
Abstract
Key message
The powdery mildew resistance gene Pm58 was traced to a 141.3-kb interval with the co-segregating marker Xkasp68500 in wheat breeding.
Abstract
Pm58 is a powdery mildew resistance gene identified in Aegilops tauschii accession TA1662 and effective in a common wheat background. To finely map Pm58, an F2 population of 676 plants derived from the cross T093 × TA1662 was used for recombinant screening. We obtained 13 recombinants that occurred between the flanking markers Xhnu670 and Xhnu186. Genotyping and phenotyping these recombinant F2:3 families delimited Pm58 to a 0.22-cM interval (Xsts20220–Xkasp61553) on chromosome arm 2DS. The region carrying the Pm58 locus was approximately 141.3-kb, which contained eight annotated genes according to the reference genome sequence of Ae. tauschii AL8/78. Haplotype analysis of 178 Ae. tauschii accessions using the candidate gene-specific markers identified a disease resistance gene AET2Gv20068500 as a candidate for Pm58. Comparative mapping of the Pm58-containing interval revealed two presence/absence variations (PAVs) between AL8/78 and common wheat Chinese Spring. PAV-1 resides in the 3′-end of AET2Gv20068500. The majority of 158 common wheat cultivars (84.8%) displayed the absence of a 14.1-kb fragment in the PAV-1 region, which was confirmed by aligning the targeted genome sequences of the other sequenced Ae. tauschii accessions and common wheat cultivars. A co-segregating marker Xkasp68500 developed from AET2Gv20068500 can distinguish TA1662 from all randomly selected common wheat cultivars and will be instrumental for tracking Pm58 in breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wheat (Triticum aestivum L.) powdery mildew, caused by the fungus Blumeria graminis (DC.) Speer f. sp. tritici emend. É. J. Marchal (Bgt), is a globally devastating disease particularly damaging in regions with high humidity and cool to moderate temperatures (Te Beest et al. 2008). This disease can cause reductions in the yield components of tiller number, grain number, and kernel weight (Bowen et al. 1991), and the resulting losses in yield are 30 to 40% in heavily epidemic years (Conner et al. 2003; Singh et al. 2016). Deployment of host resistance is the preferred choice for managing this disease and reducing pesticide dependency (Kang et al. 2020). Race-specific resistance is the basis of wheat powdery mildew resistance. Many cloned powdery mildew resistance genes encode nucleotide-binding leucine-rich-repeat-containing (NLR) immune receptors that can recognize pathogen effectors (Bourras et al. 2015, 2019; Praz et al. 2017; Hewitt et al. 2021). To prolong the usefulness of race-specific resistance, it is necessary to continuously broaden resistance gene pools, stack genes with different resistance modes, and allocate reasonable growing regions (Mundt 2002; Kang et al. 2020).
Wild relatives of common wheat (AABBDD, 2n = 6× = 42) are frequently used as sources of disease resistance despite the presences of crossing barriers and incompatibility (Kishii 2019). When a new race of the fungus Puccinia graminis f. sp. tritici (Ug99 or TTKSK) and wheat blast (Magnaporthe oryzae Triticum) appeared and threatened the global wheat production, breeders sought and deployed resistance genes in Aegilops species (Olson et al. 2013; Cruz et al. 2016). Aegilops tauschii Coss. (DD, 2n = 2x = 14), the D-genome progenitor of common wheat (Huang et al. 2002), carries several genes for resistance to powdery mildew (Lutz et al. 1995; Miranda et al. 2006, 2007; Sun et al. 2006; Li et al. 2011; Wiersma et al. 2017). Some of them, such as Pm2a, Pm19, Pm34, Pm35 and Pm58, have been transferred into common wheat through indirect or direct crossing approaches (Kishii 2019). Among these genes, only Pm2a has been cloned by mutant chromosome sequencing (Sánchez-Martín et al. 2016).
The powdery mildew resistance genes from the wild species or wheat relatives, such as Pm12 (Jia et al. 1996), Pm16 (Chen et al. 2005), Pm20 (Friebe et al. 1994), and Pm62 (Zhang et al. 2018), can usually provide durable resistance to Bgt pathogens. However, these genes are more difficult to directly use in breeding due to higher probabilities of having inferior agronomic characteristics dragged along with the target loci (Summers and Brown 2013). To eliminate the undesirable linkages associated with resistance genes, it is necessary to isolate them and develop their diagnostic markers. The releases of reference genome sequences of T. urartu (AA; Ling et al. 2018), T. turgidum ssp. dicoccoides (AABB; Avni et al. 2017), T. turgidum ssp. durum (AABB; Maccaferri et al. 2019), Ae. tauschii (DD; Luo et al. 2017), and Secale cereale (RR; Li et al. 2021) have greatly facilitated the development of new markers and map-based cloning of resistance genes from the wild relatives of wheat. Furthermore, re-sequencing of mapping parents has been used to identify single nucleotide polymorphisms (SNPs) and insertion/deletion variations (InDels) that have contributed to fine map the targeted genes (Qiu et al. 2021).
Powdery mildew resistance gene Pm58 was identified in Ae. tauschii accession TA1662. It was previously mapped to an 8.6-Mb interval flanked by Kompetitive allele-specific PCR (KASP) markers K-TP127986 and K-TP61544 using 96 BC2F4 introgression lines and was confirmed to be effective at both seedling and adult stages (Wiersma et al. 2017, 2018). The objectives of this study were to (1) further narrow down the genomic region harboring the Pm58 locus and identify its candidate genes, and (2) develop the co-segregating markers of Pm58 allowing efficiently tracking the resistance allele in the hexaploid backgrounds.
Materials and methods
Plant materials
TA1662 (carrying Pm58) was crossed with T093, which was collected from Henan Province of China and was highly susceptible to powdery mildew. T093 and TA1662 were phylogenetically clustered into two distinct lineages of Ae. tauschii, i.e., lineage 1 (L1) and lineage 2 (L2), respectively (Zhou et al. 2021). A total of 676 F2 plants and derived F2:3 families were used for fine mapping of Pm58. One hundred and seventy-eight Ae. tauschii accessions used for haplotype analysis were collected from diverse regions of China, the Middle East and Central Asia (Supplementary Table 1). Furthermore, 158 worldwide common wheat cultivars were randomly selected and used to examine the situation of a presence/absence variation (PAV) resided in the Pm58 region and detect the polymorphisms of the Pm58 candidate gene-specific markers in the hexaploid backgrounds (Supplementary Table 2).
