
Abstract
Key message
We identified 27 stable loci associated with agronomic traits in spring wheat using genome-wide association analysis, some of which confirmed previously reported studies. GWAS peaks identified in regions where no QTL for grain yield per se has been mapped to date, provide new opportunities for gene discovery and creation of new cultivars with desirable alleles for improving yield and yield stability in wheat.
Abstract
We undertook large-scale genetic analysis to determine marker-trait associations (MTAs) underlying agronomic and physiological performance in spring wheat using genome-wide association studies (GWAS). Field trials were conducted at seven sites in three countries (Sudan, Egypt, and Syria) over 2–3 years in each country. Twenty-five agronomic and physiological traits were measured on 188 wheat genotypes. After correcting for population structure and relatedness, a total of 245 MTAs distributed over 66 loci were associated with agronomic traits in individual and mean performance across environments respectively; some of which confirmed previously reported loci. Of these, 27 loci were significantly associated with days to heading, thousand kernel weight, grain yield, spike length, and leaf rolling for mean performance across environments. Despite strong QTL by environment interactions, eight of the loci on chromosomes 1A, 1D, 5A, 5D, 6B, 7A, and 7B had pleiotropic effects on days to heading and yield components (TKW, SM−2, and SNS). The winter-type alleles at the homoeologous VRN1 loci significantly increased days to heading and grain yield in optimal environments, but decreased grain yield in heat prone environments. Top 20 high-yielding genotypes, ranked by additive main effects and multiplicative interaction (AMMI), had low kinship relationship and possessed 4–5 favorable alleles for GY MTAs except two genotypes, Shadi-4 and Qafzah-11/Bashiq-1–2. This indicated different yield stability mechanisms due to potentially favorable rare alleles that are uncharacterized. Our results will enable wheat breeders to effectively introgress several desirable alleles into locally adapted germplasm in developing wheat varieties with high yield stability and enhanced heat tolerance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bread wheat (Triticum aestivum L.) is one of the world’s most important and widely consumed cereal crops. The world population is expected to reach about 9 billion by the end of the 21st century, and it has been predicted that the demand for cereals, especially wheat, will increase by approximately 50% by 2030 (Borlaug and Dowswell 2003), much of these in developing countries. Wheat adaptability and production stability in semi-arid, tropical, and subtropical climates is crucial to the attainment of the projected demands. Wheat crops in these regions experience above optimum temperature during reproductive stages of development, leading to a significant yield loss (Talukder et al. 2014). In a recent study, Ray et al. (2013) using historical data from various parts of the world reported that climate variability accounts for roughly 32–39% in yield variability with production fluctuations of ~22 million tons annually for five major crops, including wheat. Similarly, Asseng et al. (2015) reported that global wheat production is estimated to fall by 6% for each °C of further temperature increase, becoming more variable over space and time. This poses considerable challenge for wheat breeders in developing varieties targeted at heat prone environments against the backdrop of fluxes in patterns of climate change.
Phenotypic characterization over multi-environment field trials is important to assess genotype responses across target environments and the extent of genotype × environment interactions (GEI). Better understanding of the genetic control of key traits and their underlying genes/quantitative trait loci (QTL) will facilitate the estimation of gene or QTL × environment interactions to further refine crop improvement (Trethowan 2014). To date, many QTL associated with yield-related traits in bread wheat have been identified by bi-parental QTL mapping (Marza et al. 2006; Kuchel et al. 2007; Olivares-Villegas et al. 2007; McIntyre et al. 2010; Tang et al. 2011; Bennett et al. 2012; Lopes et al. 2013; Rebetzke et al. 2013; Gao et al. 2015). However, such QTL mapping studies only locate associated genomic regions with low resolution limiting their utility across diverse genetic background (Sukumaran et al. 2015). Genome-wide association studies (GWAS) based on random, high-density genotyping can help to “backfill” regions of the chromosome, where unknown genes with major effects are located (Crossa et al. 2007). The ability to survey large gene pools that are more representative of the breeding pool within any given country or geographic area lends itself to the detection and mapping of multiple traits in a single panel of genotypes (Neumann et al. 2011; Mulki et al. 2013; Jighly et al. 2016). GWAS has been bolstered by the availability of high-density molecular markers made possible by advances in the development of low-cost high-throughput genotyping resources (Wang et al. 2014; Zegeye et al. 2014). The limitations of GWAS include its inability to detect rare alleles and the effects of unaccounted (Zhao et al. 2007) or overcorrected (Segura et al. 2012) population structure. However, GWAS is still useful to detect robust QTL that have an effect across different genetic backgrounds and environments (Jannink 2007).
GWAS has been successfully used to map QTL for traits in wheat such as grain yield (GY), foliar diseases, agronomic traits, and end-use quality using different molecular marker systems (Crossa et al. 2007). Recently, Rasheed et al. (2014) and Zegeye et al. (2014) conducted GWAS to identify genomic regions that underpin grain morphology and stripe rust resistance in synthetic hexaploid wheats, respectively. In the same vain, Bentley et al. (2014) used GWAS to genetically dissect key agronomic traits in elite European wheat genotypes, and concluded that GWAS offer potential for application in both research and breeding. Similar findings have been reported with CIMMYT wheat germplasm (Edae et al. 2014), ICARDA elite spring and winter wheat germplasm (Jighly et al. 2015; Tadesse et al. 2015), elite breeding lines from the breeding programs of Bioplante and INRA (Bordes et al. 2014), and spring wheat population grown in temperate irrigated environments (Sukumaran et al. 2015).
In this study, GWAS were performed using 188 wheat genotypes grown in 15 environments and genotyped with DArT markers to identify loci associated with key agronomic and physiological traits and understand the genetic interactions between the traits.
Materials and methods
Germplasm
The plant materials for the study consisted of 188 wheat genotypes (Table S1). These genotypes were obtained from the International Centre for Agricultural Research in the Dry Areas (ICARDA), Syria, the International Maize and Wheat Improvement Centre (CIMMYT), Mexico and CSIRO Plant Industry, Australia and the Australian winter cereal collection Center (AWCC), Tamworth, Australia.
Experimental sites
Field trials were conducted in 15 environments (7 locations, 3 years, and two growing seasons, Table 1) at historical hotspots of heat occurrences during grain filling in three sites each at Egypt (Al-Matana, Kom Ombo, and Sids) and Sudan (Hudeiba, Wadmedani, and Dongola), respectively, and one in Syria (Tel-hadya) from 2010 to 2013. Detailed environment characteristics with time of sowing and harvesting are provided in Table 1. Environment names are coded by unique prefix (E1–E15), followed by abbreviation of location name and year of evaluation, e.g., E1-HUD13 refers to environment-1 at Hudeiba (Sudan) in 2013. The geographic coordinates and meteorological data, including minimum and maximum temperature, rainfall, and/or irrigation for each site during field trials, are given in Table 1 and S2. In total, data for 25 different traits were recorded; however, not all the traits were recorded in all environments (Table 2). Trials were fertilized and maintained free from weeds, insects, and diseases.
