
Abstract
Key message
SNPs identify prospective genes related to response to Colletotrichum sublineola (anthracnose) in the sorghum association panel lines.
Abstract
Sorghum association panel (SAP) lines were scored over several years for response to Colletotrichum sublineola, the causal agent of the disease anthracnose. Known resistant and susceptible lines were included each year to verify successful inoculation. Over 79,000 single-nucleotide polymorphic (SNP) loci from a publicly available genotype by sequencing dataset available for the SAP lines were used with TASSEL association mapping software to identify chromosomal locations associated with differences in disease response. When the top-scoring SNPs were mapped to the published sorghum genome, in each case, the nearest annotated gene has precedence for a role in host defense.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The disease anthracnose is considered to be a global problem in sorghum production. It can infect leaves, stalks, panicles and seeds while exhibiting extreme variability in terms of pathogenicity to host cultivars (Tesso et al. 2012). Although many sorghum cultivars have shown resistance to one or more isolates, that resistance is often fleeting due to variability in the pathogen, Colletotrichum sublineola (also often written C. sublineolum). Sutton first proposed (Sutton 1968) that appressorium shape for anthracnose isolates from sorghum justified designation as a separate species (C. sublineola) from that in maize (Colletotrichum graminicola), but it was not until ribosomal ITS DNA sequence data (Sherriff et al. 1995) became available that the separate species designations were generally accepted. As would be expected, the two genomes are overall quite similar, but significant differences have been revealed (Buiate et al. 2017).
Because PCR amplification identified portions of both MAT1 and MAT2 sequences in some C. sublineola isolates (neither was found in others) (Zanette et al. 2009), it can be assumed that there is an ascomycetous perfect stage (Glomerella sp.) as is the case for other anthracnose-causing pathogens, including C. graminicola (Politis 1975). However, as is also the case of the maize pathogen, no reports of crosses leading to ascospore production in natural situations appear in the literature. Even so, there is a considerable variation found among isolates as can be detected based on the use of host differentials that are susceptible to some pathotypes but resistant to others. Examples include identification of eight different response patterns among 12 isolates from Texas, Georgia and Puerto Rico using eight host cultivars (Cardwell 1989), 13 pathotypes among 87 isolates from Arkansas using the same eight cultivars (Moore et al. 2008), while Prom et al. (2012) recorded nine pathotypes among isolates from the USA and Puerto Rico using the same eight cultivars, but adding six additional hosts showed that all 18 isolates differed in pathogenicity factors (Prom et al. 2012). A study from Brazil, with 10 hosts, including the same eight used in prior studies revealed 22 races among 37 isolates (Valèrio et al. 2005). A later study identified 68 unique pathotypes among 480 isolates, with greater variation in virulence detected from isolates taken from plots where three-way hybrids were grown than from plots with pure lines (Costa et al. 2015). DNA sequence variation is also readily detected, as has been documented with increasingly sophisticated technology. Examples include the use of PCR with single RAPD primers (Guthrie et al. 1992; Valèrio et al. 2005), RFLPs (Rosewich et al. 1998) and AFLPs (Chala et al. 2011; Prom et al. 2012).
The sorghum association panel was developed as a community resource by including 277 lines converted for short stature and day-neutral flowering while retaining 90% of the genome of the original exotic cultivar along with 149 other breeding lines. Lines included were selected to include diversity with respect to geographic origin, morphological and genotypic/phenotypic characters that have proven useful in breeding (Casa et al. 2008). A further resource was created when the SAP lines were included in a genotyping by sequencing project that revealed locations of more than 260,000 single-nucleotide polymorphisms that are well spread throughout all ten chromosomes of sorghum (Morris et al. 2013a). The panel is being widely used for genome-wide association studies in order to locate tags for traits useful in breeding. Traits examined using the SAP SNP data for a genome-wide association study (GWAS) include traits such as plant height and inflorescence branch length (Morris et al. 2013b), flavonoid pigment traits (Morris et al. 2013b), grain polyphenol concentrations (Rhodes et al. 2014), seed size (Zhang et al. 2015), stalk rots (Adeyanju et al. 2015) and thermal stress in seedlings (Chopra et al. 2017). A GWAS study based on the 242 lines in the sorghum mini-core collection identified eight genes potentially related to anthracnose resistance in the vicinity of SNP alleles (Upadhyaya et al. 2013). Most recently, another GWAS study also examined anthracnose resistance as measured in Puerto Rico in the SAP accessions using a different analysis procedure. That study was able to identify three candidate resistance genes and showed that resistant accessions came from all four major races of sorghum (Cuevas et al. 2018). Because differences were found in the disease response of a number of the same cultivars when tested in Texas, another GWAS analysis was made using TASSEL (Bradbury et al. 2007). Here, we report finding additional genes that appear to be associated with relative resistance or susceptibility to anthracnose as it occurs in Texas.
