
Abstract
Key message
This manuscript provides genome-level analysis of disease resistance genes in four maize lines, including studies of haplotype and resistance gene number as well as selection and recombination.
Abstract
The Rp1 locus of maize is a complex resistance gene (R-gene) cluster that confers race-specific resistance to Puccinia sorghi, the causal agent of common leaf rust. Rp1 NB-LRR disease resistance genes were isolated from two Rp1 haplotypes (HRp1-B and HRp1-M) and two maize inbred lines (B73 and H95). Sixty-one Rp1 genes were isolated from Rp1-B, Rp1-M, B73 and H95 with a PCR-based approach. The four maize lines carried from 12 to 19 Rp1 genes. From 4 to 9 of the identified Rp1 genes were transcribed in the four maize lines. The Rp1 gene nucleotide diversity was higher in HRp1-B and HRp1-M than in B73 and H95. Phylogenic analysis of 69 Rp1 genes revealed that the Rp1 genes maintained in HRp1-B, HRp1-M and H95 are evolving independently of each other, while Rp1 genes in B73 and HRp1-D appear more like each other than they do genes in the other lines. The results also revealed that the analysed Rp1 R-genes were under positive selection in HRp1-M and B73. Intragenic recombination was detected in Rp1 genes maintained in the four maize lines. This demonstrates that a genetic process that has the potential to generate new resistance genes with new specificities is active at the Rp1 locus in the four analysed maize lines and that the new resistance genes may act against newly arising pathogen races that become prevalent in the pathogen population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One vital trait that affects all plants grown in agricultural or natural environments is their ability to withstand disease. Plants have evolved several mechanisms of protection in response to pathogen infection. The plant defense response is one of these systems and involves an elaborate induction process following plant recognition of a pathogen avirulence gene product by a plant resistance gene product. The most abundant resistance genes are those that encode proteins with nucleotide binding site (NB) and leucine-rich repeat (LRR) domains (Bent 1996; Hulbert et al. 2001). To date, about three-fourths of the plant disease resistance genes that have been cloned are from this class. Several conserved motifs are maintained in the NB domain and are responsible for nucleotide binding and initiating a signal transduction cascade to activate plant defenses (Tameling et al. 2002). The LRR region is typically involved in protein–protein interactions and pathogen recognition specificity (Leister and Katagiri 2000; Dangl and Jones 2001; Jiang et al. 2007).
Despite these defenses, the pathogen’s ability to change or lose avirulence genes renders the genes unrecognizable by the corresponding plant resistance gene protein, resulting in plant susceptibility. Therefore, the plant population must be able to produce new specificities in response to the changing pathogen to protect themselves and survive. More than 20 maize resistance genes were identified as Rp loci during the 1960s (Hooker and Russell 1962; Hagan and Hooker 1965; Saxena and Hooker 1968; Wilkinson and Hooker 1968). The majority of these genes mapped to a region on the short arm of chromosome 10. The Rp1 complex consists of a highly variable cluster of fourteen (Rp1-A to Rp1-F and Rp1-H to Rp1-N) NB-LRR genes (Collins et al. 1999).
Each of these genes represents a gene family that can be distinguished by the P. sorghi isolates to which it confers resistance. Extensive genetic and molecular analysis of this locus has demonstrated that unequal intragenic (new gene) or intergenic (gene reassortment) crossing over events can create new resistance specificities (Richter et al. 1995; Smith and Hulbert 2003, 2005; Smith et al. 2010). Therefore, Rp1 is a classic example of a complex disease resistance locus. New resistance specificities have been selected in a few other systems. The best characterized system is the L locus of flax. Variant alleles were first characterized in flax in the early 1970s and were molecularly characterized more recently. Similar to the Rp1 locus, it was found that the new resistances at the L locus were the result of recombination events between the two parental alleles involved in the cross-over event (Ellis et al. 1999, 2000; Luck et al. 2000). The selection of variants with new resistant phenotypes from different systems demonstrates the significance of recombination in creating novel genes or haplotypes with new resistance specificities.
Comparative analysis of resistance gene family members has provided evidence that R-genes are subject to positive selection, particularly in the LRR region. The LRR region encodes solvent-exposed residues that are predicted to interact with the corresponding Avr protein in the pathogen in a direct or indirect manner (Kobe and Deisenhofer 1995; Hu and Hulbert 1996; Hulbert and Pumphrey 2014). Mondragón-Palomino et al. (2002) demonstrated that selection has acted to diversify the LRR domain of several groups of Arabidopsis NB-LRR gene family members (Mondragón-Palomino et al. 2002). Additionally, comparative analysis of NB-LRR R-gene family members from tomato, lettuce, rice and flax has demonstrated that solvent-exposed residues of the LRRs are hypervariable (Mondragón-Palomino et al. 2002). This indicates that selective forces imposed by the pathogen incite allelic diversity (Hulbert et al. 2001; Zhang et al. 2013; Hurni et al. 2013). For this reason, the selective advantage of carrying an R-gene and the pressure imposed on the R-gene to diversify depends on the frequency of the corresponding Avr gene in the pathogen population. Investigation of R-gene variation patterns has proven to be a powerful tool to estimate R-gene abundance and selection pressure.
Molecular analysis of the Rp1 locus has contributed greatly to what is known about complex disease resistance loci and how they evolve. Analysis of R-genes in previously uncharacterized Rp1 haplotypes will help identify resistance gene haplotypes that will be potentially important for the production of disease resistant varieties. The specific objectives of this study were to analyse the haplotypic diversity of NB-LRR R-genes in previously uncharacterized Rp1 haplotypes and to postulate how these R-genes evolve. This study describes the diversity and evolution of Rp1 R-genes from three Rp1 haplotypes and two maize inbred lines. The findings provide insights into Rp1 R-gene number, transcription and diversity, thus generating a data resource for future use of this class of genes for improved maize cultivar performance.