Powdery mildew evaluations
The prevalent Chinese Bgt isolate E09 (Lu et al. 2020) was used to inoculate the mapping populations and two parents TA1662 and T093. Each F1 and F2 seed was planted in a single cell of a 72-cell tray (6 × 12), and 20 to 25 seeds from each F2:3 family were sown in five cells of a 50-cell tray (5 × 10). For fine mapping of Pm58, at least 25 F3 plants were tested to confirm the phenotypes and genotypes of the corresponding F2 recombinants. TA1662 and T093 were used as resistant and susceptible controls, respectively. All seedlings at the two-leaf stage were inoculated with fresh conidiospores and incubated under controlled chamber conditions with a daily cycle of 16 h of light at 22 °C and 8 h of darkness at 18 °C. Infection types (ITs) were recorded 5 to 7 d post-inoculation when T093 displayed severe symptoms using a scale from 0 to 4, in which 0, 0;, 1, and 2 were classified as resistant, and 3 and 4 were considered as susceptible (Lu et al. 2020).
Re-sequencing and SNP/InDel calling
The genomic DNA of TA1662 and T093 was extracted using a standard CTAB method (Huang et al. 2000), and 1 μg DNA per sample was fragmented by sonication to an average size of 300–400 bp. The libraries containing selected fragments were sequenced using a BGISEQ-500 platform with a paired-end read length of 150 bp. We filtered raw data using SOAPnuke (Chen et al. 2018) and obtained clean reads with sequencing depths of more than 20 × for each accession.
The remaining high-quality reads were mapped to the Ae. tauschii AL8/78 reference genome (Luo et al. 2017) using the Burrows-Wheeler Alignment tool (BWA, Li and Durbin 2009). The duplicated reads were marked and removed using the Genome Analysis Toolkit (GATK, McKenna et al. 2010). The genomic variants (SNPs and InDels) were identified with the GATK HaplotypeCaller module. Variants’ genic positions (e.g., intragenic, upstream, downstream, and intergenic) and associated functions were further annotated with ANNOVAR software (Wang et al. 2010) based on AL8/78 genome annotation information.
Marker development and linkage mapping
Pm58 has been previously mapped to the 13.5–22.1 Mb region on chromosome 2DS (Wiersma et al. 2017). The AL8/78 reference sequence corresponding to the target interval was used to screen for simple sequence repeat (SSR) markers with the Perl script MISA program (https://webblast.ipk-gatersleben.de/misa/). Linkage analysis with 12 newly-developed SSR markers and 179 T093/TA1662 F2 plants delimited Pm58 to the Xhnu670‒Xhnu186 interval on chromosome 2DS. Based on the re-sequencing results of the two mapping parents, the SNPs and InDels resided in the Pm58-containing interval were searched to develop KASP or sequence-tagged site (STS) markers for saturation mapping. All primers were designed with MacVector 11.0 (Accelrys, San Diego, CA, USA). Primer sequences of the markers detecting polymorphisms are listed in Supplementary Tables 3 and 4.
The SSR and STS markers were amplified in a T100™ Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA) according to the following procedures: 95 °C for 3 min; 32 cycles of 95 °C for 15 s, 52–60 °C for 20 s, and 72 °C for 50 s; and a final extension at 72 °C for 10 min. The polymerase chain reaction (PCR) profile contained 7.5 µl of 2 × Taq Plus master Mix II (Vazyme, Nanjing, China), 1.2 µl of primer mix (3 µM for each primer), 1.5 µl (150 ng) of genomic DNA, and 4.8 µl of ddH2O. The PCR products were analyzed by electrophoresis with 8% non-denaturing polyacrylamide gels or 1% agarose gels.
KASP genotyping assays
Twelve SNPs located within the Pm58 region were converted to KASP markers (Supplementary Table 4). Two different tail sequences, FAM (5′-GAAGGTGACCAAGTTCATGCT-3′) and HEX (5′-GAAGGTCGGAGTCAACGGATT-3′), were added to the 5′-end of TA1662 allele (A1)- and T093 allele (A2)-specific primers, respectively. KASP assays were performed in 96-well formats with 10 µl reactions containing 5 µl of 2 × KASP Master mix (LGC Genomics, Middlesex, UK), 0.14 µl of primer mix (12 µM for each allele-specific primer and 30 µM for a common primer), 1.5 µl (150 ng) of genomic DNA, and 3.36 µl of ddH2O. A LightCycler 480 II system (Roche, Indianapolis, IN, USA) was used for PCR thermocycling and fluorescence detection. The cycling programs were referred from Lu et al. (2020).
Physical and comparative mapping
The sequences of the polymorphic markers closely linked to Pm58 were used as queries to search against the AL8/78 reference sequence (Luo et al. 2017). Based on the physical positions of two flanking markers Xsts20220 and Xkasp61553, the Pm58 interval was anchored onto the AL8/78 reference genome. According to the gene annotations of AL8/78 (http://aegilops.wheat.ucdavis.edu/jbrowse/index.html?data=Aet%2Fdata%2F&loc), eight annotated genes residing in the Xsts20220‒Xkasp61553 interval were extracted and used to search for their homologs in the D genome of common wheat cv. Chinese Spring (CS) (IWGSC 2018), using cutoff parameters of E-value ≤ 1E−10, identity ≥ 80%, and a minimum of 100 bp match length.
In the Pm58-containing interval, two PAVs were identified by aligning the targeted genome sequences of AL8/78 and CS, which were confirmed by comparative analysis of the other sequenced Ae. tauschii accessions (Zhou et al. 2021) and common wheat cultivars (Walkowiak et al. 2020; Sato et al. 2021). We also designed two pairs of primers (Supplementary Table 5) to analyze the situation of PAV-1 in 158 common wheat cultivars and 178 Ae. tauschii accessions.
Haplotype analysis
Eight candidate gene-specific markers (Supplementary Table 4) were designed and employed for genotyping 178 Ae. tauschii accessions. For association analysis, 5–10 plants (two-leaf stage) of each Ae. tauschii accession were evaluated for responses to Bgt isolate E09 in a chamber with three replicates.
Statistical analysis
Genetic analysis was conducted using an F2 population derived from T093 × TA1662. The Chi-squared test was performed to test the hypothesis that Pm58 was a dominant gene conferring powdery mildew resistance. The genetic linkage map was constructed with JoinMap 4.0 (Van Ooijen 2006) using the maximum likelihood algorithm and the Kosambi function.