Materials and methods
Disease evaluations
In this study, 359 lines from the sorghum association panel (SAP) with SC748 as resistant check and BTx623, RTx430, Tx2911, TAM428, ATx623, Sureno, Dorado as susceptible checks were evaluated for resistance against C. sublineola. Seed from the SAP lines were obtained from the USDA-ARS, Plant Genetic Resources Conservation Unit, Griffin, Georgia. The anthracnose evaluations were conducted during the 2010, 2013, 2014 and 2015 growing seasons at the Texas AgriLife Experiment Station, near College Station, Texas. Each line was included in at least two evaluation periods. In the trials, lines were planted in a randomized complete block design, with each line replicated three times. Seeds were planted in 6 m rows at 0.31 m spacing between rows. Standard field preparation which included fall plowing and incorporation of the compound fertilizer NPK was employed during the evaluation years. To control weeds and seedling insects, a pre-emergent insecticide and herbicide were applied before planting and supplemented with hand weeding during the growing season.
The inoculation technique and disease assessment method used were previously described (Prom et al. 2009). Briefly, inoculum was prepared by inoculating autoclaved sorghum grain with a spore suspension from one of eight local isolates of C. sublineola (FSP2, FSP5, FSP7, FSP35 and FSP36, from Sorghum line BTx635, FSP46 and FSP50 from RTx 2536, and FSP53 from BSBC) that were harvested after growth on half-strength potato dextrose agar. Each isolate was grown separately on the autoclaved seeds in large, open plastic bags, with daily mixing to facilitate complete colonization. After 3–4 days of incubation, the isolates were mixed to give a uniform inoculum for the plants. Sorghum plants were inoculated 30 days after planting by placing 8–10 C. sublineola-colonized grains into the leaf whorls. Disease assessments were conducted 30 days after inoculating the plants and thereafter on a weekly basis for 3 weeks. Plants were rated on a scale of 1–5, where 1 = no symptoms or chlorotic flecks on leaves; 2 = hypersensitive reaction (reddening or red spots) on inoculated leaves, but no acervuli formation and symptoms observed on other leaves; 3 = lesions on inoculated and bottom leaves with acervuli in the center; 4 = necrotic lesions with acervuli observed on inoculated and bottom leaves with infection spreading to middle leaves; and 5 = most leaves are dead due to infection with infection on the flag leaf containing abundant acervuli. The symptom types were then categorized into two reaction classes, resistant = rated as 1 or 2 and susceptible = rated as 3, 4 or 5.
GWAS and SNP mapping
TASSEL (Bradbury et al. 2007) version 5.2.48 was used to conduct a mixed linear model (MLM) association analysis based on mean scores for disease ratings. The 2018 updated version of genome-wide SNP data for the SAP originally made available for public use (Morris et al. 2013a) (link https://www.morrislab.org/data) was used in this analysis. In order to reduce potential false associations, SNPs with greater than 20% unknown alleles were removed as were those with minor allele frequency (MAF) below 5%. Principal component analysis was used to calculate the population structure which was followed by calculation of kinship data based on the TASSEL ‘centered IBS’ method. Optimum compression level and P3D variance component estimation were used as MLM options. SNPs with high probability of contribution to anthracnose scores were tracked to the specific chromosome location based on the sorghum genome sequence, version 3.1.1 available at the JGI Phytozome 12.5.1 web site, updated in 2018 (McCormick et al. 2018). For each of the prospective genes included in Table 1, the mean disease rating score for all SAP lines with either of the two prevalent bases was determined and verified to differ significantly (P < 0.05) using JMP statistical software from SAS®, version 14.
