
Abstract
Key message
A reduction in acid detergent lignin content in oilseed rape resulted in an increase in seed oil and protein content.
Abstract
Worldwide increasing demand for vegetable oil and protein requires continuous breeding efforts to enhance the yield of oil and protein crop species. The oil-extracted meal of oilseed rape is currently mainly used for feeding livestock, but efforts are undertaken to use the oilseed rape protein in food production. One limiting factor is the high lignin content of black-seeded oilseed rape that negatively affects digestibility and sensory quality of food products compared to soybean. Breeding attempts to develop yellow seeded oilseed rape with reduced lignin content have not yet resulted in competitive cultivars. The objective of this work was to investigate the inheritance of seed quality in a DH population derived from the cross of the high oil lines SGDH14 and cv. Express. The DH population of 139 lines was tested in field experiments in 14 environments in north-west Europe. Seeds harvested from open pollinated plants were used for extensive seed quality analysis. A molecular marker map based on the Illumina Infinium 60 K Brassica SNP chip was used to map QTL. Amongst others, one major QTL for acid detergent lignin content, explaining 81% of the phenotypic variance, was identified on chromosome C05. Lines with reduced lignin content nevertheless did not show a yellowish appearance, but showed a reduced seed hull content. The position of the QTL co-located with QTL for oil and protein content of the defatted meal with opposite additive effects, suggesting that the reduction in lignin content resulted in an increase in oil and protein content.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Major breeding aims in oilseed rape (Brassica napus L.) are to continuously increase the seed oil content and to enhance protein content of the defatted meal. The meal is used as a by-product after oil extraction in feed mixtures and has the potential to be used as a source of vegetable protein in food production (Fleddermann et al. 2013; Wanasundara et al. 2016). However, compared to soybean, the protein content of the oilseed rape meal is lower and its quality is hampered by higher fibre contents (Nesi et al. 2008). Particularly, the lignin content of the seed hulls of black-seeded oilseed rape limits digestibility of the meal and may affect taste and appearance of food products. According to the system of Van Soest et al. (1991), fibre consists of NDF (neutral detergent fibre), ADF (acid detergent fibre) and ADL (acid detergent lignin). The NDF fraction consists predominantly of hemicellulose, cellulose and lignin, while the ADF fraction comprises cellulose and lignin. ADL represents the non-digestible lignin fraction. It has been tempted for a long time to develop yellow seeded cultivars with lower lignin content (Simbaya et al. 1995; Rahman and McVetty 2011), but those cultivars have not yet been shown to gain significant market shares. The reason for this remains unclear, because some publications claimed higher protein and/or oil content of yellow seeded genotypes compared to black ones (Badani et al. 2006; Yan et al. 2009; Rahman and McVetty 2011). Breeding progress in the past has been limited by the lack of large numbers of sequence informative molecular markers that allow cost efficient and high throughput analysis of genotypes. In this respect, the availability of SNP markers and their chip-based analysis represented a quantum jump forward (Clarke et al. 2016). Early work performed with RFLP markers gave insight into the complex genetic control of oil content (Ecke et al. 1995). Although only a limited number of RFLP markers were available, three QTL controlling seed oil content were detected. Two of these QTL were found to be closely associated with qualitative variation in erucic acid content. This work also clarified the positive effect of the erucic acid alleles on oil content. The increase in molecular weight through the cytoplasmic elongation of oleoyl-CoA over eicosenoyl-CoA to erucoyl-CoA explains the concomitant increase in oil content. In subsequent work increasing numbers of QTL for oil and protein content were reported for different populations (Zhao et al. 2005; Delourme et al. 2006; Qiu et al. 2006; Yan et al. 2009; Jiang et al. 2014; Teh and Möllers 2015). Conditional mapping allowed the detection of QTL for oil content independent from pleiotropic effects of other genes, e.g. those for erucic acid and seed protein content (Zhu 1995; Zhao et al. 2006). Using a refined version of their earlier map, Zhao et al. (2012) were able to map candidate genes for seed oil biosynthesis and compare their QTL positions with those of other published maps (Burns et al. 2003; Delourme et al. 2006; Qiu et al. 2006; Chen et al. 2010).
Another limitation for increasing seed oil content is the restricted genetic variation within the relatively young species B. napus compared to other crops (Sun et al. 2017). Since Chinese oilseed rape has been bred for high oil content independently from European oilseed rape, this may represent a useful genetic resource for further improvements. In previous work, the old German cultivar Sollux was crossed to the Chinese cultivar Gaoyou (Zhao et al. 2005), both having high oil contents. A doubled haploid (DH) population was developed from this cross and tested in field experiments. From this population, line SGDH14 was identified as having the highest oil content under north-western European growing conditions and a combination of all QTL alleles increasing oil content from both parental lines (Zhao et al. 2005). The objective of this work was to further investigate the inheritance of seed oil content and other seed quality traits in a new DH population derived from the cross of SGDH14 and the inbred line 617 of the German cultivar Express. Sequence informative SNP markers of the Illumina Infinium 60 K Brassica chip were used for genotyping the population to allow for identification of candidate genes for oil content using the B. napus genome sequence (Chalhoub et al. 2014; Sun et al. 2017).
Materials and methods
Plant material
The plant material consisted of 139 DH lines developed from F1 plants of the cross SGDH14 × Express617 by microspore culture (Georg-August-Universität Göttingen, Department of Crop Sciences, Division of Plant Breeding, Göttingen, Germany), further referred to as SGEDH population. SGDH14 is a DH line derived from the cross Sollux × Gaoyou (SG) analysed by Zhao et al. (2005). Sollux is an old German cultivar, whereas Gaoyou is an old Chinese cultivar. Both lines are characterized by high erucic acid and high glucosinolate content (‘++’-quality) as well as high seed oil content. SGDH14 was chosen as parental line since it showed highest oil content at the German locations tested, and characterization by molecular markers revealed SGDH14 to combine all favourable QTL alleles for oil content from Sollux and Gaoyou (Zhao et al. 2005). Marker genotype distribution of SGDH14 was generally balanced, showing 52.4% Sollux and 47.6% Gaoyou alleles in a set of 473 markers (Prof. Jianyi Zhao, Zhejiang Academy of Agricultural Sciences, Hangzhou, PR China, personal communication). Like its parental lines, SGDH14 has high erucic acid and high glucosinolate content. Express617 is an inbred line of the German winter oilseed rape cultivar Express. It was chosen as parental line due to its low erucic acid and low glucosinolate content (canola-/‘00’-quality) and high oil content. For comparison of the results the following line cultivars were also included in the experiments: NK Beauty, Krypton, Billy, Komando, Oase, Adriana, and Favorite.