Phenotyping
The experiments were laid out as an alpha lattice design with two replications. Each plot consisted of six-to-eight rows, 8 m long with a cut back to 6 m and 15 cm spacing between rows. Data were collected on physiological, yield, and yield-related traits, though some traits were not measured in some of the studied environments, as shown in Table 2. Heading days (HD), which is duration between dates of sowing and appearance of heads, was recorded at the stage when more than 50% of plants in each plot were displaying heads (Zadoks stage 59, Zadoks et al. 1974). Days to maturity (DM) data were recorded at the period between the date of sowing and the date when more than 50% of the spikes in a plot showed a total loss of green color (physiological maturity) (Zadoks stage 89, Zadoks et al. 1974). Grain filling duration (GFD) was calculated as the period between days to heading and physiological maturity. Plant height (PH) of each genotype was estimated with meter rule when all plots reached physiological maturity by measuring the distance between the base of the stem and the top of the spike excluding the awn. The number of spikes m−2 (SM−2) was measured by counting the number of spikes in 1 m2 area for each plot using a quadrat of 1 m2. Five plants per plot were randomly sampled for the peduncle length (PL) and spike lengths (SL). They were measured with meter rule. PL in cm was determined as average height of peduncle from the last node of the main stem to the initial tip of the spike at maturity. SL in cm was measured from the base of the first spikelets to the tip of terminal spikelets excluding awns at maturity. Canopy temperature (CT) was read off at the same time for each genotype like a “snapshot” with a hand-held infrared thermometer at mid-day (11 am to 1 pm) under bright sunlight and less wind movement. The device was positioned above the canopy of the plant at angle of 45° and the canopy temperature was read off for each plot. For thousand-kernel weight (TKW), the grains from ten randomly selected plants in each replicate of every genotype were bulked separately. Thousand threshed kernels were counted randomly from each bulk and weighed on electric weighing balance. For the number of kernels per spike, ten plants were sampled from each plot. The spike of the main stem was threshed manually and the numbers of kernel per spike (KPS) were counted for each genotype. Biomass was calculated as follows: a 1 m2 plot of each genotype was harvested at the time of harvest followed by the estimation of individual biomass yield. This was later converted to tons per hectare. Harvest index (HI) was calculated as the ratio of grain yield to biological yield (BIO), while threshing index (TR) was the ratio of the threshed seeds to the panicles. Chlorophyll content was estimated with a portable chlorophyll meter (SPAD-502, Minolta) at anthesis, beginning of grain filling, and mid grain filling. Wax score (WAX) and leaves rolling (LR) were estimated by visual scoring of each plot.
DNA extraction and genotyping
Genomic DNA was extracted from 2-week-old seedlings using pooled leaf samples from five individual plants, frozen in liquid nitrogen, and stored at −80 °C before DNA extraction. DNA extraction was carried out according to the procedure described in Ogbonnaya et al. (2001). The 188 genotypes were genotyped with high-density Diversity Arrays Technology (DArT®) markers from a PstI/BstNI representation (“wPt’’ markers) using 10 μl of a 100 ng μl−1 DNA of each sample sent to Triticarte Pty. Ltd. Australia (http://www.triticarte.com.au) as a commercial service provider for DArT markers.
Furthermore, the genotypes were also screened for functional genes: Ppd-D1 and three homoeologous Vrn-1 genes. The sequences of the primers and the PCR protocol for Vrn-A1, Vrn-B1, and Vrn-D1 have been described earlier (Fu et al. 2005; Yan et al. 2004). The genotyping to identify the 2 kb deletion in promoter region of Ppd-D1 was conducted using protocol and primers described by Beales et al. (2007).
Statistical analysis
Phenotypic
Restricted/residual maximum likelihood (REML) analyses were carried out on the results obtained in the alpha lattice experiments to generate the best linear unbiased estimates (BLUEs) for the multi-locational trials. These REML analyses were performed with GenStat Release 10.3DE (http://www.GenStat.co.uk). Phenotypic correlations of the studied traits were obtained with SPSS version 16.0. Estimation of variance components was performed using PROC GLM in the Statistical Analysis System (SAS Institute, 2000) for all traits, with genotypes as fixed effects, and environments, genotypes x environment interactions, and replication nested in environment effects as random. Broad sense heritability (h 2) was calculated across environments from variance components obtained from REML analysis in GenStat Release 10.3DE (http://www.GenStat.co.uk) using formula h 2 = σ 2 g /(σ 2 g + σ 2 ge /r +σ 2 ε /re), where the genetic variance σ 2 g = (MS f –MS fe )/re, genotype × environment interaction variance σ 2 ge = (MS fe –MS e )/r, error variance σ 2 ε = MS e , MS f = genotype mean square, MS fe = genotype × environment interaction mean square, MS e = error mean square, and r and e were the numbers of replicates and environments, respectively.
DArT marker analysis
The PowerMarker V3.25 software was used to estimate the allele frequency of all the DArT markers (Liu and Muse 2005). The markers with minor allele frequency (MAF) less than 5% were removed from the data set prior to the mixed linear model (MLM) association analyses to reduce false-positive outcomes (type 1 error).
Population structure, principal component analysis, and linkage disequilibrium
The population structure was estimated with the model-based Bayesian clustering software STRUCTURE version 2.33 (Pritchard et al. 2000). Forty unlinked DArT markers covering the wheat genome (one marker from each chromosome arm) were chosen for the structure analyses, except for chromosomes 4D and 6D, where only one marker was chosen to avoid physical linkage. The genetic distance between two chosen markers on the same chromosome was at least 50 cM (Mulki et al. 2013). To infer population structure among the wheat genotypes, three independent runs for each K value from 1 to 10 were performed. Both the length of burn-in period and the number of iterations were set at 100,000. The STRUCTURE was run twice with two different sets of markers. The K value reached a plateau when the minimal number of groups that best described the population substructure has been attained (Pritchard et al. 2000). The average K values were plotted against their respective logarithm of the probability of likelihood [LnP (D)]. An ad-hoc quantity statistic (∆K) based on the rate of change in the log probability of data between successive K values (Evanno et al. 2005) was used to predict the most appropriate number of subpopulations (Mulki et al. 2013). Principal component analysis (PCA) was conducted based on all markers data using TASSEL 5.0 The first three principal components were plotted against each other using ‘scatter plot’ function in Microsoft Excel 2011.
Genome-wide LD in the data set was estimated by pairwise comparisons among the genome anchored DArT markers using the TASSEL 5.0 software (Bradbury et al. 2007). LD was estimated as squared allele frequency correlations (r 2) between pairs of DArT markers according to Weir (1996). To depict the extent of LD between pairs of loci, r 2 values were plotted against inter-marker genetic distance (cM) for the whole-genome and individual genomes. Locally weighed polynomial regression (LOESS) curves were then fitted into the scatter plot using function ‘smooth.spline’ of R (R Development Core Team, 2011). Specifically, the 95th percentile in the distributions of r 2 of the selected loci was estimated as the threshold r 2 (Breseghello and Sorrells 2006) on the assumption that LD was attributable to linkage. At its points of intersection with the LD decay curves, the threshold r 2 was plotted as a horizontal line in the LD scatterplot which provided estimates of the extent of LD. LD along chromosomes was assessed by a sliding window approach with 5 cM windows at 500 positions along the chromosomes.