Results
Inheritance of the powdery mildew resistance in TA1662
Aegilops tauschii accession TA1662 was immune to Bgt isolate E09 with IT 0 and T093 was highly susceptible with IT 4 (Fig. 1). To analyze the inheritance of Pm58, we crossed TA1662 with T093 and infected their F1 plants with Bgt E09. The F1 hybrids were highly resistant (IT 0–1), indicating the dominant nature of Pm58. Among the 179 F2 plants, 142 were resistant with IT 0 to 1 and 37 were susceptible with IT 3 to 4, fitting a 3:1 ratio (χ2 = 1.79, P = 0.18). The F2:3 populations segregated as 44 homozygous resistant, 98 segregating, and 37 homozygous susceptible fitting a 1:2:1 segregation ratio (χ2 = 2.16, P = 0.34). These results suggest that Pm58 in TA1662 is a dominant powdery mildew resistance gene.

Powdery mildew reactions of resistant parent TA1662, susceptible parent T093, and their F1 hybrids to Bgt isolate E09. Representative leaves were photographed at 7 d post-inoculation. Scale bar, 5 mm
Rough mapping of Pm58 with SSR markers
Using a population of 96 BC2F4 introgression lines, Pm58 has been delimited to an 8.6-Mb interval flanked by K-TP127986 and K-TP61544 on chromosome 2DS (Wiersma et al. 2017). Based on the AL8/78 reference sequence in the targeted region, 12 SSR markers showed identical polymorphisms between the two parents and two contrasting bulks were developed (Supplementary Fig. 1). Using these newly developed SSR markers, we genotyped 179 F2 plants previously used for inheritance analysis and initially localized Pm58 into a 602.8-kb interval flanked by Xhnu670 and Xhnu186, and co-segregated with Xhnu415 (Fig. 2a).

Genetic and physical maps of the Pm58 region. a Primary mapping of Pm58. b High-density genetic map saturated with the KASP and STS markers. Maps a and b were constructed based on 179 and 676 F2 plants, respectively, derived from T093 × TA1662. Genetic distance is shown on the left side of the maps in cM. c Partial physical map of AL8/78 chromosome 2DS. The physical positions (bp) of molecular markers are indicated on the right side of the physical map
Re-sequencing of TA1662 and T093
To identify more SNPs and InDels resided in the targeted interval, we re-sequenced TA1662 and T093 using a BGISEQ-500 platform with 23.3- and 24.8-fold coverage, respectively. The high-quality reads were mapped against the AL8/78 reference genome to identify genomic variants. In total, 28,835,554 homozygous SNPs and 3,327,243 homozygous InDels were detected between T093 and TA1662, which was similar to the number of variants between T093 and AL8/78 but 3.0-fold higher than that between two L2 accessions TA1662 and AL8/78 (Supplementary Fig. 2a). The SNPs and InDels between T093 and TA1662 were distributed unevenly among the Ae. tauschii chromosomes (Supplementary Fig. 2b). The number of variants per chromosome ranged from 3,933,976 for chromosome 1D to 5,099,799 for chromosome 7D. The SNP/InDel density also varied considerably among the chromosomes and ranged from 7500 variants per Mb for chromosome 2D to 8565 variants per Mb for chromosome 4D. A total of 5295 homozygous variants (including 4577 SNPs and 718 InDels) between the two parents were identified in the targeted 602.8-kb region containing Pm58. Among them, 1694 were located within genes, 341 were within the 1-kb upstream regions of transcription start sites, 259 were within the 1-kb downstream regions of transcription stop sites, and 3001 were in the intergenic regions. These variants facilitate the development of new molecular markers linked to Pm58.
Fine mapping of Pm58 with KASP/STS markers
For saturation mapping of the Pm58 region, 12 polymorphic KASP/STS markers were developed based on the variants resided in the Xhnu670‒Xhnu186 interval (Figs. 3 and 4). These polymorphic markers were used to genotype 676 F2 plants derived from T093 × TA1662, and a high-density genetic map of the 2DS region containing Pm58 was constructed (Fig. 2b). This map was comprised of 15 marker loci and spanned 0.96 cM. The average genetic distance between adjacent markers was about 0.06 cM.

Scatter plots for 12 SNP-derived KASP markers. The blue and green triangles represent TA1662 and T093 alleles, respectively, and the red triangles represent heterozygous alleles in the F2 population derived from T093 × TA1662. The gray dots represent non-template control. Xkasp68100, Xkasp68200, Xkasp68300, Xkasp68400, Xkasp68500, Xkasp68600, and Xkasp68700 were developed according to the SNPs resided in the Pm58 candidate genes (color figure online)

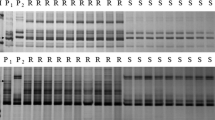
PCR amplification patterns of eight InDel-derived STS markers. R, S, and H indicate homozygous resistant, homozygous susceptible and heterozygous resistant F2 plants, respectively. M is the DNA size standard. Xsts67700 was developed according to an 8-bp InDel in the candidate gene AET2Gv20067700.32
A total of 13 recombinant F2 individuals representing 12 types of genotypes were identified between the flanking markers Xhnu670 and Xhnu186 (Table 1). The F2:3 families from these recombinants were evaluated for resistance to Bgt isolate E09, and five families were homozygous resistant, seven were segregating, and one was homozygous susceptible. With the help of recombinant types 1, 3, and 11, which were identified with Xsts20220 and Xkasp61553, Pm58 was fine mapped to a 0.22-cM Xsts20220–Xkasp61553 interval (Table 1; Fig. 2b).
Noting that only one recombinant individual (P1-390) was identified between Xsts20220 and Pm58, and only two (P1-008 and P2-106) were identified between Pm58 and Xkasp61553 (Table 1), we further checked these three critical crossovers by genotyping and phenotyping at least 40 F2:3 progenies of them. The F3 plants from three critical recombinants showed segregating responses to Bgt isolate E09, which recombined to the genotypes detected by Xsts20220 or Xkasp61553 and co-segregated with Xsts22624, Xkasp26979, Xsts24035, and Xhnu415 (Supplementary Tables 6, 7, and 8). The genotypes of the remaining ten non-critical recombinants were also checked by genotyping their corresponding F2:3 families (data not shown). Thus, we eventually delimited Pm58 to a 141.3-kb region covering 14,920,220–15,061,553 bp on chromosome 2DS (Fig. 2c).
Candidate genes for Pm58 and association analysis
According to the gene annotations of AL8/78 reference genome, eight annotated genes resided in the targeted Xsts20220–Xkasp61553 interval (Fig. 5a, b). Five of these genes encoded hypothetical or uncharacterized proteins, and another three (AET2Gv20068500, AET2Gv20068600 and AET2Gv20068700) were annotated to encode disease resistance proteins (Supplementary Table 9). Comparisons of the re-sequencing reads of TA1662 and T093 identified some SNP/InDel variants located within the candidate genes. Based on these variations, one KASP/STS marker for each gene was developed (Figs. 3 and 4) and used for genotyping all the F2 plants. As expected, these gene-specific markers displayed the consistencies between their genetic map positions and the sequence-based physical-map positions and were co-segregated with Pm58 (Fig. 5a, b).