Results
Due to the size of the collection data for anthracnose response of 359 of the 377 lines in the SAP collection, in most cases, each line was tested in replicates for at least 2 years, with some cultivars included each year to serve as controls. All of the cultivars showed evidence for active defense soon after inoculation in the form of reddish purple pigment (3-deoxyanthocyanidin) typically made by sorghum (Nicholson et al. 1987). Thus, all cultivars were scored between 2 and 5 based on the subsequent level of disease development. Year to year consistency on scoring of the controls rendered further adjustments unnecessary. Consequently, the average disease rating for each cultivar, calculated for between 2 (11 lines) and 14 replications, was used directly for identification of potential QTLs. One cultivar with only two replicates was not included based on extreme differences in scoring of the plants in years, suggesting segregation or seed contamination. Summary of anthracnose scoring data is provided in Supplementary Table 1.
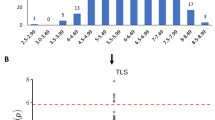
Although the genotyping data provided by Morris et al. identified 267,487 SNPs across the association panel (Morris et al. 2013a), many of the sequencing reads include a number of unidentified bases (N). Borrowing from the protocol described for stalk rots (Adeyanju et al. 2015), we eliminated all cases where there were greater than or equal to 20% ambiguous sites, leaving 79,034 SNP markers to use for anthracnose response association analysis. Eliminating minority alleles present in less than 5% of the SAP lines also means that any single gene resistance locus present in the SAP lines below that threshold would also be missed. The Manhattan plot shown in Fig. 1 based on 358 members of the SAP shows strong indication that quantitative trait loci on eight chromosomes have a role related to resistance to Colletotrichum sublineola.

Manhattan plot showing locations of SNP-detected QTLs associated with response to anthracnose on the ten chromosomes of Sorghum bicolor. All SNPs above −Log10 3.00 were examined for potential roles in disease response
A list of SNP markers that identify 18 chromosomal locations associated with disease response starting with the least likely one to represent chance association is shown in Table 1. These correspond to the TASSEL-based Manhattan plot values below 5.2 × 10−4. When mapped back to the published genome, all 18 of these SNPs are nearest to genes that have previously been implicated in various resistance responses or identified in other disease association studies. Table 1 also shows the distance in base pairs to the nearest gene, the bases that create the SNP and the fraction of the population with each SNP allele.
Discussion
With an overall genome size estimated at 732 Mb (Paterson 2008) and an overall length in terms of recombination of around 1600 cM (Mace et al. 2009), detection of 79,304 SNPs among the SAP lines suggests an average of one SNP per approximate 10 kb of sequence and 0.02 cM in map distance. However, the use of the restriction endonuclease ApeK1 that has partial DNA methylation sensitivity in the genotyping by sequencing technique helps to favor cuts and sequencing from genic regions over transposon-related repeat sequences (Elshire et al. 2011). SNP gene association was made using the latest version (3.1.1) of the sorghum genome available at the Phytozome Web site (v12.1.5) which offers greatly improved gene annotation over prior versions. The latest version took advantage of sequences now available from other cultivars and for transcriptome data from an array of tissues and conditions (McCormick et al. 2018).
On tracking the location of the SNP variants showing significant differences to the annotated genome, all of the genes nearest to the point of the SNP variant have logical roles in host defense. The highest probability of association was a series of 4 SNPs on chromosome 3 between 72,520,274 and 72,520,325 which are near a gene for a pentatricopeptide repeat (PPR). While members of this large gene family with RNA binding capacity are usually associated with organelle development, a PPR has been shown to be involved with defense against necrotrophic fungi in Arabidopsis (Laluk et al. 2011). Likewise, PPR protein-encoding sequences have also been identified as candidate QTLs for resistance to Fusarium graminearum in soybean (Cheng et al. 2017) and to Puccinia triticina, the leaf rust pathogen in wheat (Juliana et al. 2018). The top SNP tagged gene on chromosome 8 has a leucine-rich repeat motif, a feature found in the vast majority of known disease resistance genes (Sekhwal et al. 2015). The gene identified by two SNPs separated by 41,396 base pairs on chromosome 2 encodes 9-cis-epoxycarotenoid dioxygenase, an enzyme involved in ABA biosynthesis. While known primarily for a role in abiotic stress responses, ABA has been shown to increase the amount of disease for some pathogens and to reduce disease in others (Ton et al. 2009; Liao et al. 2018).