Materials and methods
Plant material
Three Rp1 haplotypes (HRp1-B, HRp1-D and HRp1-M) and two maize inbred lines (B73, H95) were selected for the analysis. The Rp1 haplotypes were selected based on resistance specificity and the predicted minimum number of Rp1 genes maintained in each haplotype. The two Rp1 haplotypes (HRp1-B, HRp1-M) differing in resistance specificity and two maize inbred lines (B73 and H95), were considered appropriate to generate the crosses needed for detailed resistance gene characterization, generation of recombinant haplotypes with new resistance specificities and improved maize cultivar performance. Additionally, Southern blot analysis indicated that HRp1-B and HRp1-M carry the fewest number of Rp1 genes among the Rp1 haplotypes that have not been subjected to sequence analysis (Collins et al. 1999). Therefore, HRp1-B and HRp1-M were selected as additional Rp1 haplotypes because fewer clones would need to be analysed to sample all of the Rp1 genes maintained in each haplotype. The Rp1-D haplotype is the best characterized Rp1 haplotype and carries only nine Rp1 genes (Sun et al. 2001). Rp1 genes from the Rp1-D haplotype were used in this study for comparative analysis because all of the Rp1 genes from this haplotype have been cloned and characterized and are a classic example of a resistance gene family. The three Rp1 haplotypes are in the H95 background, while many other previously characterized Rp1 haplotypes are maintained in the B73 background. B73 and H95 are susceptible to all known P. sorghi isolates and are homozygous recessive (rp1-rp1) at the Rp1 locus. Therefore, both H95 and B73 were selected for this work to analyse the Rp1 genes in the two maize inbred lines. The Rp1-B, -D and -M haplotypes utilized for this work are near-isogenic and carry a single dominate Rp1 gene (Rp1-B/Rp1-B; Rp1-D/Rp1-D or Rp1-M/Rp1-M). Seeds from the five maize lines were collected and planted in pots in the greenhouse to collect tissue for DNA and RNA extractions. All of the maize lines were grown in fields with high disease (P. sorghi) pressure and were maintained in the greenhouse.
Genomic DNA and total RNA was isolated from fully expanded uninoculated second leaf sections collected from the four maize lines (HRp1-B, HRp1-M, B73 and H95) using CTAB extraction methods with modifications (Murray and Thompson 1980) and Life Technologies (Rockville, MD, USA) Trizol reagent as described by the manufacturer, respectively. Four seedlings were collected for each maize line and combined for DNA and RNA extractions. The quality of the DNA and RNA samples were assessed on a 0.5 % agarose gel and the quantity was determined with the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies).
PCR amplification and cloning of Rp1 genes
A PCR-based approach was used to isolate the C-terminal half of the LRR region (1.0 kb) of Rp1 genes from the four maize lines. Rp1 genes are partially conserved in this region and highly duplicated allowing for the design of highly conserved primers that will amplify the majority if not all of the Rp1 genes in a haplotype (Sun et al. 2001; Smith et al. 2004, 2010). This method facilitates high fidelity amplification of large target lengths (0.1–48 kb) and has proven to be very efficient when amplifying the 4 kb coding region of many Rp1 genes (Sun et al. 2001; Smith et al. 2004, 2010). PCR amplification of genomic DNA templates was performed with Enhanced DNA Polymerase (Stratagene, La Jolla, CA, USA) with ~1 min of extension time for every kb of fragment size. All other parameters were performed according to the manufacturer’s suggestions. The DNAs were amplified with a conserved primer pair (Forward P19-TTGATAGGTTGGTTGTAAGTG; Reverse 4890R-CCTGAACTCTGGAGCTTCAAC) designed from the LRR and 3′ UTR region (Fig. 1). This region has been used in the design of PCR-based cloning to characterize R-genes from dicot and monocot species (Meyers et al. 2003). The resulting 1.0 kb PCR products were isolated from a 1.5 % agarose gel, purified with an Invitrogen Quick gel extraction kit (Carlsbad, CA, USA) and cloned into the Invitrogen (Carlsbad, CA, USA) TOPO TA cloning vector using the methods described by the manufacturer. One library for each Rp1 haplotype (Rp1-B and -M) and maize inbred line (H95 and B73) was generated from each cloning experiment. Ninety-six to one-hundred-and-ninety-two clones were sequenced with T3 and T7 primers per gel purification product using the Big-Dye Terminator v3.1 cycle sequencing kit (Applied Biosystems), following the manufacturer’s protocol. All sequencing was performed at the UGA sequencing facility.

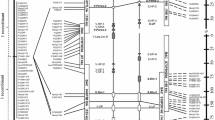
Rp1 gene structure. The line between LRRI and LRRII indicates a 284 bp region that does not conform to the LRR consensus sequence (××L×L××). Horizontal arrows indicate primer sites and directionality. The asterisk indicates a stop codon. The vertical arrow indicates a 79 bp intron in the 3′ untranslated region (UTR)
Analysis of Rp1 transcripts
RT-PCR was performed on 500 ng of total RNA isolated from fully expanded uninoculated second leaf sections taken from the two Rp1 (Rp1-B and Rp1-M) haplotypes and two maize inbred lines (B73 and H95) to indentify the transcribed Rp1 genes. Four seedlings collected from each maize line were combined for RNA extractions. Rp1 transcripts have been characterized previously from the Rp1-D haplotype. Rp1 cDNA sequences were amplified using a StrataScript First-Strand Synthesis System as described by the manufacturer (Stratagene, LaJolla, CA, USA). Following first strand synthesis with an oligo dT primer, second strand synthesis was performed using a primer pair (4065F-TGCCATGAGCAGAGGATAAGAT/4890R-CCTGAACTCTGGAGCTTCAAC) designed from a conserved region among Rp1 genes that flanks a highly polymorphic region at the 3′ end of Rp1 genes to differentiate between Rp1 sequences (Fig. 1). The primer pair also flanks a 3′ intron for detection of genomic DNA contamination. The resulting 1.2 kb cDNA product was gel purified and cloned into the Invitrogen (Carlsbad, CA, USA) TOPO TA cloning vector. Ninety-six to one-hundred-and-ninety-two cDNA clones were sequenced from each Rp1 haplotype and maize inbred line with T3 and T7 primers (Table 1). The PCR amplified genomic and cDNA clones overlap in the C-terminal half of the LRR region. Therefore, the cDNA sequences with homology to Rp1 genes were aligned to the Rp1 genomic sequences isolated from the Rp1 haplotypes and maize inbred lines to identify the transcribed Rp1 genes.