Analysis of the Pm58 candidate genes. a Genetic map of Pm58. The number of crossovers identified between the adjacent marker loci is shown above the rectangle. b Eight candidate genes annotated within the 141.3-kb targeted region according to the AL8/78 annotations. The candidate genes and their corresponding molecular markers are connected with dashed lines. The annotated genes associated with disease resistance are shown as red arrows. c Haplotype analysis of the Pm58 region. A total of 178 Ae. tauschii accessions were analyzed using eight candidate gene-specific markers. The solid and blank circles indicate TA1662 and T093 genotypes, respectively. R, resistant, and S, susceptible. The number of accessions with resistant or susceptible phenotypes to Bgt E09 is shown in parenthesis
Pm58 haplotypes in 178 Ae. tauschii accessions were examined using eight candidate gene-specific markers (Fig. 5c). A total of nine haplotypes were detected, with three major haplotypes (Hap5, Hap8, and Hap9) representing 89.3% of the accessions used in this study. Hap9, as represented by susceptible parent T093, had the highest frequency (61.8%), followed by Hap5 (19.1%) and Hap8 (8.4%). TA1662 and another four accessions (AY001, AY007, AY034 and AY049) from Azerbaijan, Tajikistan, or Iran were assigned to Hap1 and were found to be highly resistant to Bgt isolate E09 (Supplementary Table 1). Compared to Hap1 with eight TA1662 alleles, two accessions carrying Hap3 with five TA1662 alleles were susceptible, indicating the importance of Xkasp68500, Xkasp68600, and Xkasp68700 loci in powdery mildew resistance (Fig. 5c). However, multiple powdery mildew testes confirmed that Ae. tauschii AY072 (Hap4) carrying TA1662 alleles at both Xkasp68600 and Xkasp68700 loci was highly susceptible to Bgt E09 (Fig. 5c; Supplementary Table 1). Therefore, we speculate that the nonsynonymous SNP (A/G) at Xkasp68500 locus is critical for powdery mildew resistance, and AET2Gv20068500 is the most likely candidate gene for Pm58.
Comparative analysis of allelic genomic regions carrying Pm58
Alignments of the candidate genes to their homologs in the D genome of common wheat CS showed that the Pm58-containing interval was collinear to a 139.3-kb region (from 14,804,472 to 14,943,785 bp on CS chromosome 2DS) with eight annotated genes (IWGSC 2018; Supplementary Fig. 3). Except for AET2Gv20068300, which was considered as a low-confidence gene with a small size (114-bp), homologs for the Ae. tauschii genes were found in the D genome of CS. The gene synteny was considerably well in the Pm58 region, even though two PAVs were identified between Ae. tauschii and common wheat (Supplementary Fig. 3). PAV-1 was located between AET2Gv20068400/TraesCS2D02G033600LC and AET2Gv20068500/TraesCS2D02G041300. The 14.1-kb deletion in the CS genome caused truncated forms deficient in 3′-ends of both TraesCS2D02G033600LC and TraesCS2D02G041300 (Fig. 6a; Supplementary Fig. 4). PAV-2 was located within the predicted second intron of AET2Gv20068700/TraesCS2D02G041600. The 12.0-kb insertion in the CS genome involved two tandem repeat sequences with 6016 bp each (Supplementary Fig. 4).

Polymorphic patterns of Xkasp68500 between TA1662 and common wheat cultivars. a Xkasp68500 is located within the 14.1-kb PAV-1 region. Vertical dashed lines indicate the breakpoints flanking PAV-1; horizontal arrows represent the positions/directions of two predicted open reading frames; the vertical arrow indicates the position of Xkasp68500; and short horizontal lines represent the positions of PAV-1-F1/R1 and PAV-1-F2/R2. b Frequency analysis of the 14.1-kb deletion in 158 wheat cultivars and 178 Ae. tauschii accessions with two pairs of primers. c Allelic forms of Xkasp68500 in 24 wheat cultivars without the 14.1-kb deletion. Scattered triangles with different colors show clustering of alleles of TA1662, T093, and wheat cultivars on the x-(FAM) and y-(HEX) axes. Gray diamonds represent non-template controls (NTC)
To gain more insights into these two structural variations on 2DS, we examined the situations of PAV-1 and PAV-2 in another four sequenced Ae. tauschii accessions (Zhou et al. 2021) and ten common wheat cultivars (Walkowiak et al. 2020; Sato et al. 2021) in addition to AL8/78 and CS (Supplementary Fig. 4). All of the five Ae. tauschii accessions carried the 14.1-kb nucleotide sequences in the PAV-1 region, whether they belonged to lineage L1 or L2. However, the majority of the 11 genome-sequenced common wheat cultivars (90.9%) displayed extensive nucleotide sequence deletions in this region. For PAV-2, four Ae. tauschii accessions contained none of the duplicates, while XJ02 and nine common wheat cultivars contained one of the two repeat sequences. Wheat cultivar Julius had the 14.1-kb insertion and 12.0-kb deletion in the PAV-1 and PAV-2 regions, respectively, making it most similar to Ae. tauschii accessions in the Pm58 region (Supplementary Fig. 4).
Allelic analysis of the critical Xkasp68500 locus in common wheat cultivars
Due to the Xkasp68500 locus was located within the PAV-1 region, we first analyzed the frequency of the 14.1-kb deletion in 158 common wheat cultivars with two pairs of primers (Fig. 6a, b). As expected, the PCR products were successively amplified in 24 cultivars with PAV-1-F1/R1 and in the others with PAV-1-F2/R2 (Supplementary Table 2), indicating that 84.8% of the wheat cultivars did not contain this 14.1-kb fragment in the PAV-1 region. By contrast, the targeted bands were amplified in all of the 178 Ae. tauschii accessions using PAV-1-F1/R1 (Fig. 6b). This result, plus the findings depicted in Supplementary Fig. 4, demonstrates that nucleotide sequence and gene losses in the PAV-1 region indeed occurred in a large number of wheat genotypes during hexaploidization.
We further analyzed the allelic forms of Xkasp68500 in 24 wheat cultivars without the 14.1-kb deletion. Intriguingly, all of these cultivars, including ten from China and 14 from other countries, carried the susceptible alleles consistent with T093 (Fig. 6c). Therefore, Xkasp68500 can clearly distinguish TA1662 from all of the 158 common wheat cultivars and has a polymorphism rate of 100%, which is much higher than that of other candidate gene-specific markers (Supplementary Fig. 5).