Several of the SNP-identified genes have obvious potential for involvement in disease defense, including genes involved in cell wall synthesis and strengthening. Examples include SNPs near a xyloglucan endo-transglycosylase on chromosome 10 and another on chromosome 7. Levels of mRNA encoding this protein fell in Arabidopsis following inoculation with Pseudomonas syringae (Mohr and Cahill 2007). Other cell wall components, including a galacturon-binding receptor kinase located on chromosome 2, the arabinogalactan protein on chromosome 6 and a glycosyl transferase on chromosome 1, have previously been implicated in disease resistance in plants (Vorwerk et al. 2004; Krattinger et al. 2009).
RING finger proteins such as that identified by a SNP on chromosome 6 have been shown to be involved during infection of rice by Magnaporthe oryzae (Li et al. 2014).
It would make sense that energy metabolism would have an effect on host–pathogen interactions, and in fact, other subunits of vacuolar ATP synthase have long been implicated (Vera-Estrella et al. 1994) based on elicitor responses in tomato. Thus, differences in the ATP synthase subunit E identified on chromosome 3 may also affect disease responses as seen here. Similarly, a gene nearest to another SNP on chromosome 8 identifies a serine-domain gene involved in sphingolipid synthesis that was also identified as a cotton gene associated with Verticillium resistance (Li et al. 2017). Plant mannose-binding lectins (MBLs) are crucial for pepper plant defense signaling during pathogen attack by recognizing specific carbohydrates on pathogen surfaces (Hwang and Hwang 2011); two SNPs related to MBLs were found on chromosome 2. Also, a marker on chromosome 2 identifies an AAA ATPase, an enzyme known to be involved in immunity in both mammals and plants, and also potentially involved in sorghum immunity as well (Zhu et al. 2016). The first fungal Avr gene (Avr9 gene) was reported in 1991 (Shen et al. 2017); here, a SNP was detected near an Avr9 elicitor response protein. Interestingly, a member of the xanthine/uracil/vitamin C permease family was spotted on chromosome 2 in this study. When C. elegans was infected by S. aureus, xanthine/uracil/vitamin C permease was 6.9-fold induced 8 h postinoculation (Irazoqui et al. 2010). While C. elegans is not a plant, many similarities in active disease defense in plants and animals have been documented (Nürnberger et al. 2004).
RNA-binding proteins are known to take roles in plant immune responses against microbial pathogens (Woloshen et al. 2011), and on chromosome 8, a SNP near dsRNA-binding protein was detected. Finally, RNA recognition motif (RRM) gene expression has been shown to be highly induced in rice by inoculation with Xanthomonas oryzae pv. oryzicola (Zhou et al. 2010). In fact, a number of prospects further down the list than those included in Table 1 such as another leucine-rich repeat component on chromosome 8 and a peroxidase on chromosome 4 were also identified. Peroxidases are universally recognized as an early response in host defense (Hammerschmidt 1999).
Essentially, the same population was recently subjected to GWAS analysis by Cuevas et al. (2018) based on response to C. sublineola isolates from Puerto Rico. Because the C. sublineola isolates differ in the two locations, as do the climatic conditions (Prom et al. 2012), it was considered useful to explore SNP linkage to disease response to local isolates in the Texas environment. Cuevas et al. initially used an enriched mixed linear model (ECMLM) procedure with 268,289 SNP sites, but found no SNPS highly associated with resistance. A follow-up logistic regression analysis that is limited to detecting major genes revealed 3 SNPS on chromosome 5. However, when using TASSEL, we elected to eliminate all SNPs in the database that were ambiguous for 20% or more of the accessions and thus ended up with only 79,034 useful SNPS (Cuevas et al. 2018). Even though some or all of the high-scoring SNPs identified in this study could have occurred by chance, we chose to determine whether or not high-scoring SNPs in the Manhattan plot are associated with genes related to host defense. As documented, that turned out to be the case. Even though we also described two markers on chromosome 5, they were not at the same location as those described by Cuevas et al. Most of the genes described here would be expected to contribute to various aspects of host defense and thus not register as major genes for resistance. Population structure can be important in GWAS studies; however, in both studies, the observation that resistant accessions were found in each of the five races of sorghum suggested that population structure was not a factor.
We realize that other nearby genes may be even more critical, than those listed here, but the fact that each of the highest scoring SNPs can be associated with genes already known to play a role in host defense strongly suggests their potential importance for use in breeding, as well as for future tests to verify a potential role for these genes in active host defense.