Sequence diversity and recombination analysis
Rp1 genomic and cDNA sequences isolated from the two Rp1 haplotypes and two maize inbred lines were used to run a BLASTN search against the NCBI non-redundant database to verify putative homologies to known resistance genes. Sequences from genomic and cDNA clones were aligned separately to identify the overlapping region and to assemble the LRR region of each clone. The LRR region of each genomic and cDNA clone was aligned to identify redundant clones, estimate the number of Rp1 genes maintained in the four maize lines and determine how many Rp1 gene are transcribed in each maize line. Due to the polymorphic nature of the LRR region of Rp1 genes, this region has been particularly useful when distinguishing between Rp1 genes and estimating gene number (Smith et al. 2010).
The accuracy and assembly of Rp1 sequences were verified with PHRED and PHRAP bioinformatics tools, respectively (Ewing and Green 1998) as well as BioEdit (Hall 1999). The minimum value for acceptable sequences was set at 20 (q > 20) for a PHRED quality score, or an accuracy of 99.99 % (Ewing and Green 1998). The accuracy of single nucleotide polymorphisms was further verified by scrutinizing chromatograms. Additionally, two identical clones from different PCR reactions were sequenced and aligned to identify nucleotide polymorphisms introduced by PCR. Sequences were manually edited when errors were found to be introduced. The ends of sequences were trimmed when sequences did not meet the minimum value for a PHRED quality score (q > 20). All sequencing was performed in the Genomics and Bioinformatics Sequencing Facility at the University of Georgia.
DnaSP (DNA Sequence Polymorphism) software package version 5.10.01 (Librado and Rozas 2009) was used to estimate the sequence diversity of Rp1 genes by calculating the average pairwise difference between sequences, π (Tajima 1983) and the number of segregating sites in a sample, θ w (Watterson 1975). The latter parameter has expected values of 4Neμ for an autosomal gene of a diploid organism, where Ne and μ are the effective population size and the mutation rate per nucleotide site per generation, respectively. Total sequences and silent sites were considered separately for nucleotide diversity estimates (Librado and Rozas 2009). The recombination parameter (R) per gene and between adjacent sites was calculated based on the average number of nucleotide differences between pairs of sequences (Hudson et al. 1987). The recombination parameter (R) has expected values of 4Nr for an autosomal gene of a diploid organism, where N is the population size and r is the recombination rate per sequence per gene.
Tajima’s D analysis was performed using DnaSP and PAML (Yang 1997; Yang et al. 2000) to test for deviations from the neutral equilibrium model of evolution. Tajima’s D is based on the discrepancy between the mean pairwise differences (π) and Watterson’s estimator (θ w ) (Tajima 1989), and was calculated for Rp1 genes in each maize line at all sites and at silent sites separately.
A phylogenetic tree was constructed with Rp1 genomic sequences by MEGA 5 (Nei 1987; Saitou and Nei 1987; Tamura et al. 2007, 2011) using the Neighbor-Joining (NJ) method with distances represented as the number of nucleotide differences. One thousand bootstrap replicates were used to assess the confidence of the phylogeny. Neighbor-joining tree construction of Rp1 genes from each maize line facilitated the analysis of sequence diversity and the distribution of Rp1 genes into haplotypes. Sequence data from this article have been deposited in Genbank under accession numbers: Sequences have seen submitted to Genbank.
Results
Estimate of Rp1 genes and transcribed members in the four maize lines
To estimate the number of Rp1 genes maintained in the analysed maize lines and to identify the transcribed Rp1 genes, two 3′ end primer pairs (P19/4890R; 4065F/4890R) designed from conserved regions within the C-terminal end of the LRR domain of characterized Rp1 genes was used to amplify genomic and cDNA template from HRp1-B, HRp1-M, B73 and H95 (Fig. 1). The use of a conserved primer pair allows for the amplification of the majority, if not all, of the Rp1 genes in a haplotype. Additionally, the C-terminal end of the LRR region was selected for this work because PCR primers could be designed to generate an amplification product that would cover the most divergent region of Rp1 genes and where recombination is most frequently detected (Lawrence et al. 1997; Ellis et al. 2000). This would alleviate the need to sequence complete Rp1 genes that are ~4 kb in size from maize lines that likely carry a large number of Rp1 genes. A PCR amplified fragment ~1.0 kb in size corresponding to the divergent C-terminal end of the LRR region was isolated and cloned as putative Rp1 resistance genes. When Rp1 clones were sequenced from each haplotype, multiple clones corresponded to individual Rp1 genes, suggesting that the majority of the Rp1 genes in these haplotypes had been sampled efficiently. It is possible that there are more than the identified Rp1 genes in each haplotype, since genes that are identical through the sequenced ~1.0 kb region would not be differentiated.
A total of 544 genomic and cDNA clones were sequenced for the four maize lines (Table 1). From these clones, 531 were observed to be homologous to the LRR sequences of Rp1 and other NB-LRR encoding genes previously isolated from maize and other plant species. A 79 bp intron was present between the conserved primer sites and was used to detect genomic contamination in the cDNA sequences. Alignment of the genomic and cDNA sequences demonstrated that the 3′ intron had been removed from the cDNA sequences indicating the absence of genomic contamination. The Rp1 sequences were designated Rp1-B-p, Rp1-M-p, Rp1-B73-p or Rp1-H95-p. The Rp1 designation indicates the locus and is followed by the name of the maize line the Rp1 genes were isolated from. The p designation corresponds to the assigned paralog number (Table 1).
Ninety-six of the 531 PCR amplified clones were genomic clones isolated from the Rp1-B haplotype. Alignment of the Rp1-B genomic sequences and removal of redundant sequences identified 16 different sequences representing unique Rp1 genes and were designated as such (Table 1). The 16 Rp1 genes were aligned with the 96 Rp1-B cDNA sequences to identify the transcribed Rp1-B genes. The Rp1-B cDNA sequences corresponded to eight of the 16 Rp1-B sequences indicating, HRp1-B carries at least 16 unique Rp1 genes, eight of which are transcribed. The eight LRR encoding Rp1-B genes had uninterrupted ORFs after the 3′ intron was removed and were identical to their corresponding cDNA sequences. The remaining eight Rp1-B genes did not correspond to any of the cDNAs and harbored stop codons or frameshift mutations. These genes were therefore considered untranscribed Rp1 genes and are likely pseudogenes.