Field experiments
The plant material was tested in the three consecutive growing seasons 2009/2010, 2010/2011 and 2011/2012 in four to five different environments (n = 14) located in north-west Germany and south Sweden. Field experiments were conducted as group-wise randomized block design in small plots without replications. The three different groups within the experiment represented genotypes with low (L; < 10%), medium (M; 10–35%) and high (H; > 35%) erucic acid content. For each genotype seed samples were harvested at maturity from the main raceme of ten open pollinated plants. Seeds were bulked, and seed quality traits were analysed by near-infrared reflectance spectroscopy (NIRS) and gas chromatography (GC).
Analysis of seed quality traits
Near-infrared reflectance spectroscopy
If not otherwise stated NIRS spectra from seed samples from 14 environments were analysed. To determine seed quality traits, about 3 g of bulked seeds was scanned between 400 and 2498 nm by a NIRS monochromator (FOSS NIR Systems model 6500, NIRSystems, Inc., Silver Springs, MD, USA). Absorbance values log at 2 nm intervals were recorded to create a NIR spectrum for each sample. With WinISI II software (version 1.50) oil, protein, glucosinolate and moisture contents were determined using the calibration raps2012.eqa provided by VDLUFA Qualitätssicherung NIRS GmbH (Teichstr. 35, 34130 Kassel, Germany, http://h1976726.stratoserver.net/cms/). Oil (Oil) and protein (Protein) contents were determined in per cent at 91% seed dry matter. Glucosinolate content (GSL) was measured in µmol/g at 91% seed dry matter. Protein content in the defatted meal (Prot.idM) was calculated by using NIRS predicted seed oil and seed protein content (both at 91% dry matter) as: % protein in the defatted meal (PidM) = [% protein/(100 − % oil)] × 100. Neutral detergent fibre (NDFm), acid detergent fibre (ADFm) and acid detergent lignin (ADLm) in the defatted meal were determined by using a calibration developed by Dimov et al. (2012). The calibration was extended by adding 60 selected samples from SGDH14 × Express617 and in validation the standard error of prediction corrected for the bias (SEP(C)) of the extended calibration was 2.92%, 1.58% and 1.21% for NDFm, ADFm and ADLm content of the defatted meal. The hemicellulose (HCm) content was determined by subtracting ADFm from NDFm, and cellulose (Cm) content was determined by subtracting ADLm from ADFm content. Fibre content was predicted by using NIRS spectra derived from six environments.
Gas chromatography (GC)
Fatty acid profile analysis was performed by gas chromatography according to the method as described by Rücker and Röbbelen (1996) with minor modifications. Fatty acid methyl ester in isooctane was injected into a gas chromatograph (Trace GC ultra, Thermo Electron corporation) with a 25 m × 0.25 mm I.D. FFAP column (Macherey and Nagel, 0.25 µm film thickness, 210 °C, split injection 1:70, carrier gas: 150 kPa H2, injection/detector: temperature 230 °C). Palmitic (16:0), palmitoleic (16:1), hexadecadienoic (16:2), stearic (18:0), oleic (18:1), linoleic (18:2), linolenic (18:3), arachidic (20:0), eicosenoic (20:1), behenic (22:0) and erucic (22:1) acid were determined by the GC chromatogram of each sample. Fatty acids were expressed as per cent of the sum of all fatty acids.
Thousand kernel weight and seed hull proportion
Thousand kernel weight (TKW) was obtained from weight conversion of 500 seed. Seeds were counted using a Contador seed counter (Pfeuffer GmbH, D-97318 Kitzingen, http://www.pfeuffer.com). Seed samples from six environments were used to determine the seed hull proportion (in %). Hundred seeds (approx. 500 mg) per sample were dried at 105 °C for 8 h and then imbibed in water for 15–20 h. Afterwards seed hulls were carefully separated from embryos by using a dissecting needle. Both fractions were dried at 105 °C overnight, and dry weights were determined (Dimov et al. 2012).
Correction of oil and protein content considering erucic acid content
SGEDH population segregated for erucic acid content and erucic acid and oil content showed a strong positive correlation. Thus, to be able to compare the oil content of genotypes with varying erucic acid contents, oil contents were corrected by eliminating the effect of erucic acid on oil content in three different ways. (1) For the correction of oil content by regression information (RC), the linear regression between GC erucic acid content and NIRS oil content from trait means over 14 locations was calculated. The slope of this regression was used to calculate the regression corrected oil content the following way: NIRS oil content − (slope of linear regression between GC erucic acid content and NIRS oil content × GC erucic acid content). (2) Correction by molecular weight (MW): De novo fatty acid biosynthesis until oleoyl-CoA takes place in the chloroplasts. However, oleoyl-CoA elongation to erucoyl-CoA via eicosenoyl-CoA and triacylglycerol (TAG) assembly takes place in the cytoplasm. If one molecule oleoyl-CoA is elongated to eicosenoyl-CoA and subsequently to erucoyl-CoA, this leads to an increase of molecular weight of 10 and 19%, respectively. Therefore, if the number of fatty acid molecules produced by the de novo fatty acid biosynthesis remains the same, an increase in erucic acid content in the seed oil should result in a proportional increase in seed oil content. Following this theoretical assumption the molecular weight of eicosenoic acid and erucic acid can be reduced on the basis of oleic acid by 9 and 16.6% to eliminate their effects on oil content. Hence, the molecular correction of oil content was calculated from trait means over 14 locations as: NIRS oil content − [0.09 × (GC eicosenoic acid content × NIRS oil content/100) + 0.166 × (GC erucic acid content × NIRS oil content/100)]. (3) Correction by conditioning (CN): The mixed model approach for the conditional analysis of quantitative traits described by Zhu (1995) was applied to calculate oil content independent of erucic acid contents. The SGEDH population showed also strong positive correlation between erucic acid and protein content in defatted meal. To investigate protein content in defatted meal independent of the effect caused by erucic acid, correction based on the regression of protein content in defatted meal was conducted. For the correction of protein content in defatted meal by regression information, the linear regression between GC erucic acid content and protein content in defatted meal was calculated. The slope of this regression was used to calculate the regression corrected protein content in defatted meal the following way: Protein content in defatted meal − (slope of linear regression between GC erucic acid content and protein content in defatted meal × GC erucic acid content).
