
Abstract
Key message
A high-resolution genetic linkage map was constructed using the comparative genomics analysis approach and the wheat reference genome, which placed wheat powdery mildew resistance gene Pm52 in a 0.21-cM genetic interval on chromosome arm 2BL.
Abstract
The gene Pm52 confers resistance to powdery mildew and has been previously mapped on chromosome arm 2BL in winter wheat cultivar Liangxing 99. Because of its effectiveness against the disease, this study was initiated to finely map Pm52 using the comparative genomics analysis approach and the wheat reference genomic sequence. Based on the EST sequences that were located in the chromosome region flanking Pm52, four EST-SSR markers were developed, and another nine SSR markers were developed using the comparative genomics technology. These thirteen markers were integrated into a genetic linkage map using an F2:3 subpopulation of the Liangxing 99 × Zhongzuo 9504 cross. Pm52 was mapped within a 3.2-cM genetic interval in the subpopulation that corresponded to a ~40-Mb genomic interval on chromosome arm 2BL of the Chinese Spring reference genome. The Pm52-flanking markers Xicsl163 and Xicsl62 identified 344 recombinant individuals from 8820 F2 plants. Nine SSR markers generated from the Chinese Spring genomic interval were incorporated into a high-resolution genetic linkage map, which placed Pm52 in a 0.21-cM genetic interval corresponding to 5.6-Mb genomic region. The constructed high-resolution genetic linkage map will facilitate the map-based cloning of Pm52 and its marker-assisted selection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wheat powdery mildew, a foliar disease that is incited by the biotrophic fungus Blumeria graminis f. sp. tritici (Bgt), frequently occurs in many of the temperate wheat (Triticum aestivum L.) producing regions of the world. Morgounov et al. (2012) reported that the average incidence and severity of the powdery mildew in 51 countries during 1969–2010 were 54.5% and 7.5%, respectively. In China, this disease has been a serious yield constraint for wheat production since the 1970s, especially in the winter wheat-growing regions with high inputs of irrigation and fertilizers (Luo et al. 2009). The incidence of wheat powdery mildew was approximately 6 million hectares in 2018, and a similar scale of infected areas has occurred annually during the past decade (http://www.natesc.org.cn). In the future, powdery mildew is anticipated to cause greater damage to winter wheat production due to climate change, and the percentage of affected acreage has increased significantly by 8.5% per decade from 1970 to 2012 (Tang et al. 2017).
Host plant resistance and chemical control are the traditional approaches to mitigate the economic losses in wheat production caused by powdery mildew. The increase in grain yield through disease managements was estimated to be 15 million tons over the last decade (Liu et al. 2016). The discovery of resistance genes for developing resistant wheat cultivars is the preferred means to effectively and economically control wheat powdery mildew.
Various classes of molecular markers, in particular simple sequence repeat (SSR) markers, have been more efficient to identify powdery mildew (Pm) resistance genes in wheat cultivars compared to the conventional cytogenetic methods using the wheat aneuploids. However, it is difficult to develop a saturated genetic linkage map only based on SSR markers due to their limited number and scattered distribution on wheat chromosomes. The development of large numbers of chromosome-anchored wheat expressed sequence tags (ESTs) provides a valuable genomic resource (Lazo et al. 2004), which has been used to develop target gene-linked markers before the complete genome sequences of wheat become available. There exists a strong syntenic relationship among wheat and certain cereal crops and grass species that have complete genome sequences, such as rice (Oryza sativa L.) (International Rice Genome Sequencing Project, 2005), sorghum (Sorghum bicolor (L.) Moench) (Paterson et al. 2009), purple false brome (Brachypodium distachyon L.) (The International Brachypodium Initiative 2010), and barley (Hordeum vulgare L.) (The International Barley Genome Sequencing Consortium 2012). Therefore, comparative genomics analysis has been effectively applied to develop molecular markers for finely mapping Pm genes in wheat. Examples include the fine mapping of MlHLT (Wang et al. 2015), MlIW172 (Ouyang et al. 2014), and PmTm4 (Xie et al. 2017), as well as other Pm genes.
The release of the genome sequences of common wheat and its closely related species has greatly facilitated comparative genomics analysis and map-based cloning of disease resistance genes in wheat. Since the first whole-genome shotgun sequences of common wheat were available (Brenchley et al. 2012), a great effort has been made to improve the wheat genome sequence assembly from the scaffold level to the chromosome level (International Wheat Genome Sequencing Consortium 2014; Chapman et al. 2015; Zimin et al. 2017). Recently, the complete genome sequence of the wheat cultivar Chinese Spring RefSeq v.1.1 was released (The International Wheat Genome Sequencing Consortium (IWGSC) 2018). The genome sequences of the A and D genome donor species T. urartu Thumanjan Ex Gandilyan and Aegilops tauschii Coss., respectively, also have been updated (Luo et al. 2017; Ling et al. 2018). Meanwhile, the genome sequence of wild emmer wheat [T. turgidum ssp. dicoccoides (Körnicke ex Asch. & Graebn.) Thell.] (AABB genome) also was published (Avni et al. 2017). The improvement in next-generation sequencing (NGS) technologies and conversion of single nucleotide polymorphisms (SNPs) to Kompetitive allele specific PCR (KASP) markers have promoted the high-throughput discovery of DNA variants and detection of gene-linked markers. Taking the advantages of those genomic sequences, Wu et al. (2018b) confined Yr26 to a 0.003-cM interval on wheat chromosome arm 1BL and Zou et al. (2018) cloned the powdery mildew resistance gene Pm60 from T. urartu using a map-based cloning technique. It is expected that more and more genes for disease resistance and other agronomic traits will be identified and/or cloned in the future.