Ninety-six PCR amplified genomic and cDNA clones were isolated from HRp1-M. Based on sequence alignment, 19 different Rp1 genes were represented among the 96 clones (Table 1). The HRp1-M cDNA sequences corresponded to twelve of the genomic sequences (Table 1). Therefore, HRp1-M carries at least 19 unique Rp1 genes, twelve of which are transcribed. The transcribed genes contained uninterrupted ORFs and were identical to their corresponding cDNA sequences with the exception of the 3′ intron. Seven of the Rp1-M genes contained stop codons and/or frameshift mutations.
Alignment of 96 genomic and cDNA sequences isolated from B73 identified fourteen different sequences (Table 1). The B73 cDNA sequences were identical to four B73 genomic sequences indicating B73 carries at least fourteen different Rp1 genes and four of these genes are transcribed members. One-hundred-and-sixty genomic and cDNA clones were analysed for the H95 maize line. The clones represented twelve different Rp1 genes (Table 1). Eight of these genes are transcribed. All of the transcribed Rp1 genes isolated from B73 (4 genes) and H95 (8 genes) contained uninterrupted ORFs, while the untranscribed Rp1 genes harbored stop codons or frameshift mutations.
Rp1 nucleotide and haplotype diversity
The LRR region of Rp1 genes was analysed in HRp1-B, HRp1-M, B73 and H95 using DnaSP (Librado and Rozas 2009) to determine the genetic diversity of these genes at a complex disease resistance locus. The length of aligned sequence for Rp1 genes from the four maize lines varied between 696 and 754 bp and contains only coding sites. This variability was due to manual trimming of the ends of sequences that did not meet the minimum value for a PHRED quality score (q >20). HRp1-B, HRp1-M, B73 and H95 maize lines exhibited 342, 351, 52 and 371 SNPs, respectively (Table 2). Although numerous indels and SNPs were detected, 39 (52 %) of the total number of different Rp1 genes (61) identified in the four maize lines appeared fully functional and were transcribed. For the remaining 22 Rp1 genes (48 %), frameshifts or SNPs yielding premature stop codons suggested nonfunctional alleles, at levels of 50 % (8 of 16 Rp1 genes are nonfunctional), 37 % (7 of 19 Rp1 genes are nonfunctional), 71 % (10 of 14 Rp1 genes are nonfunctional) and 33 % (4 of 12 Rp1 gene are nonfunctional) in HRp1-B, HRp1-M, B73 and H95, respectively.
The average nucleotide diversity (π) for Rp1 genes in HRp1-B, HRp1-M, B73 and H95 was 2.28, 3.31, 0.51 and 1.35 %, respectively (Table 2). The number of segregating sites detected in HRp1-B, HRp1-M, B73 and H95 was 342, 351, 52, and 371, respectively. Higher nucleotide diversity was observed in the LRR of Rp1 genes maintained in the Rp1 haplotypes (Rp1-B and -M) in comparison to Rp1 genes isolated from B73 and H95. Similar estimates of diversity were also observed for HRp1-B (2.03 %), HRp1-M (2.00 %) and B73 (0.48 %) with the θ w parameter, while the Rp1 genes in H95 (2.09 %) demonstrated a higher nucleotide diversity with this measurement (Table 2). Watterson’s estimator (θ w ) is a method used for estimating population mutation rate (genetic diversity) but also takes into account the effective population size and the mutation rate per generation in the population of interest, whereas the π estimator is simply the sum of the pairwise differences divided by the number of pairs.
Neighbor-joining tree construction for Rp1 genes isolated from Rp1-B, Rp1-M, B73 and H95 facilitated the analysis of haplotypic diversity relative to the LRR region. This analysis showed the distribution of Rp1 genes from each maize line into clusters based on sequence differences. Clones were defined as belonging to a cluster (closely related family of genes) based on the nucleotide sequence identity of the dataset, when aligned sequences demonstrated at least 90 % nucleotide identity. Rp1 genes present in different clusters were less than 70 % identical. This was not surprising because R-gene homology represents true relatedness. Different clusters represent different Rp1 haplotypes (H). The Rp1 gene haplotype relationships were well-supported within the clusters indicated by the high bootstrap support for all of the clades (Fig. 2a–d). There were no distinct classes formed in any of the neighbor-joining trees based on transcribed Rp1 genes.

Neighboring joining tree of Rp1 genes in a HRp1-B, b HRp1-M, c B73 and d H95. Rp1 indicates the R-genes analysed. The next designation indicates the maize line the R-genes were isolated from (-B, -M, -B73 and -H95) and p indicates the paralog number. The asterisk indicates the transcribed paralogs. Numbers at nodes indicate the level of bootstrap support (%) with one-thousand bootstrap replicates
The Rp1 maize lines carried a relatively large number of Rp1 genes ranging from 12 (H95) to 19 (HRp1-M) unique genes and were arranged into a large number of haplotypes (Fig. 2a–d). Indels were identified in Rp1 genes isolated from each haplotype. Indel sizes in the untranscribed Rp1 genes were variable, with single nucleotide indels being the most frequent size class.
The 16 Rp1 genes identified in HRp1-B were distributed into ten distinct clusters based on nucleotide sequence identity and represented different Rp1 haplotypes (Table 2). The most distant Rp1 genes were B-p19* and B-p24 (Fig. 2a; Table 2). These two genes were separated by 365 polymorphic sites in a 707 bp region and are 48.1 % identical. Conversely, Rp1-B-p66 and Rp1-B-p65 differed by a single nucleotide substitution and share 99.8 % identity.
Similarly, the 19 Rp1-M genes formed eleven different clusters and represented different Rp1 haplotypes (Fig. 2b; Table 2). Rp1-M-p1* and Rp1-M-p14* were the most distant with 351 nucleotide differences (49.1 % identity) in a 690 bp region. Paralogs Rp1-M-p3* and Rp1-M-p10* were the most similar, separated by only one nucleotide change (99.7 % identity).