Author contribution statement
LKP managed the disease nurseries and disease ratings, EA assisted in inoculation and did the association mapping and statistics, TI prepared inoculum for field screening, and CM prepared the manuscript.
References
Adeyanju A, Little C, Yu J, Tesso T (2015) Genome-wide association study on resistance to stalk rot diseases in grain sorghum. G3 Genes Genom Genet 5:1165. https://doi.org/10.1534/g3.114.016394
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) Tassel: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Buiate EAS, Xavier KV, Moore N, Torres MF, Farman ML, Schardl CL, Vaillancourt LJ (2017) A comparative genomic analysis of putative pathogenicity genes in the host-specific sibling species colletotrichum graminicola and Colletotrichum sublineola. BMC Genom 18:67. https://doi.org/10.1186/s12864-016-3457-9
Cardwell KF (1989) Pathotypes of Colletotrichum graminicola and seed transmission of sorghum anthracnose. Plant Dis 73:255–257
Casa AM, Pressoir G, Brown PJ, Mitchell SE, Rooney WL, Tuinstra MR, Franks CD, Kresovich S (2008) Community resources and strategies for association mapping in sorghum. Crop Sci 48:30–40
Chala A, Tronsmo AM, Brurberg MB (2011) Genetic differentiation and gene flow in Colletotrichum sublineolum in Ethiopia, the centre of origin and diversity of sorghum, as revealed by AFLP analysis. Plant Pathol J 60:474–482
Cheng P, Gedling CR, Patil G, Vuong TD, Shannon JG, Dorrance AE, Nguyen HT (2017) Genetic mapping and haplotype analysis of a locus for quantitative resistance to Fusarium graminearum in soybean accession pi 567516c. Theroret Appl Genet 130:999–1010
Chopra R, Burow G, Burke JJ, Gladman N, Xin Z (2017) Genome-wide association analysis of seedling traits in diverse sorghum germplasm under thermal stress. BMC Plant Biol 17:12
Costa RV, Zambolim L, Cota LV, Silva DD, Parreira DF, Lanza FE, Souza AGC (2015) Pathotypes of Colletotrichum sublineolum in response to sorghum populations with different levels of genetic diversity in Sete Lagoas-MG. J Phytopathol 163:543–553
Cuevas HE, Prom LK, Cooper EA, Knoll JE, Ni X (2018) Genome-wide association mapping of anthracnose (Colletotrichum sublineoloum) resistance in the US sorghum association panel. Plant Genome 11:1. https://doi.org/10.3835/plantgenome2017.11.0099
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379
Guthrie PAI, Magill CW, Frederiksen RA (1992) Random amplified polymorphic DNA markers: a system for identifying and differentiating isolates of Colletotrichum graminicola. Phytopathology 82:832–835
Hammerschmidt R (1999) Induced disease resistance: how do induced plants stop pathogens? Physiol Mol Plant Pathol 55:77–84
Hwang IS, Hwang BK (2011) The pepper mannose-binding lectin Gene CaMBL1 is required to regulate cell death and defense responses to microbial pathogens. Plant Physiol 155(1):447–463
Irazoqui JE, Troemel ER, Feinbaum RL, Luhachack LG, Cezairliyan BO, Ausubel FM (2010) Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathogens 6(7):e1000982. https://doi.org/10.1371/journal.ppat.1000982
Juliana P, Singh RP, Singh PK, Poland JA, Bergstrom GC, Huerta-Espino J, Bhavani S, Crossa J, Sorrells ME (2018) Genome-wide association mapping for resistance to leaf rust, stripe rust and tan spot in wheat reveals potential candidate genes. Theor Appl Genet. https://doi.org/10.1007/s00122-018-3086-6
Krattinger SG, Lagudah ES, Spielmeyer W, Singh RP, Huerta-Espino J, McFadden H, Bossolini E, Selter LL, Keller B (2009) A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 323:1360–1363
Laluk K, AbuQamar S, Mengiste T (2011) The arabidopsis mitochondria-localized pentatricopeptide repeat protein PGN functions in defense against necrotrophic fungi and abiotic stress tolerance. Plant Physiol 156:2053–2068