In general, there are two sources of Pm genes that confer resistance to powdery mildew, i.e., common wheat and its relatives. Some of the Pm genes identified in close or distant relatives may be linked with certain deleterious genes, so a pre-breeding step is required to improve their yield and agronomic performance. This impedes the direct application of such type of Pm genes in breeding programs. The resistance genes or alleles identified from commercial cultivars are preferred by breeders as those genes can be more easily incorporated for the improvement in disease resistance in wheat. Using the molecular marker-based mapping technology, several Pm genes had been identified in commercial Chinese wheat cultivars. For example, the resistance to powdery mildew in Yumai 66 and Zheng 9754 was controlled by PmYm66 and PmHNK54 on chromosome arm 2AL, respectively (Hu et al. 2008; Xu et al. 2011); Zhoumai 22 carried the Pm gene PmHNK on chromosome arm 3BL (Xu et al. 2010).
Liangxing 99 is a winter wheat cultivar and has been grown over 300 million hectares in northern China, since it was released in 2006 (http://202.127.42.178:4000/SpeciesQuery/Details/9917). It had also served as a control cultivar in the yield trials at the provincial (Shandong province) and the national levels, since 2010 and 2012, respectively. Liangxing 99 was effective against 81.3% of the 123 Bgt isolates collected from northern China and displayed an adult plant resistance in several years of field tests (Zou et al. 2017). It even had a greater percentage of resistance (94%) against another group of 49 Chinese Bgt isolates in a separate study (Ma et al. 2018). Because of its superior agronomic performance and high level of resistance to the disease, Liangxing 99 has also been used as a parent to develop commercial wheat cultivars.
Resistance to powdery mildew in Liangxing 99 was conferred by a gene, designated Pm52 (McIntosh et al. 2014). Previous study has located Pm52 on chromosome arm 2BL (Zhao et al. 2013). A project was initiated to finely map Pm52 to facilitate its efficient and accurate detection in the breeding efforts for improving disease resistance. The objectives of this study were (1) to develop molecular markers closely linked to Pm52 using the comparative genomics analysis method taking the advantage of the existing genomic sequences of wheat and other cereal crops and (2) to construct a high-resolution linkage map.
Materials and methods
Plant materials
In 2006, Liangxing 99 (pedigree: Ji 91102/Lumai 14//PH85-16) was released by the Shandong Liangxing Seed Co. Ltd., Ningjin, Shandong province (http://www.ampcn.com/info/detail/12696.asp). To develop a saturated genetic linkage map for Pm52, the F2 and F2:3 mapping populations (consisting of 8820 members) produced by crossing Liangxing 99 to powdery mildew susceptible cultivar Zhongzuo 9504 were used throughout the study. A subpopulation consisting of 165 F2:3 families were used to examine the polymorphisms of the molecular markers developed in the present study. Zhongzuo 9504 was also used to increase Bgt isolate E20, which was avirulent on the Pm genes 1a, 1c, 2, 12, 13, 16, 17, 20, 24, 2 + 6, and 5 + 6, but virulent on the Pm genes 1e, 3a, 3b, 3d, 3e, 3f, 3g, 4a, 4b, 4c, 5a, 5e, 6, 7, 8, 11, 19, Y39, PS5A, 33, DR147, H, Mlxbd, and 1 + 2 + 9 (Li et al. 2011), for phenotyping the mapping population and served as the susceptible control in all the powdery mildew tests. The cultivars derived from Liangxing 99, i.e., Zhongxinmai 99 (pedigree: Liangxing 99/222), DH51302 (pedigree: DH6388/Lankaoaizao 8//Liangxing 99), and Heng 10-5039 (pedigree: Heng 7228/Shangdong 93-5031//Liangxing 99), were used to confirm the Pm52-linked markers. Based on the reactions to 27 Bgt isolates, Zhongxinmai 99 and DH51302 were proposed to carry Pm52; and Heng 10-5039 did not carry Pm52 (Zou et al. 2017).
Evaluation of powdery mildew resistance
The seedling reactions of the F2:3 families that were derived from the F2 plants of the subpopulation and the recombinant plants were tested using the Bgt isolate E20 to predict the genotypes of the corresponding F2 plants. Fifteen seeds of each F2:3 family were planted in plastic pots (6.5 cm × 6.5 cm × 6.5 cm in dimension) that were placed in plastic trays. Liangxing 99 and Zhongzuo 9504 were grown in each tray as the resistant and the susceptible controls, respectively. When the first leaves were unfolded, the plants were inoculated by dusting with Bgt conidiospores that were increased on Zhongzuo 9504 seedlings. After incubation in a dew chamber in the dark for 24 h, the inoculated plants were grown in a greenhouse under a daily cycle of 14 h of light at 22 ± 2 °C and 10 h of darkness at 18 ± 2 °C to promote the development of disease symptoms. Fifteen days after inoculation when the disease was fully developed on the susceptible control plants, the infection types (ITs) of all plants were determined individually on a 0–4 scale as previously described (Liu et al. 1999). Plants with ITs 0, 0;, 1, and 2 were classified in the resistant group, and ITs 3 and 4, in the susceptible group. Each F2:3 family was tested at least twice to ensure consistency of the phenotypes.
Development of molecular markers
The EST sequences in the bin 2BL2-0.36-0.50 of chromosome arm 2BL were used to search against the barley genomic sequences (http://www.barleygenome.org.uk). Based on the sequences of the collinear region identified, EST-SSR markers were designed and subjected to polymorphism analysis between Liangxing 99 and Zhongzuo 9504, as well as the resistant and susceptible DNA bulks that were separately generated by mixing equal amounts of DNA from the representative plants of ten homozygous resistant and ten susceptible F2:3 families from the subpopulation. The polymorphic EST-SSR markers were validated using the subpopulation to confirm their genetic linkage relationship with Pm52. The corresponding EST sequences of the polymorphic EST-SSR markers flanking the target resistance gene were used as queries to search the Ae. tauschii SNP database (http://probes.pw.usda.gov/WheatDMarker/), and the Brachypodium (http://mips.helmholtz-muenchen.de/plant/brachypodium/), rice (http://rice.plantbiology.msu.edu/), and sorghum (http://mips.helmholtz-muenchen.de/plant/sorghum/) genomic sequences with an E-value cutoff of 10e−10. The orthologous genomic regions were determined by comparative genomics analysis of the putatively conserved gene pairs in the genomes of Ae. tauschii, Brachypodium, rice, and sorghum. The resulting orthologous gene pairs were then used to search the Chinese Spring 454 contigs sequence (Brenchley et al. 2012) and the whole genomic assembly sequences for Chinese Spring wheat (IWGSC WGS v1.1, NRGene DeNovoMAGIC, Seq Repository of Wheat Portal on URGI, INRA, France) (The International Wheat Genome Sequencing Consortium (IWGSC) 2018) to find homologous contig or scaffold sequences that were used to develop SSR markers using BatchPrimer3 (You et al. 2008).