The fourteen Rp1 genes identified in B73 were distributed into ten Rp1 haplotypes (Fig. 2c; Table 2). The most distant Rp1 paralogs were B73-p13 and B73-p45*. These two paralogs were separated by 52 polymorphic sites in a 615 bp region and are 92 % identical. The Rp1-B73-p13 and Rp1-B73-p45 paralogs differed by two nucleotide substitutions and share 99.6 % identity.
Analysis of H95 Rp1 sequences identified twelve Rp1 genes that formed eight Rp1 haplotypes (Fig. 2d; Table 2). Rp1-H95-p1 and Rp1-H95-p50* were the most distant Rp1 genes differing by 371 single nucleotide substitutions in a 708 bp region thus are 47 % identical. However, Rp1-H95-p29* and Rp1-H95-P63 were the most similar separate by only two single nucleotide substitutions and share 99.6 % identity.
A composite neighbor-joining tree was constructed with the 70 Rp1 genes identified in the HRp1-B, HRp1-M, B73, H95 and HRp1-D (Fig. 3). The Rp1 genes were separated into four distinct clades with 33, 8, 14 and 14 genes clustering together in clade I, II, III, and IV, respectively. The majority of the Rp1 genes from HRp1-B, HRp1-M, and H95 formed clade I. Thirteen of the 16 Rp1-B genes clustered in clade I and eleven of the twelve Rp1 genes from H95 were identified in this clade. Eight of the 19 Rp1-M genes were also present in clade I, while there were no Rp1 genes from B73 identified in clade I. Similarly, clade III contained Rp1 genes from Rp1-B, -M and H95. The remaining 3, 10 and 1 Rp1 genes from HRp1-B, HRp1-M and H95 respectively, were clustered in clade III, while B73 Rp1 genes were absent from this clade. There were two monophyletic clades (II and IV) formed containing Rp1 genes from the same haplotype. Clade II contains only Rp1-D genes from HRp1-D. HRp1-D is the best characterized Rp1 haplotype and was used in this analysis to determine the evolutionary relationship of known Rp1 genes to uncharacterized Rp1 genes. Additionally, all 14 of the B73 Rp1 genes clustered in clade IV. Identical Rp1 genes were not identified in HRp1-B, HRp1-M, B73 and H95.

Neighboring-Joining phylogenetic tree of Rp1 genes isolated from HRp1-B, HRp1-M, B73 and H95. The Rp1 designation indicates the locus analysed. The -B, -M,-B73 and -H95 designation indicates the maize line the R-genes were isolated from. The p designation indicates the paralog number. The asterisk indicates the transcribed genes. Numbers at the nodes indicate the level of bootstrap support (%) with one-thousand bootstrap replicates
The most distant Rp1 genes among the five haplotypes were H95-p61 and B73-p13. These two genes were separated by 310 polymorphic sites within a 642 bp region and share 51.7 % identity. Conversely, B-p36* and M-p25* Rp1 genes in clade III differed by four nucleotide substitutions and are 99.4 % identical.
The nucleotide diversity data were also supported by phylogenetic analysis of the Rp1 genes characterized in the four maize lines (Fig. 3). As indicated, the Rp1 genes isolated from B73 all clustered on clade IV. These genes were also the least divergent of the characterized Rp1 genes based on sequence analysis. This clade was further divided into two smaller clades with short branch lengths. Similarly, Rp1 genes from H95 also demonstrated low nucleotide diversity and clustered on a single clade with Rp1 genes from HRp1-B and HRp1-M (Clade I) with the exception of paralog H95-p50*. Conversely, Rp1 genes from HRp1-B and HRp1-M, the more divergent haplotypes, did not cluster into monophyletic clades.
Detection of recombination and positive selection
The frequency of recombination in the LRR region of the Rp1 genes from the four maize lines was examined using the recombination parameter (Rm) (Hudson et al. 1987) from DnaSP (Table 3). The minimum number of recombination events between adjacent polymorphic sites for HRp1-B, HRp1-M, B73 and H95 were 19, 22, 3 and 9, respectively. The recombination frequency (between adjacent sites and per gene) was relatively low for all Rp1 haplotypes analysed with the highest values observed for HRp1-M (0.0023 and 1.5) subsequently decreasing for HRp1-B (0.0012 and 0.001), B73 (0 and 0.001), and H95 (0 and 0.001).
Patterns of nucleotide substitution in the LRR region of R-genes can be informative in assessing the type of selection pressure acting on the evolution of gene family members (Sun et al. 2001). Nucleotide diversity was detected at 0.43984 in the LRR region of the 69 Rp1 genes. Nonsynonymous (K a) and synonymous (K s) amino acid substitution rates for the 69 Rp1 genes were 1.66518 and 0.65045, respectively. Therefore, the nonsynonymous to synonymous amino acid substitution ratio (K a:K s) for the 69 Rp1 genes is >1 (2.56003) indicating that the LRR region of Rp1 genes maintained in the four maize lines are under positive selection. Tajima’s D statistics was also used to detect neutral selection. When the Rp1 genes from each maize line were tested separately with Tajima’s D tests, negative values were observed for HRp1-B and H95 with no selection detected with significant P values, indicating a relative excess of low frequency alleles compared with expectations under a stationary neutral model (Table 3). Conversely, positives values were detected for HRp1-M and B73 with Tajima’s D, with significant P values detected.
Discussion
Amplification of Rp1 genes from four maize lines
Extensive studies on plant disease resistance genes have demonstrated that resistance genes frequently occur in tightly linked clusters (Pryor 1987; Michelmore and Meyers 1998). Multiple Rp (Resistance to P uccinia sorghi) genes have been shown to confer resistance to P. sorghi in maize. This locus was designated the Rp1 complex because fourteen genetically distinct loci mapped to this locus on the short arm of chromosome 10 in maize (Hulbert 1997). Complex disease resistance clusters have also been identified in Arabidopsis (Meyers et al. 2003), rice (Song et al. 1997), barley (Wei et al. 1999) and many other species. Many of the specificities within these genetically well-defined resistance loci have been targeted for molecular cloning and analysis utilizing a PCR-based approach and/or genomic library screening methods. In this study, a PCR-based approach was used to analyse two previously uncharacterized Rp1 maize lines and two maize inbred lines by estimating Rp1 gene number and diversity, identification of transcribed Rp1 genes and detection of recombination and selection acting on these genes in the four maize lines. The PCR-based approach amplified the C-terminal end of LRR region of Rp1 genes in the four maize lines. This is the most divergent region of Rp1 genes and has been used to distinguish between Rp1 genes and to estimate R-gene number (Richter et al. 1995; Smith et al. 2004, 2010) without sequencing the full-length gene as most Rp1 genes are ~4 kb in size. This approach did not distinguish between identical genes or amplify truncated genes in this study. As a result, these types of Rp1 genes were not sampled. Hence, to study specific haplotypes that carry an abundance of large R-genes, a PCR-based approach has proven to be most appropriate, where single molecules represent pertinent regions of each haplotype.
