horvais R, Gandon B, Specel S et al (2006) Genetic control of oil content in oilseed rape (Brassica napus L.). Theor Appl Genet 113:1331–1345
Desprez T, Juraniec M, Crowell EF, Jouy H, Pochylova Z, Parcy F, Höfte H, Gonneau M, Vernhettes S (2007) Organization of cellulose synthase complexes involved in primary cell wall synthesis in Arabidopsis thaliana. Proc Natl Acad Sci USA 104:15572–15577
Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L (2010) MYB transcription factors in Arabidopsis. Trends Plant Sci 15:573–581
Fornalé S, Rencoret J, García-Calvo L, Encina A, Rigau J, Gutiérrez A, Del Río JC, Caparros-Ruiz D (2016) Changes in cell wall polymers and degradability in Maize mutants lacking 3′- and 5′-O-methyltransferases involved in lignin biosynthesis. Plant Cell Physiol 58:240–255
Fu FY, Liu LZ, Chai YR, Chen L, Yang T, Jin MY, Ma AF, Yan XY, Zhang ZS, Li JN (2007) Localization of QTLs for seed color using recombinant inbred lines of Brassica napus in different environments. Genome 50:840–854
Gigolashvili T, Berger B, Mock HP, Müller C, Weisshaar B, Flügge UI (2007) The transcription factor HIG1/MYB51 regulates indolic glucosinolate biosynthesis in Arabidopsis thaliana. Plant J 50:886–901
Guo L, Devaiah SP, Narasimhan R, Pan X, Zhang Y, Zhang W, Wang X (2012) Cytosolic glyceraldehyde-3-phosphate dehydrogenases interact with phospholipase Dδ to transduce hydrogen peroxide signals in the Arabidopsis response to stress. Plant Cell 24:2200–2212
Jiang C, Shi J, Li R, Long Y, Wang H, Li D, Zhao J et al (2014) Quantitative trait loci that control the oil content variation of rapeseed (Brassica napus L.). Theor Appl Genet 127:957–968
Liang Y, Liang Y, Li J, Chen L (2002) Difference between yellow and black seed coat in B. napus. Chin J Oil Crop Sci 24:13–18
Liu L, Stein A, Wittkop B, Sarvari P, Li J, Yan X, Dreyer F, Frauen M, Friedt W, Snowdon RJ (2012) A knockout mutation in the lignin biosynthesis gene CCR1 explains a major QTL for acid detergent lignin content in Brassica napus seeds. Theor Appl Genet 124:1573–1586
Liu L, Qu C, Wittkop B, Yi B, Xiao Y, He Y, Snowdon RJ, Li J (2013) A high-density SNP map for accurate mapping of seed fibre QTL in Brassica napus L. PLoS ONE 8:e83052
Liu J, Osbourn A, Ma P (2015) MYB transcription factors as regulators of phenylpropanoid metabolism in plants. Mol Plant 8:689–708
Newman LJ, Perazza DE, Juda L, Campbell MM (2004) Involvement of the R2R3-MYB, AtMYB61, in the ectopic lignification and dark-photomorphogenic components of the det3 mutant phenotype. Plant J 37:239–250
Persson S, Paredez A, Carroll A, Palsdottir H, Doblin M, Poindexter P, Khitrov N, Auer M, Somerville CR (2007) Genetic evidence for three unique components in primary cell-wall cellulose synthase complexes in Arabidopsis. Proc Natl Acad Sci USA 104:15566–15571
Qu C, Fu F, Lu K, Zhang K, Wang R, Xu X, Wang M, Lu J, Wan H, Zhanglin T, Li J (2013) Differential accumulation of phenolic compounds and expression of related genes in black- and yellow-seeded Brassica napus. J Exp Bot 64:2885–2898
Raboanatahiry N, Chao H, Guo L, Gan J, Xiang J, Yan M, Zhang L et al (2017) Synteny analysis of genes and distribution of loci controlling oil content and fatty acid profile based on QTL alignment map in Brassica napus. BMC Genom 18:776
Richmond TA, Somerville C (2000) The cellulose synthase superfamily. Plant Physiol 124:495–498
Rius SP, Casati P, Iglesias AA, Gomez-Casati DF (2008) Characterization of Arabidopsis lines deficient in GAPC-1, a cytosolic NAD-dependent glyceraldehyde-3-phosphate dehydrogenase. Plant Physiol 148:1655–1667