Marker analysis
Genomic DNA was isolated from leaf tissues of each F2 plant and the 165 F2:3 families of subpopulation and the cultivars derived from Liangxing 99 using the CTAB method (Saghai-Maroof et al. 1984). Each reaction mixture (10 µl) of DNA amplification was composed of 50–100 ng of template DNA, 0.4 µM the forward and reverse primers each, 0.2 U of Taq polymerase, 0.2 mM dNTPs, and 1 × PCR buffer with 2 mM Mg2+. DNA amplification was started from one denaturation cycle at 94 °C for 5 min, and then 35 cycles at 94 °C for 45 s, 50–60 °C (depending on the specific primers) for 45 s, and 72 °C for 1 min. The reaction was terminated with an extension cycle at 72 °C for 10 min. The amplification products were separated on 8% non-denaturing polyacrylamide gels (39 acrylamide/1 bisacrylamide), and the banding patterns were visualized after silver staining.
Identification of recombinant individuals from the F2 mapping population
The SSR markers developed were integrated into a linkage map using a subpopulation consisting of 165 F2:3 families of Liangxing 99 × Zhongzuo 9504 cross. Markers Xicsl62 and Xicsl163 were used to genotype 8820 F2 plants to identify recombinant plants between the two markers with the capillary electrophoresis technology (Tagliaro et al. 1998). Amplification of DNA was carried out in a 10 µl reaction mixture which included 50–100 ng of template DNA, 0.4 µM each of the forward primer added with 5-hexachloro-fluorescein (HEX) or 5-carboxy-fluorescein (FAM) joint and reverse primer, 0.2 U of Taq polymerase, 0.2 mM dNTPs, and 1 × PCR buffer with 2 mM Mg2+. DNA amplification procedure was described previously (Wu et al. 2018a). The amplification products were analyzed on a capillary electrophoresis apparatus (SMYG2016-JL028-1, SoftGenetics LLC, State College, PA) equipped with the software GeneMarker.
Construction of a high-resolution linkage map of Pm52
The software Mapmaker/Exp Version 3.0b was used to establish the linkage relationship between markers and the target resistance gene (Lincoln et al. 1993). The Kosambi function was used to determine the genetic distances. The logarithm of the odd ratio (LOD) threshold of 3.0 was used, and the maximum distance between markers was 50.0 cM.
Physical mapping and gene annotation of the Pm52 goal interval
The sequences of the markers Xicssl326 and Xicscl795 that flanked Pm52 were used as queries to search against the Chinese Spring whole genomic assembly sequences (IWGSC WGS v1.1, NRGene DeNovoMAGIC, Seq Repository of Wheat Portal on URGI, INRA, France). The information on Chinese Spring genomic sequence was used to identify the genes that were included in the interval between the two Pm52-flanking markers. Then, those genes were used as queries to blast against the genomic sequences of Ae. tauschii (http://aegilops.wheat.ucdavis.edu/ATGSP/index.php), T. turgidum ssp. dicoccoides (Avni et al. 2017), and T. urartu (http://www.mbkbase.org/Tu/).
Results
Development of polymorphic EST-SSR markers linked to Pm52
Previously, Pm52 was located in the bin 2BL2-0.36-0.50 of chromosome arm 2BL, so the EST sequences in this interval were aligned with the barley genomic sequence. It was determined that this interval was collinear with a 46.5-Mb region on barley chromosome 2. Based on the wheat EST sequences that felt in this collinear region, 83 pairs of SSR primers were designed (Table S1). Among them, four EST-SSR markers Xicsl30, Xicsl34, Xicsl54, and Xicsl62 were linked to Pm52, but they were all located on the same side of the target gene when examined using a subpopulation consisting of 165 F2:3 families from the Liangxing 99 × Zhongzuo 9504 cross (Fig. 1a).

Genetic linkage map and physical map of Chinese Spring genomic sequence flanking the Pm52 locus. a Primary genetic linkage map of Pm52 using the subpopulation. b The high-resolution genetic map of Pm52. c The corresponding physical location of the polymorphic linkage markers of Pm52, and diagram of selection physical interval to further design SSR primers. The blue oval frame shows the interval (581167165–583268161) that was used to design 176 primer pairs. The green oval frame shows the interval (586243207–588134752) that was used to design 178 primer pairs. The orange oval frame shows the interval (591820638–596417733) used to design 409 primer pairs
Development of SSR markers based on comparative genomics analysis
The coding sequences of collinear barley genomic sequence were used as queries to search against the Chinese Spring 454 contigs (Brenchley et al. 2012). Based on sequences of the homologous contigs or scaffolds on chromosome arm 2BL, 87 pairs of SSR primers were designed (Table S2). Pm52 was flanked by markers Xicsl90 and Xicsl163 as verified using the subpopulation, and the genetic distances between these markers and the target gene were 1.9 cM and 1.9 cM, respectively (Fig. 1a).
The sequences of markers Xicsl90 and Xicsl163 were used as queries to blast against the Ae. tauschii SNP database and the Brachypodium, rice, and sorghum genome sequences. This interval was collinear with Ae. tauschii chromosome 2 (AT2D1947-AT2D2020), Brachypodium chromosome 5 (Bradi5g16920–Bradi5g18910), rice chromosome 4 (Os04g45340–Os04g48416), and sorghum chromosome 6 (Sb06g023780–Sb06g025980). Based on the Chinese Spring 454 contigs or scaffold sequences of chromosome arm 2BL that corresponded with the orthologus genes, 178 SSR markers were developed (Table S3). Markers Xicsl204, Xicsl224, Xicsl234, Xicsl236, Xicsl275, Xicsl306, and Xicsl335 were linked to Pm52, and Pm52 was localized in the genetic region between markers Xicsl62 and Xicsl163 at genetic distances of 1.3 cM and 1.9 cM, respectively (Fig. 1a).