Statistical analysis
PLABSTAT software version 3A (Utz 2011) was used to perform analysis of variance (ANOVA) applying the following general model:
where Yij is the trait value of genotype i in environment j, µ is the general mean, gi is the effect of ith genotype, ej is the effect of jth environment, and geij is the interaction between ith genotype and jth environment. Genotypes were considered fixed in the analysis, whereas environments were treated as random variables. The data were tested for outliers by a modification of the Anscombe and Tukey method (1963) based on the detection of extreme residuals. After examining the list of detected outliers, the measured values of the outliers with highest standardized residual were checked for errors and the ANOVA was repeated considering missing values for extreme outliers. The adjusted results were used in the subsequent analyses.
Heritability (h2) was calculated as: \(h^{2} = \sigma_{g}^{2} /(\sigma_{g}^{2} + \sigma_{ge}^{2} /E)\) where \(\sigma_{g}^{2}\) and \(\sigma_{ge}^{2}\) are variance components for g and e. E refers to number of environments. PLABSTAT was also used to test for normality of the distribution and to calculate Spearman’s rank correlation coefficients between trait’s mean values of the genotypes across the environments. The partial correlation coefficient (rs_partial) between a trait X and Y both correlated with a trait Z was calculated using the equation of De La Fuente et al. (2004):
DNA extraction and SNP markers
The original DH population consisted of 148 genotypes which were used for DNA extraction and construction of a linkage map. For DNA extraction 90–110 mg of leaf material was collected from 3 to 4 weeks old plantlets grown in the greenhouse. After weighing, leaf tissue was directly frozen in liquid nitrogen and stored at − 20 °C until further processing. Genomic DNA was extracted using innuPREP Plant DNA Kit (Analytik Jena, Germany) following the manufactures user manual. DNA concentration was measured with a Bio-Rad Versa Fluor™ Fluorometer (http://www.bio-rad.com/en-us/product/versafluor-fluorometer) using Bio-Rad Fluorescent DNA Quantification Kit (Bio-Rad Laboratories CA, USA). The Illumina Infinium Brassica 60 K SNP array (Illumina, Inc., http://www.illumina.com) with a total number of 58,464 markers was used to genotype the SGEDH population and its parental lines at TraitGenetics GmbH, Gatersleben, Germany (http://www.traitgenetics.com). The assay data were analysed with Illumina’s GenomeStudio® Software v2011 (Illumina, Inc.) applying proprietary cluster files.
Linkage map construction
In total 15,474 SNP markers of the 58,464 markers on the chip (26.5%) were polymorphic in the SGEDH population. Heterozygous genotype calls were manually set to missing values. Markers with more than 10% missing calls were deleted. A SNP linkage map was constructed with JoinMap® 4.1, Kyazma® (Stam 1993; Van Ooijen 2011) applying the Kosambi map function. Three subsets of about 5000 markers each were analysed to identify and to delete co-segregating markers. After combining the remaining markers a map consisting of 4779 SNP markers was obtained. This map was further reduced by deleting markers with a distance < 0.5 cM. The final SNP-map used for QTL mapping consisted of 19 chromosomes with 1764 markers and a size of 2687 cM. There were only two larger gaps on A03 (26.5 cM) and A08 (22.3 cM). The mean marker distance was 1.5 cM. The assignment of the SNP markers to the different chromosomes was done according to Clarke et al. (2016) and a consensus map provided by Jörg Plieske (TraitGenetics GmbH, Gatersleben, Germany). The final map is shown in Online Resource 1.
QTL mapping and identification of candidate genes
Mean data for seed quality traits from the all environments were used for QTL mapping. QTL computation was conducted by QTL Network software version 2.1 (Yang et al. 2008) using a systematic mapping strategy to search for QTL with epistatic effects in a mixed linear model introduced by Yang et al. (2007). At first a one-dimensional genome scan is performed by a composite interval mapping (CIM) approach to identify putative QTL with the help of selected candidate marker intervals as cofactors. In a second step epistatic effects for QTL with additive main effects are detected. F test based on Henderson method III is performed to adjust for the critical threshold value to control the experiment-wise false positive rate. A permutation testing with 1000 permutations was employed to determine an empirical threshold value of the F statistic. QTL names were defined as an abbreviation explaining the trait and a continuous “QTL-number”. QTL explaining ≥ 25% of the phenotypic variance was considered as major QTL. To determine the physical position of major QTL and to identify candidate genes, sequences of flanking SNP markers were used to BLAST them against Brassica Databases. SNP marker sequences were provided by Isobel Parkin (AAFC, Saskatoon, Canada). SNP sequences were aligned against the Darmor-bzh B. napus reference genome v4.1 (http://www.genoscope.cns.fr/brassicanapus/, Chalhoub et al. 2014) and against the ZS11 reference sequence (https://www.ncbi.nlm.nih.gov/assembly/GCF_000686985.2/, Sun et al. 2017). Most likely positions were selected from the BLAST hits considering best matching and highest possible E-value as well as genetic map data information. A. thaliana candidate gene sequences were identified using the Araport11 annotation (Cheng et al. 2017). The assignment of the oilseed rape genes to the Arabidopsis genes using the protein sequences is described in Pucker et al. (2017). Identification of Reciprocal Best BLAST Hits (RBHs) between two sets of sequences (protein/DNAReciprocal) was performed as described in Pucker et al. (2016).
Results
Phenotypic analysis
Significant effects for the genotype and the environment were detected for all traits in the SGEDH population. Since the population was segregating for canola quality, large variance components were found for glucosinolate and fatty acid contents (Table 1). Because the field experiments were performed without replicates, the genotype × environment interactions included the residual error. Data obtained from the large number of environments led to high heritability estimates for all traits, ranging from 0.81 for protein content to 1.00 for the fatty acids 20:1 and 22:1. In the DH population, the oil and protein content ranged from 42.5 to 50.8% and from 16.2 to 19.5%, respectively (Table 2). The protein content in the defatted meal ranged from 30.2 to 36.9% and the sum of oil and protein content varied between 60.8 and 68.3%. Transgressive segregation was observed for most of the traits in the DH population.