Li W-T, Chen W-L, Yang C, Wang J, Yang L, He M, Wang J-C, Qin P, Wang Y-P, Ma B-T, Li S-G, Chen X-W (2014) Identification and network construction of zinc finger protein (ZFP) genes involved in the rice-‘Magnaporthe oryzae’ interaction [online]. Plant Omics 7:540–548
Li T, Ma X, Li N, Zhou L, Liu Z, Han H, Gui Y, Bao Y, Chen J, Dai X (2017) Genome-wide association study discovered candidate genes of verticillium wilt resistance in upland cotton (Gossypium hirsutum L.). Plant Biotechnol J 15:1520–1532
Liao Y, Bai Q, Xu P, Wu T, Guo D, Peng Y, Zhang H, Deng X, Chen X, Luo M, Ali A, Wang W, Wu X (2018) Mutation in rice abscisic acid2 results in cell death, enhanced disease-resistance, altered seed dormancy and development. Front Plant Sci 9:405. https://doi.org/10.3389/fpls.2018.00405
Mace ES, Rami J-F, Bouchet S, Klein PE, Klein RR, Kilian A, Wenzl P, Xia L, Halloran K, Jordan DR (2009) A consensus genetic map of sorghum that integrates multiple component maps and high-throughput diversity array technology (DaRT) markers. BMC Plant Biol 9:13. https://doi.org/10.1186/1471-2229-9-13
McCormick RF, Truong SK, Sreedasyam A, Jenkins J, Shu S, Sims D, Kennedy M, Amirebrahimi M, Weers Brock D, McKinley B, Mattison A, Morishige Daryl T, Grimwood J, Schmutz J, Mullet JE (2018) The Sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J 93:338–354
Mohr PG, Cahill DM (2007) Suppression by aba of salicylic acid and lignin accumulation and the expression of multiple genes, in arabidopsis infected with Pseudomonas syringae pv. Tomato. Funct Integr Genom 7:181–191
Moore JW, Ditmore M, TeBeest DO (2008) Pathotypes of Colletotrichum sublineolum in Arkansas. Plant Dis 92:1415–1420
Morris GP, Ramu P, Deshpande SP, Hash CT, Shah T, Upadhyaya HD, Riera-Lizarazu O, Brown PJ, Acharya CB, Mitchell SE, Harriman J, Glaubitz JC, Buckler ES, Kresovich S (2013a) Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc Natl Acad Sci USA 110:453–458
Morris GP, Rhodes DH, Brenton Z, Ramu P, Thayil VM, Deshpande S, Hash CT, Acharya C, Mitchell SE, Buckler ES, Yu J, Kresovich S (2013b) Dissecting genome-wide association signals for loss-of-function phenotypes in sorghum flavonoid pigmentation traits. G3 Genes Genom Genet 3:1. https://doi.org/10.1534/g3.113.008417
Nicholson RL, Kollipara SS, Vincent JR, Lyons PC, Cadena-Gomez G (1987) Phytoalexin synthesis by the sorghum mesocotyl in response to infection by pathogenic and nonpathogenic fungi. Proc Natl Acad Sci USA 84:5520–5524
Nürnberger T, Brunner F, Kemmerling B, Piater L (2004) Innate immunity in plants and animals: striking similarities and obvious differences. Immunol Rev 198:249–266
Paterson AH (2008) Genomics of sorghum. Int J Plant Genom 2008:1. https://doi.org/10.1155/2008/362451
Politis DJ (1975) The identity and perfect state of Colletotrichum graminicola. Mycologia 67:56–62
Prom LK, Perumal R, Erpelding J, Isakeit T, Montes-Garcia N, Magill CW (2009) A pictorial technique for mass screening of sorghum germplasm for anthracnose (Colletotrichum sublineolum) resistance. Open Agric J. https://doi.org/10.2174/1874331500903010020
Prom LK, Perumal R, Erattaimuthu SR, Little CR, No EG, Erpelding JE, Rooney WL, Odvody GN, Magill CW (2012) Genetic diversity and pathotype determination of Colletotrichum sublineolum isolates causing anthracnose in sorghum. Eur J Plant Pathol 133:671–685
Rhodes DH, Hoffmann L, Rooney WL, Ramu P, Morris GP, Kresovich S (2014) Genome-wide association study of grain polyphenol concentrations in global sorghum [Sorghum bicolor (L.) moench] germplasm. J Agric Food Chem 62:10916–10927