Identification and phenotyping of the recombinant F2 plants from the cross Liangxing 99 × Zhongzuo 9504
The Pm52-flanking markers Xicsl163 and Xicsl62 were used to identify recombinants in the F2 mapping population of the Liangxing 99 × Zhongzuo 9504 cross. Both markers were able to clearly differentiate different genotypes of the F2 plants using the capillary electrophoresis method (Fig. 2). A total of 344 recombinant plants were identified from 8820 F2 plants. The F2:3 families produced by selfing the recombinant F2 plants were phenotyped using Bgt isolate E20. Among them, 65 F2:3 families were resistant with IT 0 or 1, 130 susceptible with IT 3 or 4, and the remaining 149 were heterozygous with both resistant and susceptible plants within a family. The DNA from the corresponding recombinant F2 plants was used in the subsequent study for finely mapping Pm52.

Amplification banding types of markers Xicsl62 (a) and Xicsl163 (b) by the capillary electrophoresis apparatus. R: resistant; S: susceptible; H: heterozygous
Development of markers tightly linked to Pm52 and construction of a high-resolution genetic linkage map
The sequences of Xicsl62 and Xicsl163 were blasted against the whole genomic assembly sequences of Chinese Spring (IWGSC WGS v1.1, NRGene DeNovoMAGIC, Seq Repository of Wheat Portal on URGI, INRA, France), and the two markers spanned a region of about 40 Mb (556678110–596417752) (Fig. 1c). Then, all the genomic sequences of Chinese Spring in this interval were used to design SSR markers, and 409 SSR primer pairs were designed based on the 5-Mb (591820638–596417733) sequence that extended from Xicsl163 to Pm52 (Fig. 1c(a) and Table S4). After examining the polymorphisms between the parental cultivars as well as the contrasting DNA bulks, five polymorphic markers Xicscl395, Xicscl437, Xicscl445, Xicscl172, and Xicscl174 were located between Xicsl163 and Pm52 and were 0.46, 0.49, 0.64, 0.66, and 0.71 cM distal to Pm52, respectively (Fig. 1b).
Since Pm52 was located between Xicscl395 and Xicsl54, 163 SSR primer pairs were designed based on the 2-Mb (58401490–58632689) segment of the sequences that were 12 Mb away from marker Xicsl54 in the direction toward Pm52 (Table S5). In addition, 178 SSR primer pairs were developed based on the 2 Mb (586243207–588134752) of sequences that extended from marker Xicscl395 toward Pm52 (Fig. 1c(b) and Table S6). Polymorphic markers Xicscl795, Xicscl817, and Xicscl726 were placed 0.12, 0.17, and 0.29 cM distal to Pm52, respectively (Figs. 1b and 3a). Pm52 was then flanked by Xicscl795 and Xicsl54 at distances of 0.12 and 0.45 cM, respectively. So, 176 SSR primer pairs were designed based on the sequences starting from a site 3 Mb distant to Xicscl795, which extended 2 Mb (581167165–583268161) toward gene Pm52 (Fig. 1c(c) and Table S7). Xicssl326 was closely linked to Pm52 (Figs. 1b and 3b). Based on this high-resolution genetic linkage map, Pm52 was flanked by markers Xicssl326 and Xicscl795, at genetic distances of 0.09 and 0.12 cM, respectively (Fig. 1b). The genotypes of 130 recombinant plants in the genetic interval between markers Xicsl54 and Xicscl726 are shown in Table 1.

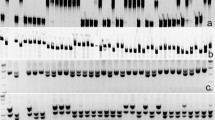
Amplification patterns of Pm52-flanked SSR markers Xicscl795 (a) and Xicssl326 (b) developed from the Chinese Spring genomic sequence in the parents and selected F2:3 families of Liangxing 99 × Zhongzuo 9504 cross in 8% silver-stained non-denaturing polyacrylamide gels. Lane M: DL2000 (Tiangen Biotech Co., Beijing, China); lane 1: Liangxing 99; lane 2: Zhongzuo 9504; lanes 3–8: homozygous resistant F2:3 families; lanes 9–14: homozygous susceptible F2:3 families; and lanes 15–20: heterozygous F2:3 families. Arrows indicate the polymorphic bands that are specific for Pm52
Physical mapping, comparative genomics analysis and the gene annotation of Pm52 goal interval
To determine the physical locations of the Pm52-linked markers, the sequences of all markers anchored in the high-resolution genetic map were used to blast against the Chinese Spring genomic sequence, and the relative physical positions of those markers were generally consistent with the genetic linkage map (Fig. 1). The genetic region of markers Xicssl326 and Xicscl795 corresponded to a 5.6-Mb (581242466–586768319) genomic interval in the Chinese Spring reference genome and contained 36 annotated protein-coding genes (Table S8, Fig. 4b).

Genetic linkage map of the powdery mildew resistance gene Pm52 (a), and genes among Chinese Spring wheat (b), Ae. tauschii (c), T. turgidum ssp. dicoccoides (d) and T. urartu (e) in the corresponding genomic interval between the Pm52-flanking markers
The corresponding orthologous genomic sequences on Ae. tauschii 2DL, T. turgidum ssp. dicoccoides 2BL and T. urartu 2AL were annotated to detect orthologous gene pairs. This interval on Chinese Spring 2BL had a collinear relationship with a 6.5-Mb (491433391–497983654) genomic region on the chromosome 2DL of Ae. tauschii with 31 orthologous genes, a 5.6-Mb (579618841–584257015) region on chromosome 2BL of T. turgidum ssp. dicoccoides with 26 orthologous genes, and a 4.8-Mb (619434655–624213456) region on chromosome 2AL of T. urartu with 22 orthologous genes; however, the genomic region of T. urartu on 2AL did not well collinear with T. aestivum 2BL with a large segment inversion (Table S8, Fig. 4).