Discussion
Aegilops tauschii, the progenitor of common wheat D genome, constitutes a reservoir of genetic diversity for improvements in resistance to rust (Olson et al. 2013; Yu et al. 2015), powdery mildew (Miranda et al. 2006, 2007; Wiersma et al. 2017), Hessian fly (Raupp et al. 1993; Zhao et al. 2006), and pre-harvest sprouting (PHS) (Zhang et al. 2017). TA1662, an important Ae. tauschii accession from Azerbaijan, confers resistance to both Ug99 stem rust (Olson et al. 2013) and powdery mildew (Wiersma et al. 2017). SrTA1662 has been cloned using association genetics with resistance gene enrichment sequencing (AgRenSeq, Arora et al. 2019), and Pm58 was previously mapped within an 8.6-Mb interval on chromosome 2DS using a population of 96 introgression lines (Wiersma et al. 2017). In this study, we narrowed down the Pm58-containing interval to 141.3 kb by analyzing 13 recombinants from the cross Ae. tauschii T093 × TA1662 (Table 1; Fig. 5).
Common wheat evolved from hybridization between tetraploid Triticum turgidum and a single lineage of Ae. tauschii (Wang et al. 2013), which led to only 25% of the genetic diversity of Ae. tauschii contributing to the initial gene flow into wheat (Gaurav et al. 2021). Collections of Ae. tauschii germplasm exhibit wide diversity for various desirable traits and can be divided into two lineages (L1 and L2) or five sublineages (L1EY, L1EX, L1W, L2W, and L2E) according to phylogenetic analyses with genome-wide SNP data (Wang et al. 2013; Zhou et al. 2021). Recent population genomic analysis revealed a rare third lineage of Ae. tauschii, which was geographically restricted to Georgia (Gaurav et al. 2021). The susceptible parent T093 used in this study belongs to L1EY, while TA1662 and AL8/78 belong to L2W (Zhou et al. 2021). Genome re-sequencing revealed three times more SNP/InDel variants between T093 and TA1662 than between TA1662 and AL8/78, and with uneven distribution (Supplementary Fig. 2). Chromosome 4D had the highest SNP/InDel density, compared to the lowest marker density of 4D in other published intervarietal genetic maps of wheat (Cui et al. 2017; Liu et al. 2018). The abundant genetic variations between T093 and TA1662 facilitate the development of new molecular markers to finely map Pm58.
A search for annotated genes in the Pm58-containing region based on the AL8/78 genome annotations (Luo et al. 2017) identified eight candidate genes (Fig. 5a, b). Among these genes, three were disease-resistance genes (Supplementary Table 9). Eight functional markers derived from these candidate genes were developed and all of them were co-segregated with Pm58 (Fig. 5a, b). A panel of 178 Ae. tauschii accessions were analyzed with these gene-specific markers, and a total of nine haplotypes were distinguished (Fig. 5c). At least one accession in each of the Hap3 to Hap9 exhibited susceptible phenotypes to Bgt E09, which was entirely consistent with the genotypes examined by Xkasp68500. AET2Gv20068500, comprised of the Xkasp68500 locus, encodes a typical NLR protein. In fact, most of the cloned powdery mildew resistance genes, such as Pm1a (Hewitt et al. 2021), Pm2 (Sánchez-Martín et al. 2016), Pm3 (Yahiaoui et al. 2004), Pm5e (Xie et al. 2020), Pm8 (Hurni et al. 2013), Pm17 (Singh et al. 2018), Pm21 (He et al. 2018), Pm41 (Li et al. 2020), and Pm60 (Zou et al. 2018), are NLR genes and confer race-specific resistance. Although pathogens can overcome a single NLR gene through rapid evolution, stacking of multiple unlinked resistance genes has proven to provide durable and broad-spectrum resistance in wheat (Luo et al. 2021). Thus, identifying the most likely candidate for Pm58 in this study may result in opportunities for broadening resistance gene pools and gene stacking. However, the association analysis presented here is preliminary because only partial sequence variations are taken into account. The next step will be to clone and sequence the candidate genes in 178 Ae. tauschii accessions and analyze the associations of all the SNPs/InDels with powdery mildew resistance.
Structural variations are typically defined as short InDels, long PAV, copy number variation (CNV), inversions and translocations (Saxena et al. 2014). Many important crops, such as oilseed rape and wheat, are polyploid species in which gene redundancies can buffer the deleterious effects of genomic variations (Walkowiak et al. 2020). Gene PAV has been implicated in the influence of many agriculturally important traits in crops, for example, stay-green and disease resistance in oilseed rape (Qian et al. 2016; Gabur et al. 2020), as well as heat tolerance, PHS and powdery mildew resistance in wheat (Lang et al. 2021; Xue et al. 2021; Zhai et al. 2021). Recently, more than 2000 PAV genes mainly related to pathogen resistance and stress adaptation have been identified, which were present in Ae. tauschii genome but absent from CS (Zhou et al. 2021). In this study, two PAVs between AL8/78 and CS were found in the Pm58-containing region, although the gene orders were very conserved (Supplementary Fig. 3). The 14.1-kb deletion in the PAV-1 region seemed widespread in common wheat cultivars (Supplementary Table 2; Supplementary Fig. 4), which greatly impacted the presences and functions of its surrounding genes TraesCS2D02G033600LC and TraesCS2D02G041300 (Fig. 6a, b). PAV-2 can be considered as an extreme CNV, where two tandem duplicates in CS were completely missing in AL8/78 (Supplementary Fig. 4). Although PAV-2 was located within the intronic region of AET2Gv20068700/TraesCS2D02G041600, further study is needed to determine whether it can change the splicing and/or expression of its residing genes.
There are two ways to utilize the useful genes of Ae. tauschii in wheat breeding, either through the synthetic hexaploid approach or the direct hybridization between T. aestivum and Ae. tauschii (Cox et al. 2017; Kishii 2019). Pm58 has been transferred into a hard winter wheat line KS05HW14 through a direct cross (Wiersma et al. 2017). Two registered wheat lines fixed for Pm58 exhibited resistant reactions to multiple Bgt isolates and good agronomic characteristics (Wiersma et al. 2018), confirming that Pm58 has significant values for resistance improvement in modern wheat cultivars. However, both introgression lines had lower yield potentials than their recurrent parent KS05HW14 in most of the tested locations (Wiersma et al. 2018). To prevent the unfavorable linkage drags of resistance genes from the wild relatives, it is best to isolate the targeted genes and use them precisely in gene transferring and pyramiding. Here, we pick up a likely candidate gene for Pm58 and provide a gene-specific KASP marker Xkasp68500 that can distinguish TA1662 from all of the 158 randomly selected common wheat cultivars (Fig. 6; Supplementary Fig. 5). The fine mapping and candidate gene predictions conducted in this study will help to accelerate the verification of the causal genes for Pm58, and the co-segregating molecular marker Xkasp68500 can be used for marker-assisted selection in wheat breeding programs.