Association analysis
Phenotypic BLUEs of all traits were used for marker-trait association analysis. The traits with broad sense heritability <0.5 were not used for association analysis. The genome association and prediction integrated tool (GAPIT) software, which uses computationally efficient and powerful methods such as EMMAX (Kang et al. 2010) and CMLM (Zhang et al. 2010), was run with the model selection option (Lipka et al. 2012). The kinship matrix was calculated by identity-by-state estimates using TASSEL 5.0 version and was used as covariate for population stratification. The equation fitted in GAPIT was
where y is the vector of observed phenotypic values of n seedlings; X is the vector of the SNPs, β is the vector of the allele effect to be estimated, P is the first 5 PCs while α represents how much each PC explains from the SNP variation to be estimated, u is the vector of the random effects for co-ancestry relations, and e is the vector of the residuals. To avoid spurious associations that could arise from population structure, we included first five principal components (PCs) derived from the genotypic data matrix (n × m) as covariates (i.e., Q matrix). The optimal number of PCs was determined by forward model selection using the Bayesian information criterion (BIC) as implemented in GAPIT. The significance of associations between markers and phenotypes was assessed using the false discovery rate (FDR) (Benjamini and Hochberg 1995) with a q value cutoff of 0.05. The P values obtained were used as an input file for a script written with minor modifications in the R software (R Development Core team 2013) to generate Manhattan plots. The proportion of the genetic variance in percentage (PG %) explained by the individual trait-associated marker was calculated as explained by (Würschum et al. 2015) by fitting all QTL simultaneously in a linear model to obtain R 2adj . The ratio P G = R 2adj /h 2, where h 2 refers to the heritability of the trait, resulted in the proportion of genotypic variance (Utz et al. 2000). The P G% values obtained for individual markers associated with relevant traits were accordingly derived from the sums of squares of the QTL (SSQTL) in the linear model as described by Würschum et al. (2015).
Results
Variations in phenotypic traits
In total, data for 25 agronomic and physiological traits were collected during field trials (Table 2). Canopy temperature before grain filling (CTBGF) was evaluated in two environments, while grain yield (GY) was evaluated in all 15 environments. The results from analysis of variance (ANOVA) for all traits indicated significant variations among genotypes, environments, and genotype × environments interactions, except for canopy temperature in middle of grain filling (CTMGF), for which genotype × environment interaction was not significant (Table S6). Phenotypic variability for the top four important agronomic traits, i.e., HD, PH, TKW, and GY across environments, is presented in the form of box plots (Fig. 1); the highest GY was recorded in E6-MAT12 (9.18 t ha−1) with a range of 4.1–12.0 t ha−1, while the lowest GY was recorded in E13-TH12L (0.86 t ha−1) with a range of 0.2–1.4 t ha−1. The two late seasons in Syria (E12-TH11HT and E13-TH12HT) had significantly lower grain yield compared to the two normal seasons, and the percentage of yield loss ranged from 58 to 88%. All six environments in Egypt (E6–E11) out-yielded the environments in Sudan and Syria.

Box plot for four important agronomic traits across all environments and data averaged for all environments (Average-AE). a Heading date, b plant height, c thousand kernel weight, and d grain yield
Broad sense heritability (h 2) for all the traits ranged between 0.19 for canopy temperature before grain filling (CTBGF) and 0.97 for HD. Heritability for PH, TKW, and GY was 0.94, 0.70, and 0.72, respectively. AMMI analysis was conducted to rank genotypes based on GY across 15 environments and 20 high-yielding genotypes were identified with average GY between 4.8 and 5.3 t ha−1. The agronomic and physiological traits that negatively correlated with GY were PH (r = −0.32), PL (r = −0.43), SL (r = −0.33), TR (r = −0.21), CTBGF (r = −0.28), CTMGF (r = −0.33), and CTLGF (r = −0.13). Contrastingly, HD (r = 0.25), KSP (r = 0.25), KM−2 (r = 0.46), SM−2 (r = 0.3), and WAX (r = 0.18) were positively correlated with GY based on average data of all environments (Figure S1). In both heat-stressed environments (E12-TH11HT and E13-TH12HT), HD was negatively correlated with GY (r = −0.4), while TKW and PH were positively correlated with GY (r = 0.22 and r = 0.38, respectively).
Environmental variability at sites of field trials
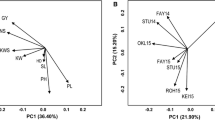
The environmental and phenotypic variability prevailing at the experimental sites were plotted based on maximum temperature (T max), minimum temperature (T min), and average temperature (T average) (Fig. 2). The average temperature during the two late seasons in Syria was 8.5 °C higher compared to the two normal growing seasons (Table S2). Similarly, the average relative humidity was 11% lower in the late seasons compared to the normal seasons. Average temperature at the other five experimental sites ranged between 16.9 °C (Sids, Egypt) and 28.1 °C (Wadmedani, Sudan). The sites in Egypt, which is considered a high-yielding environment, had lower average temperature (19.05 °C) compared to the sites in Sudan and Syria.

PCA bi-plot for environmental variability prevailing in the 15 experimental sites, in terms of minimum, maximum, and average temperature and rainfall during wheat growing seasons, along with phenotypic variability for days to heading (HD), plant height (PH), thousand kernel weight (TKW), and grain yield (GY). Codes for the sites are explained in Table 2
Population structure and linkage disequilibrium
Results using the DArT markers with MAF > 5% indicated the ΔK (Evanno et al. 2005) peaked at K = 2, providing evidence for the existence of two genetically distinct sub-groups in the GWAS panel (Figure S2a–d). The PCA also classified the population into two subpopulations, consistent with the inference utilizing the STRUCTURE software (Figure S2a). The first ten principal components (PCs) together explained about 38% of the total variability, while PC1 and PC2 together explained about 24.3% of total variation and partitioned the population into two distinct clusters. Six Australian cultivars and two CIMMYT genotypes were found to be admixture with the major clustering genotypes (n = 126) in cluster-1 (Figure S2a). On the other hand, the two check cultivars were grouped with cluster-2.
Markers in a significant LD were estimated to be maximum at 11 cM distance, while LD decay was observed beyond 11 cM (Figure S3). In total, 39.1% marker pairs were in a significant LD with 1882 marker pairs in perfect LD (r 2 = 1). No marker pairs in perfect LD were observed >50 cM (Table S3) with the highest markers in perfect LD ≤10 cM. LD pattern for all classes was higher for D genome compared to A- and B genomes with mean r 2 (0.22) and mean of r 2 > 0.2 (0.69).