Seven disease resistance-associated genes were identified in the collinear genomic regions (Table S8). Among them, three genes encode for kinase family proteins, one for the WRKY transcription factor, and the other three for cysteine-rich receptor-kinase-like proteins. The three kinase proteins (i.e., TraesCS2B02G408700.1, TraesCS2B02G409700.1, and TraesCS2B02G411000.1) on Chinese Spring 2BL have three orthologs in Ae. tauschii 2DL and T. urartu 2AL, but only two orthologs on T. turgidum ssp. dicoccoides 2BL. The WRKY transcription factor was only annotated on Chinese Spring 2BL and Ae. tauschii 2DL, but not on T. turgidum ssp. dicoccoides 2BL or T. urartu 2AL. However, the three cysteine-rich receptor-kinase-like proteins on Chinese Spring 2BL detected one and two orthologs on Ae. tauschii 2DL and one T. urartu 2AL, and none on T. turgidum ssp. dicoccoides 2BL.
Validation of the Pm52-linked markers in the cultivars derived from Liangxing 99
The two Pm52-linked markers Xicsl234 and Xicscl817 were used to analyze three wheat cultivars that were derived from Liangxing 99 and have been phenotyped previously. Zhongxinmai 99 and DH51302 produced identical banding patterns to Liangxing 99, indicating that they may carry Pm52. Heng 10-5039 amplified different banding patterns with these markers from Liangxing 99, indicating the absence of Pm52. This result is consistent with the phenotyping tests with 27 Bgt isolates in a previous study (Zou et al. 2017), and demonstrates that markers Xicsl234 and Xicscl817 are able to detect Pm52 in the progeny cultivars of Liangxing 99.
Discussion
The fine mapping of Pm52 involved the use of SSR mapping, comparative genomics analysis, and wheat reference genome sequencing. It was difficult to identify a sufficient number of polymorphic genomic SSRs linked to the target gene from the available SSR primer pairs, so four EST-SSR markers Xicsl30, Xicsl34, Xicsl54, and Xicsl62 were developed that were linked to Pm52 based on the EST sequences on the wheat chromosome arm 2BL. Then, the sequences of those markers were used as queries to blast against barley genomic sequence to find the collinear genomic region on chromosome arm 2HL. An additional nine polymorphic SSR markers linked to Pm52 were developed based on the results of comparative genomics analysis. Pm52 was placed in a 3.2-cM genetic interval using the subpopulation, which corresponded to a 40-Mb genomic interval (556678090–596417733) on the Chinese Spring 2BL genomic sequences. Based on those sequences, nine gene-linked SSR markers were developed and a new high-resolution linkage genetic map was constructed using the 344 recombinants from the mapping population of Liangxing 99 × Zhongzuo 9504 cross containing 8820 F2 plants identified by the Pm52-flanking markers Xicsl163 and Xicsl62 with genetic distance of 1.85 cM. In the newly developed genetic linkage map, Pm52 was re-localized into a 0.21-cM genetic interval between markers Xicssl326 and Xicscl795 at genetic distances of 0.09 and 0.12 cM, respectively.
Results of the present study demonstrated that the comparative genomics approach, which takes advantage of the genomic synteny among wheat and other cereal crops, is effective for developing molecular markers for the fine mapping of disease resistance genes in wheat. However, the genomic sequences of gramineous crops such as barley, rice, sorghum, as well as B. distachyon, were apparently distinct from that of hexaploid wheat. The efficiency of marker development would not be high if homologous sequences shared between these species were used as template sequences. During the saturation of the genetic linkage map of Pm52, only 13 pairs of primers were polymorphic out of 348 tested primer pairs. The release of the diploid, tetraploid, and hexaploid wheat genome sequences, in comparison with the barley, rice, sorghum, and B. distachyon genome sequences, improved comparative genomics analysis and marker development. During the marker development, the genomic region of Pm52 on 2BL was shown to have good synteny with Ae. tauschii chromosome arm 2DL and T. turgidum ssp. dicoccoides chromosome arm 2BL. So, they were useful for the further encryption of the genetic map. As Pm52 originates from common wheat cultivar Liangxing 99, it should be relatively easy to narrow the interval around Pm52 based on the improvements of the wheat reference genome sequences.
The Pm52 genomic interval was thus reduced to 5.6-Mb interval of the Chinese Spring 2BL containing 36 predicted protein-coding genes. Seven of the annotated genes associated with plant disease resistance, including kinase family proteins, WRKY transcription factor, and cysteine-rich receptor-kinase-like proteins, were observed in this genomic interval (Table S8). Zuo et al. (2014) reported the map-based cloning of qHSR1 from maize (Zea mays L.) and found its encoding product spanned the plasma membrane, potentially serving as a wall-associated kinase to perceive and transduce extracellular signals. The rice WRKY transcription factor gene OsWRKY11 was reported to regulate the defense-associated genes (Lee et al. 2018). A group of the LecRK-VI.2-responsive cysteine-rich receptor-like kinase genes CRK4, CRK6, and CRK36 enhanced a pattern-triggered immunity response that was associated with resistance of tomato (Solanum lycopersicum L.) to a virulent bacterial strain of Pseudomonas syringae pv. tomato DC3000 (Yeh et al. 2015). Previously, only five genes for wheat powdery mildew resistance have been isolated, i.e., Pm3b (Yahiaoui et al. 2004), Pm8 (Hurni et al. 2013), Pm2 (Sánchez-Martín et al. 2016), Pm60 (Zou et al. 2018), and Pm21 (He et al. 2018; Xing et al. 2018). All of them were the nucleotide-binding site and leucine-rich repeat (NBS-LRR) type of resistance genes. In addition to the NBS-LRR proteins, other genes with different mechanisms that function in resistant responses to diseases in plants have been characterized (Kourelis and van der Hoom 2018; Klymiuk et al. 2018).