Frequency distribution of oil content did not show a significant deviation from a normal distribution (data not shown). The oil content showed a mean value of 46.3% and a standard deviation of 1.8%. Plotting oil content against erucic acid content showed segregation of the DH population for erucic acid content into three groups (Fig. 1a). One group of 45 genotypes with 0–2.6% erucic acid showed oil contents of 42.5–45.2%, a second group including 61 genotypes with an erucic acid content of 16.1–34.8% had oil contents between 45.3 and 47.7% and in a third group of 33 genotypes with 39.3–50.8% erucic acid content, oil content ranged from 45.7 to 50.8%. The observed distribution did not significantly deviate from the expected 1:2:1 segregation (Chi2 = 4.2, p = 0.05) Furthermore, a clear positive correlation between oil and erucic acid content was detected. In order to eliminate the effect of the erucic acid genes, a correction was performed by regression (Fig. 1b), by conditional mapping and by considering the increased molecular weight of the erucic acid molecule. All three methods led to almost identical results (data not shown). Therefore, only the frequency distribution for the correction based on regression is shown in Fig. 2. Corrected oil contents showed a mean value of 44.5%, a standard deviation of 1.1% and the correlation coefficient was reduced to rS = − 0.01. Through the regression correction, the erucic acid-free oil content of SGDH14 was calculated to be 45.1%.

Regression between erucic acid content and oil content (Oilc) without (a) and with correction based on the regression method (b) of the DH population SGDH14 × Express617

Frequency distribution of oil content (Oilc) after correction for the effect of erucic acid content based on the regression method of the DH population SGDH14 × Express617
Contrary to common knowledge, no correlation was detected between seed oil and protein content (Table 3). More carefully looking into the data revealed that a negative correlation of − 0.52** was found for the erucic acid-free genotypes (n = 45; data not shown). Oil content of the complete population was instead positively correlated with protein content in defatted meal (rS = 0.68**), which itself was also positively correlated with erucic acid content (rS = 0.82**). Calculation of partial correlations for these three traits showed no direct effect of oil content on protein content in defatted meal (if 22:1 is kept constant, than rs_partial = 0.02), but indicated a pleiotropic effect of the erucic acid genes on the protein content in defatted meal (Fig. 3a). The calculation of the regression corrected protein content of the defatted meal resulted in no correlation to the erucic acid content (rS = 0.11; Fig. 3b).

Regression between erucic acid content and protein content in defatted meal (PidM) without (a) and with correction based on the regression method (b) of the DH population SGDH14 × Express617
Interestingly, the DH population showed a bimodal segregation for acid detergent lignin (ADLm) content (Fig. 4; Chi2 = 0.35). A bimodal segregation was also found for ADFm content (data not shown). NDFm, ADFm and ADLm contents were all closely positively correlated with each other and they were negatively correlated with cellulose (Cm) and hemicellulose (HCm) content (Table 3). Obviously, a reduction in lignin content caused a decrease in NDFm, ADFm and ADLm content. At an unchanged cellulose and hemicellulose content this led to increasing negative correlations between the three fibre fractions and hemicellulose and cellulose content. Following correction of the effect of the erucic acid alleles on oil and protein content, the negative correlations between ADLm content increased from − 0.19* to − 0.36** for oil and from − 0.20* to − 0.36** for protein content in defatted meal. Equally, the negative correlation between ADLm content and the sum of oil and protein in defatted meal increased from − 0.22 to − 0.53** (Table 3 and Fig. 5). And the correlations between hemicellulose and cellulose to the sum of corrected oil and protein contents were positive. Correlations with thousand kernel weight (TKW) were mostly loose and not significant. However, correlations between seed hull content and oil, protein content in the defatted meal, the sum of both, TKW, hemicellulose (HCm), and cellulose (Cm) content were negative, but they were positive for NDF, ADF, and ADL content. Glucosinolate content was with a negative sign only significantly correlated to NDF, ADF, ADL, and cellulose content.

Frequency distribution of the acid detergent lignin (ADLm) content in the defatted meal of the DH population SGDH14 × Express617

Regression between the acid detergent lignin content in defatted meal (ADLm) and the sum of oil and protein content in defatted meal (Oilc + PidMc) after correction based on the regression method of the DH population SGDH14 × Express617
The two groups of genotypes with low and high ADLm content were separated at 9.5% (Fig. 4), and they were compared for different traits. Results confirmed that the group of low ADLm genotypes had a 4.1% significantly lower ADLm content (Table 4). In line with this result, a significant reduction of seed hull content was also detected. Except for protein content all other traits were significantly improved in the group of low ADLm genotypes. The effect of the low ADLm trait was more enhanced on the corrected oil content (+ 1.0% for oilc) than on the corrected protein content in the defatted meal (+ 0.7% for PidMc). Following correction of the effect caused by erucic acid content, SGEDH13 was identified as the genotype with the highest oil content (46.7%) and the highest content of oil and protein in defatted meal (79.2%). Compared to a number of standard cultivars that were tested together with the DH population in the field experiments, SGEDH13 had about 2% higher contents of oil and of the sum of oil and protein content in defatted meal (Table 5).
QTL mapping
Four QTL for oil content were mapped on the linkage groups A08, C03, C05 and C07 (Table 6). Individual QTL explained between 3.8 and 46.4% of the phenotypic variance and collectively accounted for 78.5% of the total phenotypic variance. Except QTL Oil-4 on linkage group C07 all QTL showed positive additive effects, indicating that the alleles increasing oil content were derived from SGDH14. QTL Oil-1 located on linkage group A08 had the largest phenotypic effect and accounted for 46.4% of the phenotypic variance, followed by QTL-2 on linkage group C03. Epistatic main effects were detected between QTL Oil-1 and Oil-2. Positions of QTL Oil-1 and Oil-2 were almost identical with the positions for the QTL 20:1 + 22:1, indicating that the increase in oil content was due to the effect of the two erucic acid genes of SGDH14 on A08 and C03. Noteworthy, a QTL for 20:1 was mapped only on C03 and QTL for 22:1 mapped at an almost identical position on C03 and at two slightly different positions on A08. QTL Oil-3 on C05 and QTL Oil-4 on C07 explained 19% and 3.8% of the phenotypic variance. All three methods applied for the correction of the erucic acid effect resulted in almost identical results (Table 6). As expected, the QTL Oil-1 and Oil-2 on A08 and C03 disappeared following correction. QTL Oil-4 on C07 disappeared as well and two new QTLs on A10 (RCOil-1, MWOil-1 and CNOil-1) and C04 (RCOil-2, MWOil-2 and CNOil-2) were detected. The QTL on C04 had a positive sign, indicating that the allele increasing the oil content was derived from SGDH14. Through the correction of the erucic acid effect on oil content, the proportion of the explained phenotypic variance for QTL Oil-3 on C05 increased from 19 to 36.2%.