Rosewich UL, Pettway RE, McDonald BA, Duncan RR, Frederiksen RA (1998) Genetic structure and emporal dynamics of a Colletotrichum graminicola population in a sorghum disease nursery. Phytopathology 88:1087–1093
Sekhwal KM, Li P, Lam I, Wang X, Cloutier S, You MF (2015) Disease resistance gene analogs (RGAs) in plants. Int J Mol Sci 16:19248–19290
Shen Y, Liu N, Li C, Wang X, Xu X, Chen W, Xing G, Zheng W (2017) The early response during the interaction of fungal phytopathogen and host plant. Open Biol 7(5):170057. https://doi.org/10.1098/rsob.170057
Sherriff C, Whelan MJ, Arnold GM, Bailey JA (1995) RDNA sequence analysis confirms the distinction between Colletotrichum graminicola and C. sublineolum. Mycol Res 99:475–478
Sutton BC (1968) The appressoria of Colletotrichum graminicola and C. falcatum. Can J Bot 46:873–876
Tesso TRP, Little CR, Adeyanju A, Radwan GL, Prom LK, Magill CW (2012) Sorghum pathology and biotechnology—a fungal disease perspective: part II. Anthracnose, stalk rot, and downy mildew. Eur J Plant Sci Biotechnol 6:33–44
Ton J, Flors V, Mauch-Mani B (2009) The multifaceted role of ABA in disease resistance. Trends Plant Sci 14:310–317
Upadhyaya HD, Wang Y, Sharma R, Sharma S (2013) Identification of genetic markers linked to anthracnose resistance in sorghum using association analysis. Theor Appl Genet 126:649–657. https://doi.org/10.1007/s00122-013-2081-1
Valèrio H, Rèsende M, Weikert-Oliveira R, Casela C (2005) Virulence and molecular diversity in Colletotrichum graminicola from Brazil. Mycopathologia 159:449–459
Vera-Estrella R, Barkla BJ, Higgins VJ, Blumwald E (1994) Plant defense response to fungal pathogens (activation of host-plasma membrane H+-atpase by elicitor-induced enzyme dephosphorylation). Plant Physiol 104:1. https://doi.org/10.1104/pp.104.1.209
Vorwerk S, Somerville S, Somerville C (2004) The role of plant cell wall polysaccharide composition in disease resistance. Trends Plant Sci 9:203–209
Woloshen V, Huang S, Li X (2011) RNA-binding proteins in plant immunity. J Pathog 2011:278697. https://doi.org/10.4061/2011/278697
Zanette GF, Nóbrega GMA, Meirelles LDP (2009) Morphogenetic characterization of Colletotrichum sublineolum strains, causal agent of anthracnose of sorghum. Trop Plant Pathol 34:146–151
Zhang D, Li J, Compton RO, Robertson J, Goff VH, Epps E, Kong W, Kim C, Paterson AH (2015) Comparative genetics of seed size traits in divergent cereal lineages represented by sorghum Panicoidae) and rice (Oryzoidae). G3 Genes Genom Genet 5:1. https://doi.org/10.1534/g3.115.017590
Zhou Y-L, Xu M-R, Zhao M-F, Xie X-W, Zhu L-H, Fu B-Y, Li Z-K (2010) Genome-wide gene responses in a transgenic rice line carrying the maize resistance gene Rxo1 to the rice bacterial streak pathogen, xanthomonas oryzae pv. Oryzicola. BMC Genom 11:1. https://doi.org/10.1186/1471-2164-11-78
Zhu X, Yin J, Liang S, Liang R, Zhou X, Chen Z (2016) The multivesicular bodies (MVBs)-localized AAA ATPase LRD6-6 inhibits immunity and cell death likely through regulating MVBs-mediated vesicular trafficking in rice. PLoS Genet 12(9):e1006311. https://doi.org/10.1371/journal.pgen.1006311
Acknowledgements
Funding from the USDA CRIS project # 3091-22000-034-00-D.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical standards
All research has been conducted to satisfy TAMU System BSL-1 Biological Safety Standards.
Additional information
Communicated by Hai-Chun Jing.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Prom, L.K., Ahn, E., Isakeit, T. et al. GWAS analysis of sorghum association panel lines identifies SNPs associated with disease response to Texas isolates of Colletotrichum sublineola. Theor Appl Genet 132, 1389–1396 (2019). https://doi.org/10.1007/s00122-019-03285-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03285-5