Pm52 may use a different regulation mechanism from the NBS-LRR proteins for powdery mildew resistance. The final cloning of Pm52 is required to ensure a full understanding of its underlying mechanism. Another possibility is that the target gene Pm52 was not present in the genomic sequence of the Chinese Spring 2BL corresponding region. In that case, gene editing technology that clusters regularly interspaced short palindromic repeats-associated system (CRISPR-Cas) can be used to verify whether the gene exists in the final candidate interval (Li et al. 2013). Alternatively, construction and sequencing of a bacterial artificial chromosome (BAC) library and sequencing of the target gene region of BAC physical map contigs also can further predict the candidate genes. A new technology, MutChromSeq on the basis of flow sorting and sequencing of mutant chromosomes, has been used to isolate Pm2 (Sánchez-Martín et al. 2016). All these technologies will facilitate the map-based cloning of Pm52.
The utilization of a resistance gene in breeding and agriculture depends on not only its effectiveness against the pathogen, but also the genetic background of its host cultivar. Some genes, such as those that reside in lines with poor agronomic performance, need a pre-breeding step to break the linkages between the target genes and the deleterious ones to optimize their genetic backgrounds by hybridization, repeated backcrossing, and marker-assisted selection (MAS). A time-consuming procedure is required to overcome the linkage drag before a resistance gene can be used by breeders in developing disease-resistant wheat cultivars (Summers and Brown 2013). Other sources of resistance, such as those that are identified in commercial wheat cultivars, are associated with desirable traits with promising agronomic performance and quality property. This circumvents the steps for improving the genetic background of a resistance gene.
A number of cultivars have been commercialized in different provinces, for example, Shi 4366 (Liangxing 99/Shiyou 17, 2015), Zhongxinmai 99 (Liangxing 99/222, 2016), Ji 738 (Gao 9618/Liangxing 99, 2016), Wanfeng 126 (Liangxing 99/Yannong 21, 2018), and Xitu 555 (Heng 4164/Han 6172//Liangxing 99, 2018) in Hebei province, Liangxing 68 (Liangxing 872/Liangxing 99, 2018) and Xinruimai 29 (Liangxing 99/Yan 5072, 2018) in Shandong province, and Wankenmai 1221 (Liangxing 99/Hui 0208, 2016) in Anhui province. Some of the cultivars developed using Liangxing 99 as a parent, i.e., Zhongxinmai 99, may carry Pm52 as revealed by molecular marker detection and tests for disease resistance (Zou et al. 2017). However, cultivars derived from Liangxing 99 do not necessarily carry Pm52. Zou et al. (2017) reported that four of the ten Liangxing 99-derived cultivars may carry Pm52, but the other six cultivars may not carry this gene. The identification of molecular markers will facilitate the identification of Pm52 in the Liangxing 99-derived cultivars.
Author contribution statement
HJL, PW, and JL conceived and designed the study. PW and JH conducted the experiments. PW, JL, and ZL analyzed the data. JZ, DQ, YQ, YL, TL, HZ, LY, and HWL performed the phenotypic tests and other researches involved in this study. PW and HJL wrote the manuscript with contributions from YZ, ZL, and ZZ.
References
Avni R, Nave M, Barad O, Baruch K, Twardziok SO, Gundlach H, Hale I, Mascher M, Spannagl M, Wiebe K, Jordan KW, Golan G, Deek J, Ben-Zvi B, Ben-Zvi G, Himmelbach A, MacLachlan RP, Sharpe AG, Fritz A, Ben-David R, Budak H, Fahima T, Korol A, Faris JD, Hernandez A, Mikel MA, Levy AA, Steffenson B, Maccaferri M, Tuberosa R, Cattivelli L, Faccioli P, Ceriotti A, Kashkush K, Pourkheirandish M, Komatsuda T, Eilam T, Sela H, Sharon A, Ohad N, Chamovitz DA, Mayer KFX, Stein N, Ronen G, Peleg Z, Pozniak CJ, Akhunov ED, Distelfeld A (2017) Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357:93–97
Brenchley R, Spannagl M, Pfeifer M, Barker GLA, D’Amore R, Allen AM, McKenzie N, Kramer M, Kerhornou A, Bolser D, Kay S, Waite D, Trick M, Bancroft I, Gu Y, Huo NX, Luo M-C, Sehgal S, Gill B, Kianian S, Anderson O, Kersey P, Dvorak J, McCombie WR, Hall A, Mayer KFX, Edwards KJ, Bevan MW, Hall N (2012) Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491:705–710
Chapman JA, Mascher M, Buluç A, Barry K, Georganas E, Session A, Strnadova V, Jenkins J, Sehgal S, Oliker L, Schmutz J, Yelick KA, Scholz U, Waugh R, Poland JA, Muehlbauer GJ, Stein N, Rokhsar DS (2015) A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome. Genome Biol 16:26
He HG, Zhu SY, Zhao RH, Jiang ZN, Ji YY, Ji J, Qiu D, Li HJ, Bie TD (2018) Pm21, encoding a typical CC-NBS-LRR protein, confers broad-spectrum resistance to wheat powdery mildew disease. Mol Plant 11:879–882
Hu TZ, Li HJ, Liu ZJ, Xie CJ, Zhou YL, Duan XY, Jia X, You MS, Yang ZM, Sun QX, Liu ZY (2008) Identification and molecular mapping of the powdery mildew resistance gene in wheat cultivar Yumai 66. Acta Agron Sin 34:545–550
Hurni S, Brunner S, Buchmann G, Herren G, Jordan T, Krukowski P, Wicker T, Yahiaoui N, Mago R, Keller B (2013) Rye Pm8 and wheat Pm3 are orthologous genes and show evolutionary conservation of resistance function against powdery mildew. Plant J 76:957–969