For the seed protein content (Pro-) four QTL were detected that explained 43% of the phenotypic variance (Table 7). Three of the four QTL alleles increasing protein content were derived from SGDH14, whereas QTL allele Pro-3 was derived from Express617. Using the protein content data on basis of defatted meal (PidM-) led to the detection of 8 QTL that together explained 80% of the observed phenotypic variance. The largest QTL PidM-2 on A08 mapped close to QTL Pro-2 and at the identical position as QTL 20:1 + 22:1. Compared to QTL Pro-2, the proportion of the explained phenotypic variance for PidM-2 increased from 9.9 to 47.1%. All three QTL had a positive sign of the additive effect, underpinning the increasing effect of the SGDH14 allele. A second QTL PidM-4 on C03 mapped close to the QTL 20:1 + 22:1 on C03, suggesting that the erucic alleles at A08 and C03 are causative for increase of the protein content in the defatted meal. Following correction of the erucic acid alleles on protein content in defatted meal (PidMC-), only three QTL were left over. QTL PidMC-1 on A09 mapped at a similar position as QTL PidM-3. PidMC-2 on C05 mapped nearby PidM-6. Assuming that both QTL are identical, the proportion of the explained variance increased from 2.7% for PidM-6 to 11.1% for PidMC-2. QTL PidMC-2 mapped also close to QTL Oil + Pro-3, Oil-3 and RCOil-3, suggesting that this QTL allele did not only increase protein content of the defatted meal but also oil content of the seed. For the sum of corrected oil and protein content in the defatted meal (RCOil + PidMc), only one QTL was detected on C05 at 30.2 cM. Compared to QTL Oil + Pro-3 the fraction of the explained phenotypic variance increased from 16.3 to 43.2%. QTL for glucosinolate content mapped on five linkage groups and together explained 76.5% of the observed variance. Neither, the major QTL GSL-5 on C09 nor one of the others did co-locate with any of the QTL for the other traits.
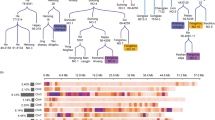
For fibre content of the defatted meal, three co-locating QTL for NDFm, ADFm and ADLm were detected on C05 at 34.4 cM which explained between 46.3 and 80.7% of the phenotypic variance (Table 8, Fig. 6). The size of the additive effect of the QTL and their proportion of explained phenotypic variance increased from NDFm over ADFm to ADLm, pointing at a reduction of lignin content as possible cause for decreasing NDFm, ADFm and ADLm contents. Two QTL for seed hull content were mapped on chromosomes A08 and C05. The major QTL at C05 mapped closed to the QTL for NDFm, ADFm and ADLm. The additive effects of the QTL for the three fibre components and seed hull content had a negative sign, showing that the allele decreasing the fibre content was derived from SGDH14. Furthermore, the additive effects of QTL for Hemicellulose (HCm) and Cellulose (Cm) were positive. The position of these QTL was similar to the QTL positions for Oil-3, RCOil-3, PidMC-2, and Oil + Pro-3, but the co-locating QTL for oil and protein content on C05 had a positive additive effect, suggesting that reduction of the lignin content concomitantly led to an increase in oil, protein, cellulose and hemicellulose content.

Depiction of the genetic and physical region of chromosome C05 with positions for a number of QTL. (−) and (+) indicate the additive effect of the SGDH14 allele
Identification of candidate genes
In order to check the accuracy of QTL mapping results, the genetic positions of the erucic acid loci were compared with their physical positions. On A08 and C03 the genetic position of the two erucic acid loci were determined by the flanking SNP markers. Using the sequence of the fae1 gene of A08 (HM362915.1), the fae1 sequence was found 184,699 bp upstream of the flanking marker Bn-A08-p12699181. Using the fae1 sequence of C03 (GU325719.1), the fae1 sequence was found 235,424 bp downstream of the flanking marker Bn-scaff_15794_3-p147011. Hence, the fae1 genes were detected close to the SNP markers flanking the QTL for 20:1 + 22:1 on both chromosomes. The two SNP markers Bn-scaff_17441_1-p950045 and Bn-scaff_17441_3-p28950 flanking the QTL for ADFm, ADLm and HCm content on C05 were located at a distance of 0.76 cM from each other. The positions of the two markers in the Brassica Darmor-bzh genome were 40,064,397 bp (Start) and 40,322,708 bp (End). The physical distance between the two markers was 258,311 bp, which corresponds to 340,000 bp per cM. The physical region between 39,000,000 and 41,000,000 of the Darmor-bzh (Online Resource 2) and the ZS11 sequence (Online Resource 3) was inspected for putative candidate genes known to be involved in the phenylpropanoid and lignin biosynthesis. The region between the two flanking markers and the adjoining markers 1 cM up- and downstream of the Darmor-bzh sequence were more carefully scrutinized. In the 3 cM region (39,713,411–40,616,480 bp; 903,069 bp) around the QTL peak six putative candidate genes were identified that may explain the low ADLm phenotype (Fig. 6). Those comprised three MATE efflux family proteins (BnaC05g44030D, BnaC05g43790D, BnaC05g43800D), two myb domain proteins (BnaC05g44010D, BnaC05g43530D) and one phenylalanine ammonia-lyase gene (PAL; BnaC05g42780D). Outside this 3 cM region no putative candidate gene was identified that could explain the observed phenotype (c.f. Online Resource 2). In the ZS11 sequence (Online Resource 3) only the SNP marker sequence of Bn-scaff_17441_3-p28950 on C05 was detected at 40,983,516 bp. Searching the region down- and upstream for candidate genes resulted in the detection of five Myb or Myb-like domain proteins (BnC05g0729210, BnC05g0728520, BnC05g0728330, BnC05g0728150, BnC05g0727930) and two multi-antimicrobial extrusion proteins (BnC05g0729230, BnC05g0728950).