From this work, 544 PCR amplified sequences corresponding to 61 R-genes isolated from HRp1-B, HRp1-M, H95 and B73 were homologous to the LRR sequences of Rp1 and other NB-LRR encoding genes. The number of Rp1 genes maintained in each maize line is relatively large ranging from at least 12 to 19 genes. Based on Southern blot analysis and characterization of Rp1 genes in various Rp1 maize lines, most Rp1 lines carry from 15 to 25 Rp1 genes and are considered large haplotypes (Ayliffe et al. 2000, 2004; Sun et al. 2001; Smith et al. 2010). Additionally, genomic sequence analysis of maize BAC clones from a B73 maize inbred line identified 15 Rp1 genes (Ramakrishna et al. 2002), whereas this study identified 14 Rp1 genes in different B73 maize inbred line using a PCR-based approach. Analysis of Rp1 genes in the HRp1-A and HRp1-K, using a similar PCR-based approach, identified more than 50 Rp1 genes in these haplotypes, while HRp1-A188 carries a single Rp1 gene (Smith and Hulbert 2003). This suggests that similar to most maize lines that carry Rp1 genes, HRp1-B, HRp1-M, H95 and B73 also maintain a relatively large number of Rp1 genes. Extensive studies in maize, flax and tomato have suggested that the large number of resistance genes maintained in tandem at complex disease resistance loci play a central role in the diversity and evolution of new specificities (Parniske et al. 1997; Ellis et al. 1999; Luck et al. 2000; Chin et al. 2001).
Rp1 nucleotide and haplotype diversity in four maize lines
Sixty-one Rp1 genes were sampled from four maize lines. Various diversity patterns were detected within the LRR region of the Rp1 genes maintained in the four maize lines. The LRR region of Rp1 genes from B73 and H95 harbored the lowest diversity, while Rp1 genes from HRp1-B and HRp1-M demonstrated the highest level of diversity between the four maize lines. Many factors can affect genetic diversity including, population size relevant for all genes and pathogen populations that are specific to each R-gene. Therefore, the higher diversity observed in HRp1-B and HRp1-M may partly reflect increased diversity in the LRR region as a mechanism of adaptive plasticity for disease resistance and responses to other environmental variables (Clay and Kover 1996).
Identification of transcribed Rp1 genes
A total of 61 Rp1 genes were identified in the four maize lines, thirty-two of these genes are transcribed. Presumably, the maize lines with large numbers of Rp1 genes have more genes that are transcribes, but this has not been demonstrated. HRp1-B and HRp1-M carry the most Rp1 genes, whereas B73 and H95 carry the fewest. Interestingly, the majority of the Rp1 genes in each of the maize lines are transcribed with the exception of B73. All untranscribed Rp1 genes harbored stop codons or frameshift mutations. This is a novel observation that has been observed at the Rp1 locus with a few other haplotypes including HRp1-E, HRp1-I and HRp1-K (Smith et al. 2004). Based on the co-evolution of plant R-genes and pathogen Avr genes, selection pressure is imposed on the pathogen to evolve new genotypes (Avr gene) that can avoid detection by the corresponding plant R-gene protein. This implies that the untranscribed Rp1 alleles were potentially generated as a result of the complex plant-pathogen co-evolutionary dynamics and selection.
It has also been shown that the nonfunctional resistance genes participate in recombination creating new resistance genes (Hulbert 1997; Hulbert et al. 2001; Smith et al. 2004; Joshi and Nayak 2013). For example, recombinants selected for complete or partial loss of Rp1-D resistance resulted from unequal crossing over that occurred mostly within coding regions. The Rp1-D gene was altered or lost in all recombinants. The majority of recombination events involved the same untranscribed paralog with the functional Rp1-D gene. One recombinant with a complete LRR from Rp1-D, but the amino-terminal portion from an untranscribed paralog, conferred the Rp1-D specificity but with a reduced level of resistance. This indicates the potential usefulness of the nonfunctional genes in the creation of new resistance specificities at the Rp1 locus.
Phenotypic analysis of HRp1-B and HRp1-M indicates that these Rp1 haplotypes confer race-specific resistance to a different set of Puccinia sorghi rust isolates (Richter et al. 1995). Therefore, Rp1 haplotypes are designated by the genes they carry with detectable phenotypes. However, B73 and H95 are susceptible to all known P. sorghi isolates but still carry transcribed rp1 paralogs. This suggests that although Rp1 lines typically carry a large number of Rp1 genes that are often transcribed, most of these genes do not confer a resistance phenotype to any known rust isolate. This is also demonstrated in the Rp1-D haplotype. HRp1-D is the best characterized Rp1 haplotype and is considered one of the most meiotically stable of the fourteen different Rp1 haplotypes (Collins et al. 1999). This haplotype contains nine paralogs, seven of which are transcribed, including a truncated gene. However, only one paralog (Rp1-D) is phenotypically detectable and is located on the most distal end of the array. It is plausible that the transcribed paralogs in Rp1 haplotypes are involved in other aspects of resistance that are not phenotypically detectable or these genes once conferred resistance to a P. sorghi isolate that no longer exists in the pathogen population but are still transcribed (Jullien and Berger 2009).