Marker-trait association for agronomic stability
In total, 1785 DArT markers with MAF threshold >0.05 were used to identify MTAs. A significant positive correlation was observed between heritability (h 2) and number of MTAs identified for each trait (r = 0.72). In total, 245 MTAs were identified that were distributed over 66 loci (Table S4). MTAs were identified on all wheat chromosomes except 4D. For environment-specific MTAs, 16 loci were consistently identified over two or more environments and were referred to as consistent MTAs (Table 3; Table S3). The distribution of MTAs along wheat chromosomes and their co-localization with other traits are summarized in Table 3. Maximum numbers of MTAs were identified for DM which were distributed over 16 loci, followed by GY on 13 different loci. The minimum numbers of MTAs identified were four each for GWPS and TR with observed phenotypic variation (R 2) of 6–7.6% (Table S4). The proportion of genotypic variance (P G%) explained by each marker ranged from 0.02% by marker wPt-732556 on chromosome 1D for SNS to 27.62% for marker wPt-8172 on chromosome 1A for SW (Table 4). Maximum numbers of loci linked to traits were identified on A genome (112), followed by B genome (97), and the D genome (36), while 46 unmapped MTAs could not be assigned to any genome and were removed from further analysis. On the genetic map, the interval between 9 and 24 cM on chromosome 6A appeared to be the most important in the current study and was associated with multiple agronomic traits, including HD, DM, GY, and TKW. Of the 13 loci associated with GY, only three loci, wPt-6832 on chromosome 1B at 40 cM, wPt-7883 on 2B at 34 cM, and wPt-664276 on 6B at 15 cM, were unique, while the others co-localized and/or were pleiotropic with other important traits. These three loci which represent yield per se accounted for 5.7, 7.46, and 7.55% of the phenotypic variation, respectively.
Out of 245 MTAs, 48 MTAs identified on 27 loci were associated with traits averaged across environments (AE) and were distributed on all chromosomes except 3A, 3B, 4D, 5B, and 6D (Table 4).
Two physiological traits, canopy temperature (CT) and relative chlorophyll contents (in terms of SPAD values), were evaluated before, mid and after grain filling periods. However, both traits were not used for association mapping due to their low heritability.
Allelic effects of functional genes on flowering time and grain yield
Only two cultivars were found to have photoperiod sensitive allele (Ppd-D1b), while all other genotypes carried Ppd-D1a allele associated with photoperiod insensitivity. All three Vrn-1 loci (Vrn-A1, Vrn-B1, and Vrn-D1) formed eight haplotypes. The effect of Vrn-1 loci on HD and GY can be ranked as Vrn-A1 < Vrn-B1 < Vrn-D1. The incidence of one or combination of winter-type alleles (vrn-A1, vrn-B1, and vrn-D1) increased HD and GY in optimal environments; however, these alleles significantly decreased GY in heat-stressed environments (Fig. 3).

Allelic effects of Vrn-1 haplotypes on heading days (HD) and grain yield (GY). X-axis represents the combinations of Vrn-A1, Vrn-B1, and Vrn-D1 alleles and values in parenthesis represent the frequency of haplotypes. Left side Y-axis represents HD and two Y-axis on right side represents the GY in optimal and heat stress (HS) environments
Allelic effects on four important agronomic traits
Allelic effects were simulated for HD, PH, TKW, and GY to identify the relationship between desirable or unfavored alleles on phenotype averaged across environments (AE). For this purpose, only one marker associated with more than two environments and averaged data across all environments which accounted for the highest phenotypic variation on each locus was used (Fig. 4). The patterns of relationship were similar for four traits (GY, TKW, PH, and HD), where favored allele additively increased GY (R 2 = 0.14) and TKW (R 2 = 0.08), but decreased PH (R 2 = 0.081) and HD (R 2 = 0.02). Contrastingly, undesirable allele additively decreased GY (R 2 = 0.03) and TKW (R 2 = 0.04), but increased PH (R 2 = 0.21) and HD (R 2 = 0.11). Maximum number of varieties (35) had five favorable alleles, while maximum numbers of favored alleles (6) were observed in 26 varieties for HD (Fig. 4a). For PH, maximum favorable alleles (3) were present in 80 varieties, while maximum numbers of undesirable alleles (7) were found in four varieties (Fig. 4b). For TKW, maximum numbers of favored alleles (8) were observed in three varieties, while maximum numbers of unfavored alleles (8) were observed in 45 varieties (Fig. 4c). For GY, maximum numbers of favored alleles (5) were observed in 79 varieties, while maximum numbers of undesirable alleles (5) were observed in 10 varieties (Fig. 4d).

Allelic effects of favored and unfavored alleles based on linear regression, and frequency of favored and unfavored alleles for four important agronomic traits. a Heading days (HD), b plant height (PH), thousand kernel weight (TKW), and grain yield (GY)
Discussions
The relatively detailed phenotyping experiment conducted here is important to understand the relationship among different traits, their stability across environments (Lopes et al. 2012a, b), and their contribution towards accurate identification of stable genomic regions controlling trait stability under different environments. This data set resulted in identification of large number of MTAs; however, the major focus of the discussion was on MTAs identified for traits averaged across environments (Table 4).
Phenotypic and environmental plasticity
The diversity panel used in this study represents the spring wheat germplasm grown under Mediterranean climates, but targeted for tropical and subtropical environments aimed at enhancing wheat adaptability and yield improvement in heat prone environments. Arguably, this represents desirable panel to identify QTL underlying agronomic and physiological traits showing stability over a wide range of environments.
All the environments were characterized as arid-to-semi-arid tropics and sub-tropics, where six environments (E2-DON11, E3-DON12, E4-WAD11, E5-WAD12, E12-TH11HT, and E13-TH12HT) experienced average maximum temperature >30 °C, including two late sown trials in Syria. Desirable optimum temperature required during wheat reproductive phase is reported to be 12–22 °C and several reports confirmed that increase in temperature during reproductive phase significantly reduced GY (Saini and Aspinall 1982; Farooq et al. 2011 and literatures cited therein). In the present study, late sown environments in Syria, E12-TH11HT, and E13-TH12HT resulted in 26–66% yield reduction compared to the other environments, which is a result of the higher number of days with maximum temperature >35 °C in late sown environments. Similar trends were observed for other important agronomic traits (Fig. 1). In this study, the GWAS panel was characterized for 25 agronomic and physiological traits, potentially relevant in breeding and selection in trait-based crossing aimed at developing improved and higher yielding advanced lines. This is considered essential in identifying potentially valuable traits which are stable and widely expressed, and, therefore, can be used in crosses, knowing that positive alleles for each trait can be introduced into new genetic backgrounds. The traits that are used in breeding should be easy and inexpensive to measure, heritable, not result in penalties when conditions are favorable, nor be associated with negative pleiotropic effects on other important agronomic attributes.