Discussion
The expression of the erucic acid alleles in oilseed rape has been found to affect other seed quality traits like oil, sinapic acid ester and phytosterol content (Ecke et al. 1995; Amar et al. 2008). Conditional QTL mapping has been applied in oilseed rape to identify QTL for oil content which is independent from QTL for protein content (Zhu 1995; Zhao et al. 2006). In the present work conditional mapping has been applied to correct for effects of erucic acid alleles on oil content. In addition to the conditional mapping method, a correction based on the regression between erucic acid content and oil content and on considering the increase in molecular weight of erucoyl-CoA compared to oleoyl-CoA has been applied. The disadvantage of the conditional method is that it calculates oil content to an average erucic acid content of all genotypes, which is problematic if oil contents need to be compared with other erucic acid-free Canola-type genotypes. All three correction methods applied in the present work led to identical results in QTL mapping (Table 6). The correction by considering the molecular weight confirms the earlier hypothesis that the increase in oil content is caused only by cytoplasmic chain length extension. The regression-based method was furthermore applied to correct for the effect of the erucic acid alleles on protein content in defatted meal, which was identified in the population. Other populations need to be analysed to confirm the effect of the erucic acid alleles on seed protein content. Unexpected and in contrast to general findings, there was no negative correlation between oil and protein content in the DH population. This may have been caused by the erucic acid alleles segregating in the population, since in the subset of erucic acid-free genotypes of the population the usual negative correlation was found.
An improvement of the meal quality of oilseed rape is not yet a relevant breeding goal, because the financial return for the farmers depends primarily on the oil yield per hectare. Therefore, improvement of the meal quality is only meaningful, if the oil yield is simultaneously enhanced. Theoretically, a reduction of seed fibre content in oilseed rape could result in an enhanced oil and protein content of the seed. Fibre consists of hemicellulose, cellulose and lignin. Of those three, genetically reducing lignin content is considered to be more important, because it affects digestibility of the meal (Jung et al. 1997). Most of the published work deals about genotypes with reduced lignin content, which usually is associated with the appearance of yellow or at least brown yellowish seed phenotypes. Although a large number of major QTL for reduced lignin content have been mapped on different chromosomes (Rahman and McVetty 2011; Wang et al. 2015, 2017 and references therein), no improved cultivar with reduced fibre content has been successfully established in the market. The reasons for this remain unclear. It has been discussed that yellow seeded genotypes may suffer from lower germination and vigour and increased pre-harvest germination. Seed coat compounds may protect the embryo against damage by biotic and abiotic stress factors (Boerjan et al. 2003) and may influence seed dormancy and seed longevity (Nagel et al. 2011; Schatzki et al. 2013). Considering the large number of known genes that may affect the phenylpropanoid and lignin biosynthetic pathway (Taylor-Teeples et al. 2015), it appears plausible to assume that pleiotropic effects of QTL alleles for reduced lignin content on seed oil and protein content may depend on the type of gene affected as well as on the locus in the allopolyploid oilseed rape genome. Beside the structural genes, a large number of transcription factors have been identified that can up- or down-regulate the lignin biosynthetic pathway (for a review see Liu et al. 2015). Despite the large number of major QTL affecting seed lignin content in oilseed rape, there are few reports about QTL mapped to chromosome C05. In a genome wide association study Wang et al. (2015) identified a number of SNP markers in the region 35,235,636–40,371,177 bp that were associated with seed ADL content. Candidate gene BnaC05g43530D (BnaC.MYB83) was found to be most highly associated with a SNP marker at position 40,322,746 bp (Wang et al. 2015). Wang et al. (2017) identified SNP marker Bn-scaff_18181_1-p1267885 to be associated with seed coat colour on C05 in the region 41,022,878 bp.
In the present study, the SNP markers flanking the major ADLm-1 QTL were located on C05 at 34.4 cM with a physical position from 40,064,397 to 40,322,708 bp. The confidence interval for the QTL was 32.2–36.1 cM. To narrow down the region, the accuracy of the QTL mapping was analysed using the two fae1 loci at A08 and C03. Results showed that these QTL were detected within 1 cM of the QTL peak. This is consistent with earlier observations that in many cases genes producing a major QTL have been identified in a region less than 1 cM around the QTL peak (Price 2006). Taking this into account, the region scanned for the presence of candidate genes was delimited to 3 cM (39,713,411–40,616,480 bp) around the QTL peak. For most of the candidate genes listed by Wang et al. (2015; c.f. Online Resource 2) a direct relationship to lignin biosynthesis could not be seen. However, most likely candidate genes for the observed seed phenotype in the present study are BnaC05g42780D (phenylalanine ammonia-lyase 4), BnaC05g43790D and BnaC05g43800D (both MATE efflux proteins; Fig. 6). BnaC05g43530D (myb domain protein 3r-3 has been cited by Wang et al. 2015 as candidate gene, however, to date there is no proof that R1R2R3-MYB genes are involved in regulation of the phenylpropanoid or lignin biosynthetic pathway (Liu et al. 2015). Additional candidate genes are BnaC05g44010D (myb domain protein 83; see AtMYB83 in Liu et al. (2015)) and BnaC05g44030D (MATE efflux family protein). Phenylalanine ammonia-lyase is the central structural enzyme of the general phenylpropanoid pathway (Liu et al. 2015). A mutation in this gene should affect not only the lignin content but also the biosynthesis of flavonoids and phenolic acids. MYB proteins are transcription factors that have been identified as key regulators of the phenylpropanoid biosynthetic pathway (Liu et al. 2015). MATEs are multidrug and toxic compound extrusion transporter proteins involved in seed coat pigmentation (Chai et al. 2009). In Arabidopsis, the TRANSPARENT TESTA 12 (AtTT12) encodes a MATE secondary transporter that is specifically expressed in the developing seed coat. Functional AtTT12 leads to an accumulation of proanthocyanidine pigments in seed coat cells (Chai et al. 2009). Proanthocyanidins (PAs) belong to the flavonoids and are related to lignins by sharing the precursor 4-coumaroyl CoA in the phenylpropanoid pathway. Marles and Gruber (2004) noted that lignin is deposited normally within the cell wall, while proanthocyanidins are accumulated in the vacuoles. Co-location of QTL for fibre components and seed hull as found in the present study allows inferring that low ADL content is associated with reduced seed coat thickness (Wittkop et al. 2012). Further studies are necessary to evaluate whether the major QTL for low ADLm content has pleiotropic effect on seed germination, vigour, longevity, pre-harvest germination as well as lodging and disease resistance and seed yield. It is presently not known whether the QTL is caused by a complete or partial knock-out of the responsible candidate gene and whether the gene is expressed only or predominantly in the seed coat. Despite the significant reduction in ADLm content, the seeds of SGDH14 did not show a brown or yellowish appearance (Online Resource 4) which suggests only a limited effect on the proanthocyanidine content of the seeds. Obviously, there is a stronger effect on the seed hull content (c.f. Table 4). Additional research including the analysis of segregating bulks for the major QTL on C05 by genotyping by sequencing, RNAseq and genome editing is necessary to identify the immediate cause of the observed phenotype.