Data availability
All data generated during this study are included in this published article and its supplementary information files.
References
Arora S, Steuernagel B, Gaurav K, Chandramohan S, Long Y, Matny O, Johnson R, Enk J, Periyannan S, Singh N et al (2019) Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat Biotechnol 37:139–143
Avni R, Nave M, Barad O, Baruch K, Twardziok SO, Gundlach H, Hale I, Mascher M, Spannagl M, Wiebe K et al (2017) Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357:93–97
Bourras S, McNally KE, Ben-David R, Parlange F, Roffler S, Praz CR, Oberhaensli S, Menardo F, Stirnweis D, Frenkel Z, Schaefer LK, Flückiger S, Treier G, Herren G, Korol AB, Wicker T, Keller B (2015) Multiple avirulence loci and allele-specific effector recognition control the Pm3 race-specific resistance of wheat to powdery mildew. Plant Cell 27:2991–3012
Bourras S, Kunz L, Xue M, Praz CR, Müller MC, Kälin C, Schläfli M, Ackermann P, Flückiger S, Parlange F, Menardo F, Schaefer LK, Ben-David R, Roffler S, Oberhaensli S, Widrig V, Lindner S, Isaksson J, Wicker T, Yu D, Keller B (2019) The AvrPm3-Pm3 effector-NLR interactions control both race-specific resistance and host-specificity of cereal mildews on wheat. Nat Commun 10:2292
Bowen KL, Everts KL, Leath S (1991) Reduction in yield of winter wheat in North Carolina due to powdery mildew and leaf rust. Phytopathology 81:503–511
Chen XM, Luo YH, Xia XC, Xia LQ, Chen X, Ren ZL, He ZH, Jia JZ (2005) Chromosomal location of powdery mildew resistance gene Pm16 in wheat using SSR marker analysis. Plant Breed 124:225–228
Chen Y, Chen Y, Shi C, Huang Z, Zhang Y, Li S, Li Y, Ye J, Yu C, Li Z, Zhang X, Wang J, Yang H, Fang L, Chen Q (2018) SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7:1–6
Conner RL, Kuzyk AD, Su H (2003) Impact of powdery mildew on the yield of soft white spring wheat cultivars. Can J Plant Sci 83:725–728
Cox TS, Wu J, Wang S, Cai J, Zhong Q, Fu B (2017) Comparing two approaches for introgression of germplasm from Aegilops tauschii into common wheat. Crop J 5:355–362
Cruz CD, Peterson GL, Bockus WW, Kankanala P, Dubcovsky J, Jordan KW, Akhunov E, Chumley F, Baldelomar FD, Valent B (2016) The 2NS translocation from Aegilops ventricosa confers resistance to the Triticum pathotype of Magnaporthe oryzae. Crop Sci 56:990–1000
Cui F, Zhang N, Fan X, Zhang W, Zhao C, Yang L, Pan R, Chen M, Han J, Zhao X (2017) Utilization of a Wheat660K SNP array-derived high-density genetic map for high-resolution mapping of a major QTL for kernel number. Sci Rep 7:3788
Friebe B, Heun M, Tuleen N, Zeller FJ, Gill BS (1994) Cytogenetically monitored transfer of powdery mildew resistance from rye into wheat. Crop Sci 34:621–625
Gabur I, Chawla HS, Lopisso DT, von Tiedemann A, Snowdon RJ, Obermeier C (2020) Gene presence-absence variation associates with quantitative Verticillium longisporum disease resistance in Brassica napus. Sci Rep 10:4131
Gaurav K, Arora S, Silva P, Sánchez-Martín J, Horsnell R, Gao L, Brar GS, Widrig V, Raupp WJ, Singh N et al (2021) Population genomic analysis of Aegilops tauschii identifies targets for bread wheat improvement. Nat Biotechnol. https://doi.org/10.1038/s41587-021-01058-4
He H, Zhu S, Zhao R, Jiang Z, Ji Y, Ji J, Qiu D, Li H, Bie T (2018) Pm21, encoding a typical CC-NBS-LRR protein, confers broad-spectrum resistance to wheat powdery mildew disease. Mol Plant 11:879–882
Hewitt T, Müller MC, Molnár I, Mascher M, Holušová K, Šimková H, Kunz L, Zhang J, Li J, Bhatt D, Sharma R, Schudel S, Yu G, Steuernagel B, Periyannan S, Wulff B, Ayliffe M, McIntosh R, Keller B, Lagudah E, Zhang P (2021) A highly differentiated region of wheat chromosome 7AL encodes a Pm1a immune receptor that recognizes its corresponding AvrPm1a effector from Blumeria graminis. New Phytol 229:2812–2826
Huang X, Zeller FJ, Hsam SLK, Wenzel G, Mohler V (2000) Chromosomal location of AFLP markers in common wheat utilizing nullitetrasomic stocks. Genome 43:298–305
Huang S, Sirikhachornkit A, Su X, Faris J, Gill B, Haselkorn R, Gornicki P (2002) Genes encoding plastid acetyl-CoA carboxylase and 3-phosphoglycerate kinase of the Triticum/Aegilops complex and the evolutionary history of polyploid wheat. Proc Natl Acad Sci USA 99:8133–8138
Hurni S, Brunner S, Buchmann G, Herren G, Jordan T, Krukowski P, Wicker T, Yahiaoui N, Mago R, Keller B (2013) Rye Pm8 and wheat Pm3 are orthologous genes and show evolutionary conservation of resistance function against powdery mildew. Plant J 76:957–969
International Wheat Genome Sequencing Consortium (IWGSC) (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361:661
Jia J, Devos KM, Chao S, Miller TE, Reader SM, Gale MD (1996) RFLP-based maps of the homoeologous group-6 chromosomes of wheat and their application in the tagging of Pm12, a powdery mildew resistance gene transferred from Aegilops speltoides to wheat. Theor Appl Genet 92:559–565
Kang Y, Zhou M, Merry AM, Barry KM (2020) Mechanisms of powdery mildew resistance of wheat: a review of molecular breeding. Plant Pathol 69:601–617
Kishii M (2019) An update of recent use of Aegilops species in wheat breeding. Front Plant Sci 10:585