The high GY levels of AMMI-based top-ranking genotypes were comparable to elite cultivars from Australia, ICARDA and CIMMYT used as checks. The heritability of four important agronomic traits (HD, PH, TKW, and GY) indicated a high level of robustness, where heritability for GY was relatively lower than other traits, but it was high enough to suggest an accurate experimental design. Relatively lower levels of heritability observed for physiological traits such as CT and SPAD during different growth phases indicated a relatively high error variance in measurements at different sites.
Genome coverage, population structure, and linkage disequilibrium
In general, DArT markers have good genome coverage with exceptional under-representation and gaps on D genome and in particular chromosomes 4D and 5D. Similar results on density of DArT markers and their use in GWAS experiments have previously been reported (Mulki et al. 2013; Rasheed et al. 2014). Subsequently, several high-density SNP array have become available in wheat, including Illumina infinium 9 K (Cavanagh et al. 2013) and 90 K (Wang et al. 2014) arrays. However, there are limited studies integrating both marker systems limiting the potential to accurately compare results from the deployment of both platforms.
Population structure inferred by STRUCTURE and PCA gave consistent results and indicated that two subpopulations were appropriate in delineating the structure in the association panel. The delineation into two subpopulations based on significant (P < 0.001) population differentiation was 0.38, which not only reaffirmed the identification of two subpopulations, but also indicated limited admixture. The Australian check cultivars (Drysdale, Gladius, and Wyalkatchem) were in admixture with ICARDA germplasm. The unstructured expression and low admixture of association panel were expected, because substantial numbers of genotypes do not share the same parents and indicated higher diversity among association panel. This trend was observed previously when population structure was determined in germplasm belonging to wider geographies (Zhang et al. 2013) and multiple breeding programs (Zanke et al. 2014a, b).
The presence of LD is a pre-requisite for association mapping with LD reportedly decaying more rapidly in cross-pollinated species than self-pollinated species (Brazauskas et al. 2011). A rapid LD decay indicates that more recombination exists within a short distance, and as a result, more markers are required to capture the high frequency of recombination. Almost 28% more pairs were in a significant LD on D genome as compared to A- and B genomes, and similar pattern was observed for higher average LD on D genome. The higher LD in D genome has been linked to episodes of recent introgression and population bottlenecks accompanying the origin of hexaploid wheat (Chao et al. 2010).
Marker-trait associations for agronomic and physiological traits
The identification of several QTL associated with yield and yield stability across diverse environments on different wheat chromosomes as is the case in this study has previously been reported. Recently, Acuna-Galindo et al. (2014) reported meta-QTLs (MQTL) from 30 different studies identified in drought and heat stress environments, hence was used as a reference to compare MTAs identified in this study. They identified group-1 chromosomes as having meta-QTLs for several important physiological and morphological traits, of which MQTL2 (chromosome 1A, 53–69 cM) and MQTL5 (chromosome 1B, 40 cM) appear similar to that identified in this study for some yield components.
An important locus on chromosome 2B between 6 and 11 cM interval had a pleiotropic effect on SL and LR. LR is an important physiological trait, known to reduce transpiration by cooling canopy temperature, providing avoidance mechanism for drought and heat stress (Ayeneh et al. 2002). This region was previously identified as MQTL14 associated with many yield-related traits (Acuna et al. 2014). The yield-related MTAs identified on chromosome 2D between 96 and 104 cM in our study was previously identified as stable MTA for GY using 9 K SNP markers within the same region (Edae et al. 2014). We also identified three DArT markers (2D; 114 cM) in adjacent region associated with LR. Given the extent of LD within D genome, this region was also regarded as the same locus, and is reported to be a meta-QTL (MQTL22) harboring loci for yield and associated traits (Acuna et al. 2014).
A stable locus on 3D at 11–16 cM for SW identified in this study, also had a pleiotropic effect on LR in some environments. This is likely to be a new QTL, since no information has previously been reported in the literature for this genomic region. Likewise, the environment-specific locus for WAX on chromosome 3A at 96 cM. WAX is an important physiological trait, which can be observed visually and known to decrease radiation load on leaf surface, therefore, reducing evapotranspiration rate (Dudnikov 2011). Two other loci on chromosome 3B between 61 and 67 cM and 113 cM were associated with important yield-related traits, including TKW, GY, DM, and GWPS. These results are in agreement with the earlier report of Edae et al. (2014) and Acuna-Galindo et al. (2014) who also identified MTAs and meta-QTLs for TKW and heading time within the same genomic regions, respectively.
A stable locus was found on chromosome 5A at 111 cM which had a pleiotropic effect on PH. This MTA was also associated with GY in E13-TH12L, which was considered heat-stress environment; therefore, this heat specific MTA could be important to maintain GY under heat stress. This locus was also associated with TKW; this region has previously been implicated in conferring heat/drought tolerance. Lopes et al. (2015) and Sukumaran et al. (2015) previously identified stable yield MTAs on chromosome 5A in CIMMYT germplasm. However, it is difficult to align findings from both studies to the current study due to the use of different marker systems (SNP 9 and 90 K, respectively). Two loci on chromosome 5B associated with GY and DM at 38–41 cM are probably the meta-QTL (MQTL43 and 45) reported by Acuna-Galindo et al. (2014) which were associated with several yield traits in heat prone environments consistent with previously yield-related MTA identified using DArT markers by Edae et al. (2014).
Chromosome 6A has previously been implicated to carry stable QTLs for yield-related traits in many studies; some of which were recently validated using near-isogenic lines (Simmonds et al. 2014). Four stable loci on chromosomes 6A and 6B that underpin HD, GY, and PH were identified in our study. Edae et al. (2014) and Sukumaran et al. (2015) reported similar results and suggested targeting these regions for QTL pyramiding and further validation. Acuna-Galindo et al. (2014) reported MQTL48-50 for TKW and photosynthesis in similar genomic regions, which have been consistently observed in several studies that evaluated germplasm for drought and heat tolerance (Yang et al. 2007; Acuna-Galindo et al. 2014). A single locus on chromosome 6A mapped in the confidence interval of 9 and 16 cM was associated with HD, and had a pleiotropic effect on KPS, TKW, and KM−2 and probably an important locus for GY and related traits. Acuna-Galindo et al. (2014) reported a meta-QTL in the loci adjacent to that in the preset study and most likely represent the same loci.
Two stable loci were identified on chromosome 7A, one on 7B and one on 7D. The 7A locus between 158 and 160 cM had a pleiotropic effect on PH and HD. An important locus on 7A between 11 and 13 cM associated with PL, SM−2, SNS, LR, and DM was also reported to be associated with stay green, an important trait contributing to GY in heat prone environments (Acuna-Galindo et al. 2014). The other environment-specific loci on 7A were associated with GWPS, PL, and SL; these were also previously identified by Acuna-Galindo et al. (2014) as meta-QTL for TKW, stay green, and carbon isotope discrimination. The locus on chromosome 7D between 171 and 173 cM associated with KPS, SM−2 and SNS accords with the result of earlier findings reported by Edae et al. (2014) for yield components and canopy temperature.