In comparison to a range of standard line cultivars, SGEDH13 showed 2% higher oil content and a 2% higher content of the sum of oil and protein in defatted meal (Table 5). Since oil content is negatively correlated with protein content, it was not surprising that there were a number of line cultivars with a higher seed protein content compared to SGEDH13. Following correction of the effect of the erucic acid alleles on oil content, the two positive QTL alleles on C04 and C05 for oil content were derived from SGDH14 and the positive allele on A10 was derived from Express. The number of detected QTL for oil content is relatively small and can be explained by the limited population size of only 139 DH lines. Very similar results were obtained using the same data and performing composite interval mapping (CIM) with WinQTL Cartographer software version 2.5 (WinQTLCart 2.5; Wang et al. 2012) as in the present study (N. Behnke, in preparation). This confirms earlier results by Ravi et al. (2011) who used in his studies both programs in parallel. SGEDH13 appears to be a valuable DH line in further studies aimed at increasing oil and protein content and decreasing fibre content in oilseed rape.
Author contribution statement
CM designed the experiment and developed the microspore-derived DH mapping population. NB and ES performed the experiments. NB, ES and CM analysed the data. NB and CM wrote the manuscript, and all three authors agreed on the final manuscript.
References
Amar S, Ecke W, Becker HC, Möllers C (2008) QTL for phytosterol and sinapate ester content in Brassica napus L. collocate with the two erucic acid genes. Theor Appl Genet 116:1051–1061
Anscombe FJ, Tukey JWd (1963) The examination and analysis of residuals. Technometrics 5:141–160
Badani AG, Snowdon RJ, Wittkop B, Lipsa FD, Baetzel R, Horn R, Haro A, Font R, Lühs W (2006) Colocalization of a partially dominant gene for yellow seed colour with a major QTL influencing acid detergent fibre (ADF) content in different crosses of oilseed rape (Brassica napus). Genome 49:1499–1509
Boerjan W, Ralph J, Baucher M (2003) Lignin biosynthesis. Annu Rev Plant Biol 54:519–546
Burns MJ, Barnes SR, Bowman JG, Clarke MHE, Werner CP, Kearsey MJ (2003) QTL analysis of an intervarietal set of substitution lines in Brassica napus: (i) seed oil content and fatty acid composition. Heredity 90:39–48
Chai YR, Lei B, Huang HL, Li JN, Yin JM, Tang ZL, Wang R, Chen L (2009) TRANSPARENT TESTA 12 genes from Brassica napus and parental species: cloning, evolution, and differential involvement in yellow seed trait. Mol Genet Genom 281:109–123. https://doi.org/10.1007/s00438-008-0399-1
Chalhoub B, Denoeud F, Liu S et al (2014) Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345:950–953. https://doi.org/10.1126/science.1253435
Chen G, Geng J, Rahman M et al (2010) Identification of QTL for oil content, seed yield, and flowering time in oilseed rape (Brassica napus). Euphytica 175:161–174. https://doi.org/10.1007/s10681-010-0144-9
Cheng CY, Krishnakumar V, Chan AP, Thibaud-Nissen F, Schobel S, Town CD (2017) Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J 89:789–804. https://doi.org/10.1111/tpj.13415
Clarke WE, Higgins EE, Plieske J, Wieseke R, Sidebottom C, Khedikar Y et al (2016) A high-density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of single-locus markers in the allotetraploid genome. Theor Appl Genet 129:1887–1899
De La Fuente A, Bing N, Hoeschele I, Mendes P (2004) Discovery of meaningful associations in genomic data using partial correlation coefficients. Bioinformatics 20:3565–3574
Delourme R, Falentin C, Huteau V et al (2006) Genetic control of oil content in oilseed rape (Brassica napus L.). Theor Appl Genet 113:1331–1345. https://doi.org/10.1007/s00122-006-0386-z
Dimov Z, Suprianto E, Hermann F, Möllers C (2012) Genetic variation for seed hull and fibre content in a collection of European winter oilseed rape material (Brassica napus L.) and development of NIRS calibrations. Plant Breed 131:361–368
Ecke W, Uzunova M, Weissleder K (1995) Mapping the genome of rapeseed (Brassica napus L.). II. Localization of genes controlling erucic acid synthesis and seed oil content. Theor Appl Genet 91:972–977
Fleddermann M, Fechner A, Rößler A, Bähr M, Pastor A, Liebert F, Jahreis G (2013) Nutritional evaluation of rapeseed protein compared to soy protein for quality, plasma amino acids, and nitrogen balance—a randomized cross-over intervention study in humans. Clin Nutr 32:519–526
Jiang C, Shi J, Li R, Long Y, Wang H, Li D, Zhao J, Meng J (2014) Quantitative trait loci that control the oil content variation of rapeseed (Brassica napus L.). Theor Appl Genet 127:957–968
Jung HG, Mertens DR, Payne AJ (1997) Correlation of acid detergent lignin and Klason lignin with digestibility of forage dry matter and neutral detergent fiber. J Dairy Sci 80:1622–1628
Liu J, Osbourn A, Ma P (2015) MYB transcription factors as regulators of phenylpropanoid metabolism in plants. Mol Plant 8:689–708
Marles MA, Gruber MY (2004) Histochemical characterisation of unextractable seed coat pigments and quantification of extractable lignin in the Brassicaceae. J Sci Food Agric 84:251–262
Nagel M, Rosenhauer M, Willner E, Snowdon RJ, Friedt W, Börner A (2011) Seed longevity in oilseed rape (Brassica napus L.)—genetic variation and QTL mapping. Plant Genet Res 9:260–263. https://doi.org/10.1017/S1479262111000372
Nesi N, Delourme R, Bregeon M, Falentin C, Renard M (2008) Genetic and molecular approaches to improve nutritional value of Brassica napus L. seed. CR Biol 331:763–771. https://doi.org/10.1016/j.crvi.2008.07.018
Price AH (2006) Believe it or not, QTLs are accurate! Trends Plant Sci 11:213–216