Evidence of positive selection and recombination
The LRR region of Rp1 genes from the maize lines were determined to be under positive selection. Sequences of the Rp1 genes corresponded to the C-terminal end of the LRR region and ranged from 696 to 754 bp in size. Tajima’s D tests were also applied separately to the LRR region of Rp1 genes from each maize line and indicated that positive selection had occurred in the C-terminal half of the LRR region of Rp1 genes from HRp1-M and B73. This suggests that selection pressure favors diversifying selection in the C-terminal half of the LRR region of Rp1 genes from HRp1-M and B73, which is consistent with the predicted function of the LRR domain of R-genes. There were no sites detected as under positive selection for HRp1-B and H95 Rp1 genes. A relatively, high level of nucleotide diversity was observed for Rp1-B genes. This haplotype also confers race-specific resistance to different P. sorghi isolates, yet positive selection was not detected in the C-terminal end of LRR region, suggesting that regions other than the LRR of Rp1-B genes may be under positive selection and contribute to the resistance specificity.
The LRR domain is well-documented as a functional structure involved in protein–protein interactions binding pathogen-derived avr factors directly or indirectly (Kobe and Deisenhofer 1995; Ellis et al. 1999, 2000). Over the past 10 years, numerous studies have demonstrated that the LRR of R-genes are subject to positive selection and is where diversifying selection plays a role in the generation of new resistance specificities (Hu and Hulbert 1996; Parniske et al. 1997; Ellis et al. 1999; Hulbert and Drake 2000). There are also examples of regions other than the LRR that contribute to resistance specificity (Stirnweis et al. 2014). One of the best-characterized examples is the L locus in flax. Analysis of this locus indicated that the TIR (Toll Interleukin-1 Receptor) domain contributes to resistance specificity and may be under positive selection (Luck et al. 2000). To date, R-genes with a TIR domain have not yet been identified in grasses. Further evidence suggests that selection pressure acts differently on different regions of the LRR domain (Jiang et al. 2007). Variability in the frequency of recombination events between the Rp1 genes in the different maize lines was detected. This indicates that there are different histories of sequence exchange between Rp1 genes in the different maize lines. Evidence has accumulated suggesting that unequal recombination is a major mechanism in diversifying R-gene sequences, especially at complex disease resistance gene loci (Sudupak et al. 1993; Parniske et al. 1997; Dixon et al. 1998; Hulbert et al. 2001; Ramakrishna et al. 2002; Nagy and Bennetzen 2008; Baurens et al. 2010). Recombination has been shown to contribute significantly in the creation of genetic diversity at the Rp1 rust resistance locus (Hulbert et al. 1997; Smith et al. 2010). For example, phenotypic and genetic analyses of several recombinant Rp1 haplotypes demonstrated the creation of novel recombinant Rp1 genes and race specificities (Smith and Hulbert 2005; Smith et al. 2010). The generation of recombinant resistance genes that presumably create novel specificities has also been observed in flax, lettuce and tomato suggesting recombination plays a pivotal role in the evolution of new specificities (Parniske et al. 1997; Ellis et al. 1999; Luck et al. 2000; Chin et al. 2001).
Phylogenetic analysis of Rp1 genes
A neighbor-joining tree was constructed for the Rp1 genes isolated from each maize line to analyse the relationship between Rp1 genes. From eight to eleven different Rp1 haplotypes were detected for the four maize lines. However, only two distinct clades were observed for each maize line. The B73 and H95 clades demonstrated short branches within and between the different clades, while Rp1-B and Rp1-M branches were long in both instances. This suggests that the LRR region of Rp1 genes is more diverse in HRp1-B and HRp1-M in comparison to B73 and H95. It has been shown in numerous studies that the LRR region is typically the most diverse domain in an NB-LRR resistance gene due to its involvement in pathogen recognition and specificity (Jiang et al. 2007; Sela et al. 2009). HRp1-B and HRp1-M confer resistance to different P. sorghi isolates. Therefore, these two haplotypes carry the appropriate Rp1 gene(s) that recognizes the corresponding Avr protein in the pathogen population (Hammond-Kosack and Jones 1997). Conversely, B73 and H95 are maize inbred lines that are susceptible to all known P. sorghi isolates lacking the appropriate Rp1 gene. It is possible that the most diverse Rp1 genes observed in HRp1-B and HRp1-M arose most recently from recombination events in haplotypes with divergent Rp1 arrays. However, the least diverse Rp1 genes identified in B73 and H95 may be due to cyclical amplification and deletion events that would homogenize haplotypes, especially in inbred populations, small populations or populations where a very successful haplotype became more prevalent.
A composite neighbor-joining tree was generated using the 70 Rp1 genes from the five maize lines to evaluate the evolutionary relationship between the genes from the different lines. This analysis included Rp1 genes from the Rp1-D haplotype. Previous work at the Rp1 locus demonstrated that Rp1 genes characterized in different haplotypes vary in their evolutionary relationships and that their evolutionary patterns can be used to predict how the Rp1 genes are evolving in individual haplotypes. For this study, Rp1 genes from the five maize lines formed four distinct clades. Rp1 genes from HRp1-B, HRp1-M and H95 shared two clades and were distributed between clades I and III. This suggests that Rp1 genes maintained in HRp1-B, HRp1-M and H95 are more similar to each other than to Rp1 genes within each individual maize line. Therefore, the Rp1 genes maintained in HRp1-B, HRp1-M and H95 are evolving independently of each other.
Rp1 genes isolated from HRp1-D and B73 each formed a monophyletic clade. This indicates that the Rp1 genes maintained in B73 are more similar to each other than to Rp1 genes in the other four maize lines. The same is true for the Rp1 genes maintained in HRp1-D. Therefore, the Rp1 genes in HRp1-D and B73 are evolving in a concerted manner. This type of evolutionary pattern was also observed when Ramakrishna et al. (2002) sequenced 4 of the 15 Rp1 genes from a different B73 haplotype and found that 2 of the genes differed by a single nucleotide change. It has been demonstrated that in most gene families, orthologs from different haplotypes are often more similar in sequence than are paralogs in the same haplotype (Meyers et al. 1999) as is the case in this study for HRp1-B, HRp1-M and H95 but not for HRp1-D and B73. It is not apparent as to why some haplotypes show more evidence of within haplotype homogenization than others. Perhaps haplotypes similar to Rp1-D and B73 evolved from an extended period in the population with limited variation at the Rp1 locus, while haplotypes like HRp1-B, HRp1-M and H95 evolved during a period of high genetic diversity.