Effect of Vrn-1 loci on heading date and grain yield
Within the association panel, least average flowering time (77 days) was exhibited by 21 genotypes with dominant spring-type alleles across Vrn-1 loci, followed by 66 genotypes (79 days) with only one winter-type vrn-D1 allele, and were, respectively, lowest in GY in optimal environments. Likewise, stacking the winter-type alleles at Vrn-1 loci in varieties led to significant yield losses in heat stress environments, and could be attributed to the fact that winter-type alleles delayed flowering. Hence, varieties with winter-type alleles are exposed to heat stress for longer period, with peak temperature coinciding with grain filling period compared to spring-type alleles depending on the location/environment. These results indicated that breeding strategies should be devised to replace the winter-type alleles, especially at Vrn-A1 and Vrn-D1 loci, to develop early-flowering cultivars. This mechanism has been illustrated under diverse environments stress (Zhang et al. 2014), and will likely result in shortening the flowering time, hence time to maturity for the development of cultivars tailored for stressed environments.
Allelic effects of four important agronomic traits and residual effect on grain yield
The ultimate breeding objective is the development of cultivars with higher grains yield, a complexly inherited trait that is strongly influenced by time of flowering (HD) and PH in wheat (Lopes et al. 2015). QTL for yield per se are difficult to identify due to the confounding effects of HD, PH and the interwoven complexity of other traits/genes involved. Two strategies which can minimize their confounding effect include i) screening of the germplasm for major genes controlling HD (Vrn-1 and Ppd-1) and PH (Rht-1) and using this data as covariate in GWAS (Bentley et al. 2014; Sukumaran et al. 2015) or ii) filtering all the HD and PH QTLs co-localized with yield QTLs and assessing the residual effect of yield-related QTL. This association panel was not screened for all major HD and PH genes; therefore, the later strategy was used to assess the residual effect of yield-related MTAs. It was observed that three loci for GY on chromosome 1B (40 cM), 2B (34 cM), and 6B (12 cM) exclusively represent grain yield per se, and their residual effects were independent of other traits. Similarly, minor additive effects of five favorable and eight unfavorable alleles for GY were simulated. Similar results were observed for HD, PH, and TKW. The top 20 high-yielding genotypes, ranked by the AMMI, had very low kinship relationship and had 4–5 favorable alleles (Fig. 5) except for Shadi-4 and Qafzah-11/Bashiq-1–2, which do not have any of the detected favorable allele. This indicated the presence of rare favorable alleles in those varieties or their yield stability may be due to favorable alleles of other traits except yield per se. Recently, several studies were conducted on dissecting the genetic regions in European elite winter wheat for HD (Griffiths et al. 2009; Zanke et al. 2014a), PH (Griffiths et al. 2012; Zanke et al. 2014b), and also in CIMMYT germplasm (Edae et al. 2014; Sukumaran et al. 2015). These studies in combination with findings from our studies will be helpful in future studies to dissect the confounding effect of HD and PH QTL and facilitate the identification of QTL solely representing GY.

Kinship relationship among 20 top yielding varieties identified by AMMI analysis
Makumburage and Stapleton (2011) demonstrated that favorable alleles in a single environment show little improvement in multiple-stress environments. We also observed similar phenomenon in the current study, where environment-specific MTAs and MTAs for average environments do not significantly overlap. Differences in loci controlling stability and environment-specificity suggest that there may be separate evolutionary trajectories for them. Some of the loci identified in this study have additive effects, which are useful for selective breeding. To understand the genetic control of stability, it would also be useful to examine epistatic interactions, especially those interactions that are important only when both alleles are present (loci with no marginal effect). This may be especially important if good additive alleles were already fixed.
In the nutshell, the following genomic regions appear of prime importance for MAS-based QTL pyramiding: (1) genomic regions on chromosome 1B, 2B, 3A, 3B, 6A, 7A, and 7B are co-localized with yield associated traits and (2) aforementioned three loci on chromosome 1B, 2B, and 6B solely identified as genomic regions conferring GY per se after dissecting the confounding effects of PH and HD. Conclusively, this study demonstrated that GWAS can effectively detect stable and environment-specific QTL for multiple physio-agronomic traits. Based on results on the extent of LD across three sub-genomes, these loci may prove effective for pyramiding favorable alleles to develop germplasm with improved yield for heat prone environments.
Author contribution statement
FCO and TW designed the study. ECO, MIU, CUA, FM, AR, AJ, and AH performed the experiment and analyzed data. AR and FCO wrote the paper.
References
Acuna TLB, Rebetzke GJ, He X, Maynol E, Wade LJ (2014) Mapping quantitative trait loci associated with root penetration ability of wheat in contrasting environments. Mol Breed 34:631–642
Acuna-Galindo MA, Mason RE, Subrahmanyam NK, Hays D (2014) Meta-analysis of wheat QTL regions associated with adaptation to drought and heat stress. Crop Sci 55:477–492
Asseng S, Ewert F, Martre P, Rötter RP, Lobell DB et al (2015) Rising temperatures reduce global wheat production. Nat Clim Change 5:143–147
Ayeneh A, van Ginkel M, Reynolds MP, Ammar K (2002) Comparison of leaf, spike, peduncle and canopy temperature depression in wheat under heat stress. Field Crop Res 79:173–184
Beales J, Turner A, Griffiths S, Snape JW, Laurie DA (2007) A pseudo-response regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Theor Appl Genet 115:721–733
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B57:289–300
Bennett D, Reynolds M, Mullan D, Izanloo A, Kuchel H, Langridge P, Schnurbusch T (2012) Detection of two major grain yield QTL in bread wheat (Triticum aestivum L.) under heat, drought and high yield potential environments. Theor Appl Genet 125:1473–1485
Bentley AR, Scutari M, Gosman N, Faure S, Bedford F, Howell P, Cockram J, Rose GA, Barber T, Irigoyen J, Horsnell R, Pumfrey C, Winnie E, Schacht J, Beauchene K, Praud S, Greenland A, Balding D, Mackay IJ (2014) Applying association mapping and genomic selection to the dissection of key traits in elite European wheat. Theor Appl Genet 127:2619–2633