Lang J, Fu Y, Zhou Y, Cheng M, Deng M, Li M, Zhu T, Yang J, Guo X, Gui L, Li L, Chen Z, Yi Y, Zhang L, Hao M, Huang L, Tan C, Chen G, Jiang Q, Qi P, Pu Z, Ma J, Liu Z, Liu Y, Luo MC, Wei Y, Zheng Y, Wu Y, Liu D, Wang J (2021) Myb10-D confers PHS-3D resistance to pre-harvest sprouting by regulating NCED in ABA biosynthesis pathway of wheat. New Phytol 230:1940–1952
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Li T, Zhang Z, Hu Y, Duan X, Xin Z (2011) Identification and molecular mapping of a resistance gene to powdery mildew from the synthetic wheat line M53. J Appl Genet 52:137–143
Li M, Dong L, Li B, Wang Z, Xie J, Qiu D, Li Y, Shi W, Yang L, Wu Q, Chen Y, Lu P, Guo G, Zhang H, Zhang P, Zhu K, Li Y, Zhang Y, Wang R, Yuan C, Liu W, Yu D, Luo MC, Fahima T, Nevo E, Li H, Liu Z (2020) A CNL protein in wild emmer wheat confers powdery mildew resistance. New Phytol 228:1027–1037
Li G, Wang L, Yang J, He H, Jin H, Li X, Ren T, Ren Z, Li F, Han X et al (2021) A high-quality genome assembly highlights rye genomic characteristics and agronomically important genes. Nat Genet 53:574–584
Ling HQ, Ma B, Shi X, Liu H, Dong L, Sun H, Cao Y, Gao Q, Zheng S, Li Y et al (2018) Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 557:424–428
Liu J, Luo W, Qin N, Ding P, Zhang H, Yang C, Mu Y, Tang H, Liu Y, Li W, Jiang Q, Chen G, Wei Y, Zheng Y, Liu C, Lan X, Ma J (2018) A 55 K SNP array-based genetic map and its utilization in QTL mapping for productive tiller number in common wheat. Theor Appl Genet 131:2439–2450
Lu N, Lu MX, Liu P, Xu HX, Qiu XL, Hu SS, Wu YN, Bai SL, Wu JZ, Xue SL (2020) Fine mapping a broad-spectrum powdery mildew resistance gene in Chinese landrace Datoumai, PmDTM, and its relationship with Pm24. Plant Dis 104:1709–1714
Luo MC, Gu YQ, Puiu D, Wang H, Twardziok SO, Deal KR, Huo N, Zhu T, Wang L, Wang Y et al (2017) Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551:498–502
Luo M, Xie L, Chakraborty S, Wang A, Matny O, Jugovich M, Kolmer JA, Richardson T, Bhatt D, Hoque M, Patpour M, Sørensen C, Ortiz D, Dodds P, Steuernagel B, Wulff BBH, Upadhyaya NM, Mago R, Periyannan S, Lagudah E, Freedman R, Lynne Reuber T, Steffenson BJ, Ayliffe M (2021) A five-transgene cassette confers broad-spectrum resistance to a fungal rust pathogen in wheat. Nat Biotechnol 39:561–566
Lutz J, Hsam SLK, Limpert E, Zeller FJ (1995) Chromosomal location of powdery mildew resistance genes in Triticum aestivum L. (common wheat). 2. Genes Pm2 and Pm19 from Aegilops squarrosa L. Heredity (edinb) 74:152–156
Maccaferri M, Harris NS, Twardziok SO, Pasam RK, Gundlach H, Spannagl M, Ormanbekova D, Lux T, Prade VM, Milner SG et al (2019) Durum wheat genome highlights past domestication signatures and future improvement targets. Nat Genet 51:885–895
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Miranda LM, Murphy JP, Marshall D, Leath S (2006) Pm34: a new powdery mildew resistance gene transferred from Aegilops tauschii Coss. to common wheat (Triticum aestivum L.). Theor Appl Genet 113:1497–1504
Miranda LM, Murphy JP, Marshall D, Cowger C, Leath S (2007) Chromosomal location of Pm35, a novel Aegilops tauschii derived powdery mildew resistance gene introgressed into common wheat (Triticum aestivum L.). Theor Appl Genet 114:1451–1456
Mundt CC (2002) Use of multiline cultivars and cultivar mixtures for disease management. Annu Rev Phytopathol 40:381–410
Olson EL, Rouse MN, Pumphrey MO, Bowden RL, Gill BS, Poland JA (2013) Simultaneous transfer, introgression, and genomic localization of genes for resistance to stem rust race TTKSK (Ug99) from Aegilops tauschii to wheat. Theor Appl Genet 126:1179–1188
Praz CR, Bourras S, Zeng F, Sánchez-Martín J, Menardo F, Xue M, Yang L, Roffler S, Böni R, Herren G, McNally KE, Ben-David R, Parlange F, Oberhaensli S, Flückiger S, Schäfer LK, Wicker T, Yu D, Keller B (2017) AvrPm2 encodes an RNase-like avirulence effector which is conserved in the two different specialized forms of wheat and rye powdery mildew fungus. New Phytol 213:1301–1314
Qian L, Voss-Fels K, Cui Y, Jan HU, Samans B, Obermeier C, Qian W, Snowdon RJ (2016) Deletion of a stay-green gene associates with adaptive selection in Brassica napus. Mol Plant 9:1559–1569
Qiu L, Liu N, Wang H, Shi X, Li F, Zhang Q, Wang W, Guo W, Hu Z, Li H, Ma J, Sun Q, Xie C (2021) Fine mapping of a powdery mildew resistance gene MlIW39 derived from wild emmer wheat (Triticum turgidum ssp. dicoccoides). Theor Appl Genet 134:2469–2479
Raupp WJ, Amri A, Hatchett JH, Gill BS, Wilson DL, Cox TS (1993) Chromosomal location of hessian fly-resistance genes H22, H23, and H24 derived from Triticum tauschii in the D genome of wheat. J Hered 84:142–145
Sánchez-Martín J, Steuernagel B, Ghosh S, Herren G, Hurni S, Adamski N, Vrána J, Kubaláková M, Krattinger SG, Wicker T, Doležel J, Keller B, Wulff BB (2016) Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol 17:221
Sato K, Abe F, Mascher M, Haberer G, Gundlach H, Spannagl M, Shirasawa K, Isobe S (2021) Chromosome-scale genome assembly of the transformation-amenable common wheat cultivar 'Fielder'. DNA Res 28:dsab008
Saxena RK, Edwards D, Varshney RK (2014) Structural variations in plant genomes. Brief Funct Genomics 13:296–307
Singh RP, Singh PK, Rutkoski J, Hodson DP, He X, Jørgensen LN, Hovmøller MS, Huerta-Espino J (2016) Disease impact on wheat yield potential and prospects of genetic control. Annu Rev Phytopathol 54:303–322