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
International Wheat Genome Sequencing Consortium (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345:1251788
Klymiuk V, Yaniv E, Huang L, Raats D, Fatiukha A, Chen SS, Feng LH, Frenkel Z, Krugman T, Lidzbarsky G, Chang W, Jääskeläinen MJ, Schudoma C, Paulin L, Laine P, Bariana H, Sela H, Saleem K, Sørensen CK, Hovmøller MS, Distelfeld A, Chalhoub B, Dubcovsky J, Korol AB, Schulman AH, Fahima T (2018) Cloning of the wheat Yr15 resistance gene sheds light on the plant tandem kinase-pseudokinase family. Nat Commun 9:3735
Kourelis J, van der Hoom RAL (2018) Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell 30:285–299
Lazo GR, Chao S, Hummel DD, Edwards H, Crossman CC, Lui N, Matthews DE, Carollo VL, Hane DL, You FM, Butler GE, Miller RE, Close TJ, Peng JH, Lapitan NLV, Gustafson JP, Qi LL, Echalier B, Gill BS, Dilbirligi M, Randhawa HS, Gill KS, Greene RA, Sorrells ME, Akhunov ED, Dvořák J, Linkiewicz AM, Dubcovsky J, Hossain KG, Kalavacharla V, Kianian SF, Mahmoud AA, Miftahudin Ma X-F, Conley EJ, Anderson JA, Pathan MS, Nguyen HT, McGuire PE, Qualset CO, Anderson OD (2004) Development of an expressed sequence tag (EST) resource for wheat (Triticum aestivum L.). Genetics 168:585–593
Lee H, Cha J, Choi C, Choi N, Ji HS, Park SR, Lee S, Hwang DJ (2018) Rice WRKY11 plays a role in pathogen defense and drought tolerance. Rice 11:5–16
Li HJ, Wang XM, Song FJ, Wu CP, Wu XF, Zhang N, Zhou Y, Zhang XY (2011) Response to powdery mildew and detection of resistance genes in wheat cultivars from China. Acta Agron Sin 37:943–954
Li DL, Qiu ZW, Shao YJ, Chen YT, Guan YT, Liu MZ, Li YM, Gao N, Wang LR, Lu XL, Zhao YX, Liu MY (2013) Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biol 31:681–683
Lincoln SE, Daly MJ, Lander ES (1993) Constructing linkage maps with MAPMAKER/Exp version 3.0: a tutorial reference manual, 3rd edn. Whitehead Institute for Medical Research, Cambridge
Ling HQ, Ma B, Shi XL, Liu H, Dong LL, Sun H, Cao YH, Gao Q, Zheng SS, Li Y, Yu Y, Du HL, Qi M, Li Y, Lu HW, Yu H, Cui Y, Wang N, Chen CL, Wu HL, Zhao Y, Zhang JC, Li YW, Zhou WJ, Zhang BR, Hu WJ, van Eijk MJT, Tang JF, Witsenboer HMA, Zhao SC, Li ZS, Zhang AM, Wang DW, Liang CZ (2018) Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 557:424–447
Liu Z, Sun Q, Ni Z, Yang T (1999) Development of SCAR markers linked to the Pm21 gene conferring resistance to powdery mildew in common wheat. Plant Breed 118:215–219
Liu WC, Liu ZD, Huang C, Lu MH, Liu J, Yang QP (2016) Statistics and analysis of crop yield losses caused by main disease and insect pests in recent 10 years. Plant Protect 42:1–9
Luo PG, Luo HY, Chang ZJ, Zhang HY, Zhang M, Ren ZL (2009) Characterization and chromosomal location of Pm40 in common wheat: a new gene for resistance to powdery mildew derived from Elytrigia intermedium. Theor Appl Genet 118:1059–1064
Luo M-C, Gu YQ, Puiu D, Wang H, Twardziok SO, Deal KR, Huo NX, Zhu TT, Wang L, Wang Y, McGuire PE, Liu SY, Long H, Ramasamy RK, Rodriguez JC, Van SL, Yuan LX, Wang ZZ, Xia ZQ, Xiao LC, Anderson OD, Ouyang SH, Liang Y, Zimin AV, Pertea G, Qi P, Bennetzen JL, Dai XT, Dawson MW, Müller HG, Kugler K, Rivarola-Duarte L, Spannagl M, Mayer KFX, Lu FH, Bevan MW, Leroy P, Li PC, You FM, Sun QX, Liu ZY, Lyons E, Wicker T, Salzberg SL, Devos KM, Dvořák J (2017) Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551:498–502
Ma PT, Xu HX, Xu YF, Song LP, Liang SS, Sheng Y, Han GH, Zhang XT, An DG (2018) Characterization of a powdery mildew resistance gene in wheat breeding line 10V-2 and its application in marker-assisted selection. Plant Dis 102:925–931
McIntosh RA, Dubcovsky J, Rogers WJ, Morris C, Appels R, Xia XC (2014) Catalogue of gene symbols for wheat: 2013–2014 supplement. http://shigen.nig.ac.jp/wheat/komugi/genes/macgene/supplement2013.pdf. Accessed 10 Apr 2018
Morgounov A, Tufan HA, Sharma R, Akin B, Bagci A, Braun H-J, Kaya Y, Keser M, Payne TS, Sonder K, McIntosh R (2012) Global incidence of wheat rusts and powdery mildew during 1969–2010 and durability of resistance of winter wheat variety Bezostaya 1. Eur J Plant Pathol 132:323–340
Ouyang SH, Zhang D, Han J, Zhao X, Cui Y, Song W, Huo NX, Liang Y, Xie JZ, Wang ZZ, Wu QH, Chen YX, Lu P, Zhang DY, Wang LL, Sun H, Yang T, Keeble-Gagnere G, Appels R, Doležel J, Ling HQ, Luo MC, Gu YQ, Sun QX, Liu ZY (2014) Fine physical and genetic mapping of powdery mildew resistance gene MlIW172 originating from wild emmer (Triticum dicoccoides). PLoS ONE 9:e100160
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang HB, Wang XY, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, WangY Zhang LF, Carpita NC, Freeling M, Gingle AR, Hash CT, Keller B, Klein P, Kresovich S, McCann MC, Ming R, Peterson DG, Mehboob-ur-Rahman Ware D, Westhoff P, Mayer KFX, Messing J, Rokhsar DS (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
Sánchez-Martín J, Steuernagel B, Ghosh S, Herren G, Hurni S, Adamski N, Vrána J, Kubaláková M, Krattinger SG, Wicker T, Doležel J, Keller B, Wulff BBH (2016) Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol 17:221–227