Bordes J, Goudemand E, Duchalais L, Chevarin L, Oury FX, Heumez E, Lapierre A, Perretant MR, Rolland B, Beghin D, Laurent V, Le Gouis J, Storlie E, Robert O, Charmet G (2014) Genome-wide association mapping of three important traits using bread wheat elite breeding populations. Mol Breed 33:755–768
Borlaug NE, Dowswell CR (2003) Feeding a world of ten billion people: a 21st century challenge. In: Proceedings of the international congress in the wake of the double helix: from the green revolution to the gene revolution, pp 27–31
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brazauskas G, Lenk I, Pedersen MG, Studer B, Lübberstedt T (2011) Genetic variation, population structure, and linkage disequilibrium in European elite germplasm of perennial ryegrass. Plant Sci 181:412–420
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Cavanagh CR, Chao SM, Wang SC, Huang BE, Stephen S, Kiani S, Forrest K, Saintenac C, Brown-Guedira GL, Akhunova A, See D, Bai GH, Pumphrey M, Tomar L, Wong DB, Kong S, Reynolds M, da Silva ML, Bockelman H, Talbert L, Anderson JA, Dreisigacker S, Baenziger S, Carter A, Korzun V, Morrell PL, Dubcovsky J, Morell MK, Sorrells ME, Hayden MJ, Akhunov E (2013) Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc Nat Acad Sci USA 110:8057–8062
Chao SM, Dubcovsky J, Dvorak J, Luo MC, Baenziger SP, Matnyazov R, Clark DR, Talbert LE, Anderson JA, Dreisigacker S, Glover K, Chen JL, Campbell K, Bruckner PL, Rudd JC, Haley S, Carver BF, Perry S, Sorrells ME, Akhunov ED (2010) Population- and genome-specific patterns of linkage disequilibrium and SNP variation in spring and winter wheat (Triticum aestivum L.). BMC Genom 11:727
Crossa J, Burgueno J, Dreisigacker S, Vargas M, Herrera-Foessel SA, Lillemo M, Singh RP, Trethowan R, Warburton M, Franco J, Reynolds M, Crouch JH, Ortiz R (2007) Association analysis of historical bread wheat germplasm using additive genetic covariance of relatives and population structure. Genetics 177:1889–1913
Dudnikov AJ (2011) Waxiness in Aegilops tauschii: its occurrence in natural habitats of the species. Cereal Res Commun 39:283–288
Edae EA, Byrne PF, Haley SD, Lopes MS, Reynolds MP (2014) Genome-wide association mapping of yield and yield components of spring wheat under contrasting moisture regimes. Theor Appl Genet 127:791–807
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Farooq M, Bramley H, Palta JA, Siddique KHM (2011) Heat stress in wheat during reproductive and grain-filling phases. Crit Rev Plant Sci 30:1–17. doi:10.1080/07352689.2011.615687
Fu DL, Szucs P, Yan LL, Helguera M, Skinner JS, von Zitzewitz J, Hayes PM, Dubcovsky J (2005) Large deletions within the first intron in VRN-1 are associated with spring growth habit in barley and wheat. Mol Genet Genom 274:442–443
Gao F, Wen W, Liu J, Rasheed A, Yin G, Xia X, Wu X, He Z (2015) Genome-wide linkage mapping of Qtl for yield components, plant height and yield-related physiological traits in the chinese wheat cross Zhou 8425b/Chinese Spring. Front Plant Sci 6:1099
Griffiths S, Simmonds J, Leverington M, Wang Y, Fish L, Sayers L, Alibert L, Orford S, Wingen L, Herry L, Faure S, Laurie D, Bilham L, Snape J (2009) Meta-QTL analysis of the genetic control of ear emergence in elite European winter wheat germplasm. Theor Appl Genet 119:383–395
Griffiths S, Simmonds J, Leverington M, Wang YK, Fish L, Sayers L, Alibert L, Orford S, Wingen L, Snape J (2012) Meta-QTL analysis of the genetic control of crop height in elite European winter wheat germplasm. Mol Breed 29:159–171
Jannink JL (2007) Identifying quantitative trait locus by genetic background interactions in association studies. Genetics 176:553–561
Jighly A, Oyiga BC, Makdis F, Nazari K, Youssef O, Tadesse W, Abdalla O, Ogbonnaya FC (2015) Genome wide DArT and SNP scan for QTL associated with resistance to stripe rust (Puccinia striiformis f. sp. tritici) in elite ICARDA Wheat (Triticum aestivum L.) germplasm. Theor Appl Genet 128:1277–1295
Jighly A, Alagu M, Makdis F, Singh M, Singh S, Emebiri LC, Ogbonnaya FC (2016) Genomic regions conferring resistance to multiple fungal pathogens in synthetic hexaploid wheat. Mol Breeding 36:127. doi:10.1007/s11032-016-0541-4
Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, Sabatti C, Eskin E (2010) Variance component model to account for sample structure in genome-wide association studies. Nat Genet 42:348–354
Kuchel H, Williams KJ, Langridge P, Eagles HA, Jefferies SP (2007) Genetic dissection of grain yield in bread wheat. I. QTL analysis. Theor Appl Genet 115:1029–1041
Lipka AE, Feng T, Qishan WJP, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang Z (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28:2397–2399
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Lopes MS, Reynolds MP, Jalal-Kamali MR, Moussa M, Feltaous Y, Tahir ISA, Barma N, Vargas M, Mannes Y, Baum M (2012a) The yield correlations of selectable physiological traits in a population of advanced spring wheat lines grown in warm and drought environments. Field Crop Res 128:129–136
Lopes MS, Reynolds MP, Manes Y, Singh RP, Crossa J, Braun HJ (2012b) Genetic yield gains and changes in associated traits of CIMMYT spring bread wheat in a “historic” set representing 30 years of Breeding. Crop Sci 52:1123–1131
Lopes MS, Reynolds MP, McIntyre CL, Mathews KL, Jalal Kamali MR, Moussa M, Feltaous Y, Tahir ISA, Chatrath R, Ogbonnaya FC, Baum M (2013) QTLs for yield and associated traits in the Seri/Babax population grown across several environments in Mexico, in the West Asia, North Africa, and South Asia regions. Theor Appl Genet 126:971–984. doi:10.1007/s00122-012-2030-4
Lopes MS, Dreisigacker S, Pena RJ, Sukumaran S, Reynolds MP (2015) Genetic characterization of the wheat association mapping initiative (WAMI) panel for dissection of complex traits in spring wheat. Theor Appl Genet 128:453–464
Makumburage GB, Stapleton A (2011) Phenotype uniformity in combined-stress environments has a different genetic architecture than in single-stress treatments. Front Plant Sci 2:12
Marza F, Bai GH, Carver BF, Zhou WC (2006) Quantitative trait loci for yield and related traits in the wheat population Ning7840 × Clark. Theor Appl Genet 112:688–698
McIntyre CL, Mathews KL, Rattey A, Chapman SC, Drenth J, Ghaderi M, Reynolds M, Shorter R (2010) Molecular detection of genomic regions associated with grain yield and yield-related components in an elite bread wheat cross evaluated under irrigated and rainfed conditions. Theor Appl Genet 120:527–541