Singh SP, Hurni S, Ruinelli M, Brunner S, Sanchez-Martin J, Krukowski P, Peditto D, Buchmann G, Zbinden H, Keller B (2018) Evolutionary divergence of the rye Pm17 and Pm8 resistance genes reveals ancient diversity. Plant Mol Biol 98:249–260
Summers RW, Brown JKM (2013) Constraints on breeding for disease resistance in commercially competitive wheat cultivars. Plant Pathol 62:115–121
Sun XL, Liu D, Zhang HQ, Huo NX, Zhou RH, Jia JZ (2006) Identification and mapping of two new genes conferring resistance to powdery mildew from Aegilops tauschii (Coss.) Schmal. J Integr Plant Biol 48:1204–1209
Te Beest DE, Paveley ND, Shaw MW, van den Bosch F (2008) Disease–weather relationships for powdery mildew and yellow rust on winter wheat. Phytopathology 98:609–617
Van Ooijen JW (2006) JoinMap® 4, software for the calculation of genetic linkage maps in experimental populations. Wageningen, Kyazma BV
Walkowiak S, Gao L, Monat C, Haberer G, Kassa MT, Brinton J, Ramirez-Gonzalez RH, Kolodziej MC, Delorean E, Thambugala D et al (2020) Multiple wheat genomes reveal global variation in modern breeding. Nature 588:277–283
Wang J, Luo MC, Chen Z, You FM, Wei Y, Zheng Y, Dvorak J (2013) Aegilops tauschii single nucleotide polymorphisms shed light on the origins of wheat D-genome genetic diversity and pinpoint the geographic origin of hexaploid wheat. New Phytol 198:925–937
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164
Wiersma AT, Pulman JA, Brown LK, Cowger C, Olson EL (2017) Identification of Pm58 from Aegilops tauschii. Theor Appl Genet 130:1123–1133
Wiersma AT, Whetten RB, Zhang G, Sehgal SK, Kolb FL, Poland JA, Mason RE, Carter AH, Cowger C, Olson EL (2018) Registration of two wheat germplasm lines fixed for Pm58. J Plant Regist 12:270–273
Xie J, Guo G, Wang Y, Hu T, Wang L, Li J, Qiu D, Li Y, Wu Q, Lu P, Chen Y, Dong L, Li M, Zhang H, Zhang P, Zhu K, Li B, Deal KR, Huo N, Zhang Y, Luo MC, Liu S, Gu YQ, Li H, Liu Z (2020) A rare single nucleotide variant in Pm5e confers powdery mildew resistance in common wheat. New Phytol 228:1011–1026
Xue SL, Lu MX, Hu SS, Xu HX, Ma YY, Lu N, Bai SL, Gu AY, Wan HS, Li SP (2021) Characterization of PmHHXM, a new broad-spectrum powdery mildew resistance gene in Chinese wheat landrace Honghuaxiaomai. Plant Dis 105:2089–2096
Yahiaoui N, Srichumpa P, Dudler R, Keller B (2004) Genome analysis at different ploidy levels allows cloning of the powdery mildew resistance gene Pm3b from hexaploid wheat. Plant J 37:528–538
Yu G, Zhang Q, Friesen TL, Rouse MN, Jin Y, Zhong S, Rasmussen JB, Lagudah ES, Xu SS (2015) Identification and mapping of Sr46 from Aegilops tauschii accession CIae 25 conferring resistance to race TTKSK (Ug99) of wheat stem rust pathogen. Theor Appl Genet 128:431–443
Zhai H, Jiang C, Zhao Y, Yang S, Li Y, Yan K, Wu S, Luo B, Du Y, Jin H, Liu X, Zhang Y, Lu F, Reynolds M, Ou X, Qiao W, Jiang Z, Peng T, Gao D, Hu W, Wang J, Gao H, Yin G, Zhang K, Li G, Wang D (2021) Wheat heat tolerance is impaired by heightened deletions in the distal end of 4AL chromosomal arm. Plant Biotechnol J 19:1038–1051
Zhang D, He J, Huang L, Zhang C, Zhou Y, Su Y, Li S (2017) An advanced backcross population through synthetic octaploid wheat as a “bridge”: development and QTL detection for seed dormancy. Front Plant Sci 8:2123
Zhang R, Fan Y, Kong L, Wang Z, Wu J, Xing L, Cao A, Feng Y (2018) Pm62, an adult-plant powdery mildew resistance gene introgressed from Dasypyrum villosum chromosome arm 2VL into wheat. Theor Appl Genet 131:2613–2620
Zhao HX, Liu XM, Chen MS (2006) H22, a major resistance gene to the Hessian fly (Mayetiola destructor), is mapped to the distal region of wheat chromosome 1DS. Theor Appl Genet 113:1491–1496
Zhou Y, Bai S, Li H, Sun G, Zhang D, Ma F, Zhao X, Nie F, Li J, Chen L, Lv L, Zhu L, Fan R, Ge Y, Shaheen A, Guo G, Zhang Z, Ma J, Liang H, Qiu X, Hu J, Sun T, Hou J, Xu H, Xue S, Jiang W, Huang J, Li S, Zou C, Song CP (2021) Introgressing the Aegilops tauschii genome into wheat as a basis for cereal improvement. Nat Plants 7:774–786
Zou S, Wang H, Li Y, Kong Z, Tang D (2018) The NB-LRR gene Pm60 confers powdery mildew resistance in wheat. New Phytol 218:298–309
Acknowledgements
We thank Dr. J. Jia of the Chinese Academy of Agricultural Sciences for providing common wheat cultivars for this study. This study was partially supported by National Natural Science Foundation of China program (31971888), Key Technology R&D Program of Henan Province of China (192102110013), and Opening Foundation of Key Laboratory of Wheat Biology and Genetic Improvement on Southwestern China program (2020zws01).
Funding
National Natural Science Foundation of China, 31971888, Shulin Xue.
Author information
Authors and Affiliations
Contributions
SX, SH, XC, and YM conducted marker development, genotyping and data analysis; ML constructed the population; SB conducted bioinformatic analysis; XW, TS, YW, HW, and XA conducted phenotyping; SL supervised the project; SX and SL wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Thomas Miedaner.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xue, S., Hu, S., Chen, X. et al. Fine mapping of Pm58 from Aegilops tauschii conferring powdery mildew resistance. Theor Appl Genet 135, 1657–1669 (2022). https://doi.org/10.1007/s00122-022-04061-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-022-04061-8