Pucker B, Holtgräwe D, Sörensen TR, Stracke R, Viehöver P, Weisshaar B (2016) A de novo genome sequence assembly of the Arabidopsis thaliana accession Niederzenz-1 displays presence/absence variation and strong synteny. PLoS ONE 11:e0164321
Pucker B, Holtgräwe D, Weisshaar B (2017) Consideration of non-canonical splice sites improves gene prediction on the Arabidopsis thaliana Niederzenz-1 genome sequence. BMC Res Notes 10:667
Qiu D, Morgan C, Shi J et al (2006) A comparative linkage map of oilseed rape and its use for QTL analysis of seed oil and erucic acid content. Theor Appl Genet 114:67–80. https://doi.org/10.1007/s00122-006-0411-2
Rahman M, McVetty P (2011) A review of Brassica seed color. Can J Plant Sci 91:437–446. https://doi.org/10.4141/CJPS10124
Ravi K, Vadez V, Isobe S et al (2011) Identification of several small main-effect QTLs and a large number of epistatic QTLs for drought tolerance related traits in groundnut (Arachis hypogaea L.). Theor Appl Genet 122:1119–1132. https://doi.org/10.1007/s00122-010-1517-0
Rücker B, Röbbelen G (1996) Impact of low linolenic acid content on seed yield of winter oilseed rape (Brassica napus L.). Plant Breed 115:226–230
Schatzki J, Schoo B, Ecke W, Herrfurth C, Feussner I, Becker HC, Möllers C (2013) Mapping of QTL for seed dormancy in a winter oilseed rape doubled haploid population. Theor Appl Genet 126:2405–2415
Simbaya J, Slominski BA, Rakow G, Campbell LD, Downey RK, Bell JM (1995) Quality characteristics of yellow-seeded Brassica seed meals: protein, carbohydrates, and dietary fiber components. J Agr Food Chem 43:2062–2066
Stam P (1993) Construction of integrated genetic linkage maps by means of a new computer package: JoinMap. Plant J 3:739–744
Sun F, Fan G, Hu Q, Zhou Y, Guan M, Tong C et al (2017) The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J 92:452–468
Taylor-Teeples M, Lin L, De Lucas M, Turco G, Toal TW, Gaudinier A et al (2015) An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature 517(7536):571
Teh L, Möllers C (2015) Genetic variation and inheritance of phytosterol and oil content in a doubled haploid population derived from the winter oilseed rape Sansibar × Oase cross. Theor Appl Genet 129:181–199. https://doi.org/10.1007/s00122-015-2621-y
Utz HF (2011) PLABSTAT, Computerprogramm zur statistischen Analyse von pflanzenzüchterischen Experimenten, Version 3Awin 14. Juni 2011. Institut für Pflanzenzüchtung, Saatgutforschung und Populationsgenetik der Universität Hohenheim
Van Ooijen JW (2011) Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet Res 93:343–349
Van Soest PJ, Robertson JB, Lewis BA (1991) Methods for dietary fiber, neutral detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J Dairy Sci 74:3583–3597. https://doi.org/10.3168/jds.S0022-0302(91)78551-2
Wanasundara JP, McIntosh TC, Perera SP, Withana-Gamage TS, Mitra P (2016) Canola/rapeseed protein-functionality and nutrition. OCL 23:D407
Wang S, Basten, CJ, Zeng Z-B (2012) Windows QTL cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm. Accessed 23 Aug 2018
Wang J, Jian H, Wei L, Qu C, Xu X, Lu K, Qian W, Li J, Li M, Liu L (2015) Genome-wide analysis of seed acid detergent lignin (ADL) and hull content in rapeseed (Brassica napus L.). PLoS ONE 10(12):e0145045. https://doi.org/10.1371/journal.pone.0145045
Wang J, Xian X, Xu X, Qu C, Lu K, Li J, Liu L (2017) Genome-wide association mapping of seed coat color in Brassica napus. J Sci Food Agric 65:5229–5237. https://doi.org/10.1021/acs.jafc.7b01226
Wittkop B, Snowdon RJ, Friedt W (2012) New NIRS calibrations for fiber fractions reveal broad genetic variation in Brassica napus seed quality. J Agri Food Chem 60:2248–2256
Yan XY, Li JN, Fu FY et al (2009) Co-location of seed oil content, seed hull content and seed coat color QTL in three different environments in Brassica napus L. Euphytica 170:355–364. https://doi.org/10.1007/s10681-009-0006-5
Yang J, Zhu J, Williams RW (2007) Mapping the genetic architecture of complex traits in experimental populations. Bioinformatics 23:1527–1536
Yang J, Hu C, Hu H, Yu R, Xia Z, Ye X, Zhu J (2008) QTLNetwork: mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 24:721–723
Zhao J, Becker HC, Zhang D, Zhang Y, Ecke W (2005) Oil content in a European × Chinese rapeseed population: QTL with additive and epistatic effects and their genotype-environment interactions. Crop Sci 45:51–59
Zhao J, Becker HC, Zhang D et al (2006) Conditional QTL mapping of oil content in rapeseed with respect to protein content and traits related to plant development and grain yield. Theor Appl Genet 113:33–38. https://doi.org/10.1007/s00122-006-0267-5
Zhao J, Huang J, Chen F et al (2012) Molecular mapping of Arabidopsis thaliana lipid-related orthologous genes in Brassica napus. Theor Appl Genet 124:407–421. https://doi.org/10.1007/s00122-011-1716-3
Zhu J (1995) Analysis of conditional genetic effects and variance components in developmental genetics. Genetics 141:1633–1639
Acknowledgements
This study was funded by the Fachagentur für Nachwachsende Rohstoffe (FNR e.V.) on behalf of the German Federal Ministry of Food and Agriculture and by the GFPi e.V. (FKZ 22008009). Special thanks to the oilseed rape breeding companies of the GFPi e.V. for performing field experiments and to Gunda Asselmeyer, Carmen Mensch, Rosemarie Clemens, and Uwe Ammermann for their excellent technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest.
Additional information
Communicated by Isobel AP Parkin.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Behnke, N., Suprianto, E. & Möllers, C. A major QTL on chromosome C05 significantly reduces acid detergent lignin (ADL) content and increases seed oil and protein content in oilseed rape (Brassica napus L.). Theor Appl Genet 131, 2477–2492 (2018). https://doi.org/10.1007/s00122-018-3167-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-018-3167-6