Mulki MA, Jighly A, Ye GY, Emebiri LC, Moody D, Ansari O, Ogbonnaya FC (2013) Association mapping for soilborne pathogen resistance in synthetic hexaploid wheat. Mol Breed 31:299–311
Neumann K, Kobiljski B, Dencic S, Varshney RK, Borner A (2011) Genome-wide association mapping: a case study in bread wheat (Triticum aestivum L.). Mol Breed 27:37–58
Ogbonnaya FC, Seah S, Delibes A, Jahier J, Lopez-Brana I, Eastwood RF, Lagudah ES (2001) Molecular-genetic characterisation of a new nematode resistance gene in wheat. Theor Appl Genet 102:623–629
Olivares-Villegas JJ, Reynolds MP, McDonald GK (2007) Drought-adaptive attributes in the Seri/Babax hexaploid wheat population. Func Plant Biol 34:189–203
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rasheed A, Xia X, Ogbonnaya F, Mahmood T, Zhang Z, Mujeeb-Kazi A, He Z (2014) Genome-wide association for grain morphology in synthetic hexaploid wheats using digital imaging analysis. BMC Plant Biol 14:128
Ray DK, Mueller ND, West PC, Foley JA (2013) Yield trends are insufficient to double global crop production by 2050. PLoS One 8:e66428
Rebetzke GJ, Rattey AR, Farquhar GD, Richards RA, Condon AG (2013) Genomic regions for canopy temperature and their genetic association with stomatal conductance and grain yield in wheat. Funct Plant Biol 40:14–33
Saini HS, Aspinall D (1982) Abnormal sporogenesis in wheat (Triticum aestivum L.) induced by short periods of high temperature. Ann Bot 49:835–846
Segura V, Vilhjalmsson BJ, Platt A, Korte A, Seren U, Long Q, Nordborg M (2012) An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat Genet 44:825–830
Simmonds J, Scott P, Leverington-Waite M, Turner AS, Brinton J, Korzun V, Snape J, Uauy C (2014) Identification and independent validation of a stable yield and thousand grain weight QTL on chromosome 6A of hexaploid wheat (Triticum aestivum L.). BMC Plant Biol 14:191. doi:10.1186/s12870-014-0191-9
Sukumaran S, Dreisigacker S, Lopes MS, Chavez P, Reynolds MP (2015) Genome-wide association study for grain yield and related traits in an elite spring wheat population grown in temperate irrigated environments. Theor Appl Genet 128:353–363
Tadesse W, Ogbonnaya FC, Jighly A, Sanchez-Garcia M, Sohail Q, Rajaram S, Baum M (2015) Genome-wide association mapping of yield and grain quality traits in winter wheat genotypes. PLoS One 10:e0141339
Talukder S, Babar M, Vijayalakshmi K, Poland J, Prasad P, Bowden R, Fritz A (2014) Mapping QTL for the traits associated with heat tolerance in wheat (Triticum aestivum L.). BMC Genet 15:97
Tang Y-L, Li J, Wu Y-Q, Wei H-T, Li C-S, Yang W-Y, Chen F (2011) Identification of QTLs for yield-related traits in the recombinant inbred line population derived from the cross between a synthetic hexaploid wheat-derived variety Chuanmai 42 and a Chinese eilte variety Chuannong 16. Agric Sci China 10:1665–1680
Trethowan RM (2014) Delivering drought tolerance to those who need it: from genetic resource to cultivar. Crop Pasture Sci 65:645–654
Utz HF, Melchinger AE, Schön CC (2000) Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics 154:1839–1849
Wang SC, Wong DB, Forrest K, Allen A, Chao SM, Huang BE, Maccaferri M, Salvi S, Milner SG, Cattivelli L, Mastrangelo AM, Whan A, Stephen S, Barker G, Wieseke R, Plieske J, Lillemo M, Mather D, Appels R, Dolferus R, Brown-Guedira G, Korol A, Akhunova AR, Feuillet C, Salse J, Morgante M, Pozniak C, Luo MC, Dvorak J, Morell M, Dubcovsky J, Ganal M, Tuberosa R, Lawley C, Mikoulitch I, Cavanagh C, Edwards KJ, Hayden M, Akhunov E, Sequencing IWG (2014) Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol J 12:787–796
Weir BS (1996) Genetic data analysis II. Sinauer Associates, Sunderland
Würschum T, Langer SM, Longin CFH (2015) Genetic control of plant height in European winter wheat cultivars. Theor Appl Genet 128:865–874
Yan L, Helguera M, Kato K, Fukuyama S, Sherman J, Dubcovsky J (2004) Allelic variation at the VRN-1 promoter region in polyploid wheat. Theor Appl Genet 109:1677–1686
Yang DL, Jing RL, Chang XP, Li W (2007) Quantitative trait loci mapping for chlorophyll fluorescence and associated traits in wheat (Triticum aestivum). J Integr Plant Biol 49:646–654
Zadoks JC, Chang TT, Konzak CF (1974) A decimal code for the growth stages of cereals. Weed Res 14:415–421
Zanke C, Ling J, Plieske J, Kollers S, Ebmeyer E, Korzun V, Argillier O, Stiewe G, Hinze M, Beier S, Ganal MW, Roder MS (2014a) Genetic architecture of main effect QTL for heading date in European winter wheat. Front Plant Sci 5:217
Zanke CD, Ling J, Plieske J, Kollers S, Ebmeyer E, Korzun V, Argillier O, Stiewe G, Hinze M, Neumann K, Ganal MW, Roder MS (2014b) Whole genome association mapping of plant height in winter wheat (Triticum aestivum L.). PLoS One 9:e113287
Zegeye H, Rasheed A, Makdis F, Badebo A, Ogbonnaya FC (2014) Genome-wide association mapping for seedling and adult plant resistance to stripe rust in synthetic hexaploid wheat. PLoS ONE 9(8):e105593. doi:10.1371/journal.pone.0105593
Zhang Z, Ersoz E, Lai C-Q, Todhunter RJ, Tiwari HK, Gore MA, Bradbury PJ, Yu J, Arnett DK, Ordovas JM, Buckler ES (2010) Mixed linear model approach adapted for genome-wide association studies. Nat Genet 42:355–360
Zhang K, Wang J, Zhang L, Rong C, Zhao F, Peng T, Li H, Cheng D, Liu X, Qin H, Zhang A, Tong Y, Wang D (2013) Association analysis of genomic loci important for grain weight control in elite common wheat varieties cultivated with variable water and fertiliser supply. PLoS One 8:e57853
Zhang JJ, Dell B, Biddulph B, Khan N, Xu YJ, Luo H, Appels R (2014) Vernalization gene combination to maximize grain yield in bread wheat (Triticum aestivum L.) in diverse environments. Euphytica 198:439–454
Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C, Tang C, Toomajian C, Zheng H, Dean C, Marjoram P, Nordborg M (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet 3:e4
Acknowledgements
Funding was provided by Grains Research and Development Corporation and ICARDA (Grant No. ICA00009).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interest.
Additional information
Communicated by Andreas Graner.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ogbonnaya, F.C., Rasheed, A., Okechukwu, E.C. et al. Genome-wide association study for agronomic and physiological traits in spring wheat evaluated in a range of heat prone environments. Theor Appl Genet 130, 1819–1835 (2017). https://doi.org/10.1007/s00122-017-2927-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-017-2927-z