Summers RW, Brown JKM (2013) Constraints on breeding for disease resistance in commercially competitive wheat cultivars. Plant Pathol 62:115–121
Tagliaro F, Manetto G, Crivellent F, Smith FP (1998) A brief introduction to capillary electrophoresis. Forensic Sci Int 92:75–88
Tang XL, Cao XR, Luo Y, Ma ZH, Xu XM, Jiang YY, Fan JR, Zhou YL (2017) Effects of climate change on epidemics of powdery mildew in winter wheat in China. Plant Dis 101:1753–1760
The International Barley Genome Sequencing Consortium (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491:711–717
The International Brachypodium Initiative (2010) Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463:763–768
The International Wheat Genome Sequencing Consortium (IWGSC) (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:eaar7191
Wang ZZ, Li HW, Zhang DY, Guo L, Chen JJ, Chen YX, Wu QH, Xie JZ, Zhang Y, Sun QX, Dvorak J, Luo MC, Liu ZY (2015) Genetic and physical mapping of powdery mildew resistance gene MlHLT in Chinese wheat landrace Hulutou. Theor Appl Genet 128:365–373
Wu PP, Xie JZ, Hu JH, Qiu D, Liu ZY, Li JT, Li MM, Zhang HJ, Yang L, Liu HW, Zhou Y, Zhang ZJ, Li HJ (2018a) Development of molecular markers linked to powdery mildew resistance gene Pm4b by combining SNP discovery from transcriptome sequencing data with bulked segregant analysis (BSR-seq) in wheat. Front Plant Sci 9:95
Wu JH, Zeng QD, Wang QL, Liu SJ, Yu SZ, Mu JM, Huang S, Sela H, Distelfeld A, Huang LL, Han DJ, Kang ZS (2018b) SNP-based pool genotyping and haplotype analysis accelerate fine-mapping of the wheat genomic region containing stripe rust resistance gene Yr26. Theor Appl Genet 131:1481–1496
Xie JZ, Wang LL, Wang Y, Zhang HZ, Zhou SH, Wu QH, Chen YX, Wang ZZ, Wang GX, Zhang DY, Zhang Y, Hu TZ, Liu ZY (2017) Fine mapping of powdery mildew resistance gene PmTm4 in wheat using comparative genomics. J Integr Agric 16:540–550
Xing LP, Hu P, Liu JQ, Witek K, Zhou S, Xu JF, Zhou WH, Gao L, Huang ZP, Zhang RQ, Wang XE, Chen PD, Wang HY, Jones JDG, Karafiátová M, Vrána J, Bartoš J, Doležel J, Tian YC, Wu YF, Cao AZ (2018) Pm21 from Haynaldia villosa encodes a CC-NBS-LRR protein conferring powdery mildew resistance in wheat. Mol Plant 11:874–878
Xu WG, Li CX, Hu L, Zhang L, Zhang JZ, Dong HB, Wang GS (2010) Molecular mapping of powdery mildew resistance gene PmHNK in winter wheat (Triticum aestivum L.) cultivar Zhoumai 22. Mol Breed 26:31–38
Xu WG, Li CX, Hu L, Wang HW, Dong HB, Zhang JZ, Zan XC (2011) Identification and molecular mapping PmHNK54: a novel powdery mildew resistance gene in common wheat. Plant Breed 130:603–607
Yahiaoui N, Srichumpa P, Dudler R, Keller B (2004) Genome analysis at different ploidy levels allows cloning of the powdery mildew resistance gene Pm3b from hexaploid wheat. Plant J 37:528–538
Yeh YH, Chang YH, Huang PY, Huang JB, Zimmerli L (2015) Enhanced Arabidopsis pattern-triggered immunity by overexpression of cysteine-rich receptor-like kinases. Front Plant Sci 6:322–333
You FM, Huo NX, Gu YQ, Luo MC, Ma YQ, Hane D, Lazo GR, Dvorak J, Anderson OD (2008) BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics 9:253–265
Zhao ZH, Sun HG, Song W, Lu M, Huang J, Wu LF, Wang XM, Li HJ (2013) Genetic analysis and detection of the gene MlLX99 on chromosome 2BL conferring resistance to powdery mildew in the wheat cultivar Liangxing 99. Theor Appl Genet 126:3081–3089
Zimin AV, Puiu D, Hall R, Kingan S, Clavijo BJ, Salzberg SL (2017) The first near-complete assembly of the hexaploid bread wheat genome, Triticum aestivum. GigaScience 6:1–7
Zou JW, Qiu D, Sun YL, Zheng CX, Li JT, Wu PP, Wu XF, Wang XM, Zhou Y, Li HJ (2017) Pm52: effectiveness of the gene conferring resistance to powdery mildew in wheat cultivar Liangxing 99. Acta Agron Sin 43:332–342
Zou SH, Wang H, Li YW, Kong ZS, Tang DZ (2018) The NB-LRR gene Pm60 confers powdery mildew resistance in wheat. New Phytol 218:298–309
Zuo WL, Chao Q, Zhang N, Ye JR, Tan GQ, Li BL, Xing YX, Zhang BQ, Liu HJ, Fengler KA, Zhao J, Zhao XR, Chen YS, Lai JS, Yan JB, Xu ML (2014) A maize wall-associated kinase confers quantitative resistance to head smut. Nat Genet 47:151–159
Acknowledgements
The authors thank Dr. Robert L. Conner, Morden Research and Developmental Center, Agriculture and Agri-Food Canada, for his critical review of the manuscript. The financial support provided by the National Natural Science Foundation of China (31471491, 31871621, and 31501310), the National Key Research and Development Program of China (2017YFD0101000), the Scientific and Technological Research Project of Henan Province of China (172102110110), and the CAAS Innovation Team is gratefully appreciated.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Evans Lagudah.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wu, P., Hu, J., Zou, J. et al. Fine mapping of the wheat powdery mildew resistance gene Pm52 using comparative genomics analysis and the Chinese Spring reference genomic sequence. Theor Appl Genet 132, 1451–1461 (2019). https://doi.org/10.1007/s00122-019-03291-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03291-7