Rotmistrovsky K, Jang W, Schuler GD (2004) A web server for performing electronic PCR. Nucleic Acids Res 32:108–112
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Shi J, Li R, Qiu D, Jiang C, Long Y, Morgan C, Bancroft I, Zhao J, Meng J (2009) Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 182:851–861
Simbaya J, Slominski BA, Rakow G, Campbell LD, Downey RK, Bell JM (1995) Quality characteristics of yellow-seeded Brassica seed meals-protein, carbohydrates, and dietary fiber components. J Agric Food Chem 43:2062–2066
Snowdon RJ, Wittkop B, Rezaidad A, Hasan M, Lipsa F, Stein A, Friedt W (2010) Regional association analysis delineates a sequenced chromosome region influencing antinutritive seed meal compounds in oilseed rape. Genome 53:917–928
Stein A, Wittkop B, Liu L, Obermeier C, Friedt W, Snowdon RJ (2013) Dissection of a major QTL for seed colour and fibre content in Brassica napus reveals colocalization with candidate genes for phenylpropanoid biosynthesis and flavonoid deposition. Plant Breed 132:382–389
Sun F, Fan G, Hu Q, Zhou Y, Guan M, Tong C et al (2017) The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J 92:452–468
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M (2004) Mapman: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37:914–939
Wang S, Basten CJ, Zeng ZB (2012) Windows QTL cartographer 2.5. Department of statistics, North Carolina State University, Raleigh
Wang X, Wang H, Long Y, Li D, Yin Y, Tian J, Chen L, Liu L, Zhao W, Zhao Y, Yu L, Li M (2013) Identification of QTLs associated with oil content in a high-oil Brassica napus cultivar and construction of a high-density consensus map for QTLs comparison in B. napus. PLoS ONE 8:e80569
Wang J, Jian H, Wei L, Qu C, Xu X, Lu K, Qian W, Li J, Li M, Liu L (2015a) Genome-wide analysis of seed acid detergent lignin (ADL) and hull content in Rapeseed (Brassica napus L.). PLoS ONE 10:e0145045
Wang X, Wang H, Long Y, Liu L, Zhao Y, Tian J, Zhao W, Li B, Chen L, Chao H, Li M (2015b) Dynamic and comparative QTL analysis for plant height in different developmental stages of Brassica napus L. Theor Appl Genet 128:1175–1192
Wittkop B, Snowdon RJ, Friedt W (2009) Status and perspectives of breeding for enhanced yield and quality of oilseed crops for Europe. Euphytica 170:131–140
Zou C, Wang PX, Xu YB (2016) Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol J 14:1941–1955
Acknowledgements
This work was supported by the National Basic Research Program of China (2015CB150205), National Natural Science Foundation of China (31671721) and New Century Talents Support Program of the Ministry of Education of China (NCET110172).
Author information
Authors and Affiliations
Contributions
LM carried out the QTL analysis and wrote the manuscript. HC and HL analyzed the data. LC, WZ and BL participated in the field experiment. HW, LZ and BW provided helpful suggestions for the manuscript. ML designed, led and coordinated the overall study.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical standards
The authors declare that the experiments comply with the current laws of the country in which they were performed.
Additional information
Communicated by Heiko C. Becker.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Miao, L., Chao, H., Chen, L. et al. Stable and novel QTL identification and new insights into the genetic networks affecting seed fiber traits in Brassica napus. Theor Appl Genet 132, 1761–1775 (2019). https://doi.org/10.1007/s00122-019-03313-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03313-4