Identical Rp1 genes were not identified in HRp1-B, HRp1-M, B73 and H95 when compared. This in conjunction with the fact that most maize lines appear to have different Rp1 haplotypes when compared by Southern blot analysis indicates that maize germplasm carries many hundreds of different Rp1 genes with the potential to create new R-genes with new resistance specificities as a result of recombination and diversifying selection. Only a few of the Rp1 haplotypes have been characterized in detail. Analysis of the haplotypic diversity of NB-LRR R-genes in previously uncharacterized Rp1 haplotypes provides insights into Rp1 R-gene number, transcription and diversity. This type of analysis is necessary to create a data resource for future use of this class of R-genes and to select the appropriate Rp1 parental haplotypes used for crosses. This study characterized two Rp1 haplotypes (HRp1-B, HRp1-M) differing in resistance specificity and two maize inbred lines (B73 and H95), that would be appropriate to generate the crosses needed for detailed resistance gene characterization, generation of recombinant haplotypes with new resistance specificities and improved maize cultivar performance.
Author contribution statement
The first author SC is a graduate student and conducted all of the research on the project. The second author JG is a technician in the principle investigator’s laboratory. JG provided assistance with sequence analysis. The third author SMS is the project principle investigator. SMS developed and directed all aspects of the research project.
References
Ayliffe MA, Collins NC, Ellis JG, Pryor A (2000) The maize Rp1 rust resistance genes identifies homologues in barley that have been subject to diversifying selection. Theor Appl Genet 100:1144–1154
Ayliffe MA, Steinau M, Park RF, Rooke L, Pacheco MG et al (2004) Aberrant mRNA processing of the maize Rp1-D rust resistance gene in wheat and barley. Mol Plant Microbe Interact 17:853–864
Baurens FC, Bocs S, Rouard M, Matsumoto T, Miller R et al (2010) Mechanisms of haplotype divergence at the RGA08 nucleotide-binding leucine-rich repeat gene locus in wild banana (Musa balbisiana). BMC Plant Biol 10 PMC3017797
Bent A (1996) Plant disease resistance genes: function meets structure. Plant Cell 8:1757–1771
Chin DB, Arroyo-Garcia R, Ochoa OE, Kesseli RV, Lavelle DO et al (2001) Recombination and spontaneous mutation at the major cluster of resistance genes in lettuce (Lactuca sativa). Genetics 157:831–849
Clay K, Kover P (1996) Evolution and stasis in plant-pathogen associations. Ecology 77:997–1003
Collins N, Drake J, Ayliffe M, Sun Q, Ellis J et al (1999) Molecular characterization of the maize Rp1-D rust resistance haplotype and its mutants. Plant Cell 11:1365–1376
Dangl JL, Jones JDG (2001) Plant pathogens and integrated defence responses to infection. Nature 411:826–833
Dixon MS, Hatzixanthis K, Jones DA, Harrison K, Jones JDG (1998) The tomato Cf-5 disease resistance gene and six homologs show pronounced allelic variation in leucine-rich repeat copy number. Plant Cell 10:1915–1925
Ellis JG, Lawrence GJ, Luck JE, Dodds PN (1999) Identification of regions in alleles of the flax rust resistance gene L that determine differences in gene-for- gene specificity. Plant Cell 11:495–506
Ellis J, Dodds P, Pryor T (2000) The generation of plant disease resistance gene specificities. Trends Plant Sci 5:373–379
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194
Hagan WL, Hooker AL (1965) Genetics of reaction to Puccinia sorghi in eleven corn inbred lines from Central and South America. Phytopathology 55:193–197
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hammond-Kosack KE, Jones JDG (1997) Plant disease resistance genes. Annu Rev Plant Biol 48:575–607
Hooker AL, Russell WA (1962) Inheritance of resistance to Puccinia sorghi in six corn inbred lines. Phytopathology 52:122–128
Hu G, Hulbert SH (1996) Construction of ‘compound’ rust resistance genes in maize. Euphytica 87:45–51
Hudson RR, Kreitman M, Aguade M (1987) A test of neutral molecular evolution based on nucleotide data. Genetics 116:153–159
Hulbert SH (1997) Structure and evolution of the rp1 complex conferring rust resistance in maize. Annu Rev Phytopathol 35:293–310
Hulbert SH, Drake JA (2000) Rust-resistant sh2 sweet corn populations. Hortscience 35:145–146
Hulbert SH, Pumphrey M (2014) A time for more booms and less busts? Unraveling cereal-rust interactions. Mol Plant Microbe Interact 27:207–214
Hulbert SH, Hu G, Drake JA (1997) Kansas rust-resistant sweet corn populations A and B. Hortscience 32:1130–1131
Hulbert SH, Webb CA, Smith SM, Sun Q (2001) Resistance gene complexes: evolution and utilization. Annu Rev Phytopathol 39:285–312
Hurni S, Brunner S, Buchmann G, Herren G, Jordan T, Krukowski P, Wicker T, Yahiaoui N, Mango R, Keller B (2013) Rye Pm8 and wheat Pm3 are orthologous genes and show evolutionary conservation of resistance function against powdery mildew. Plant J 76:957–969
Jiang H, Wang C, Ping L, Tian D, Yang S (2007) Pattern of LRR nucleotide variation in plant resistance genes. Plant Sci 173:253–261
Joshi RK, Nayak S (2013) Perspectives of genomic diversification and molecular recombination towards R-gene evolution in plants. Physiol Mol Biol Plants 19:1–9
Jullien PE, Berger F (2009) Gamete-specific epigenetic mechanisms shape genomic imprinting. Curr Opin Plant Biol 12:637–642
Kobe B, Deisenhofer J (1995) Proteins with leucine-rich repeats. Curr Opin Struct Biol 5:409–416
Lawrence GJ, Morrish BC, Ayliffe MA, Finnegan EJ, Ellis JG (1997) Inactivation of the flax rust resistance gene M associated with loss of a repeated unit within the leucine-rich repeat coding region. Plant Cell 9:641–651
Leister RT, Katagiri F (2000) A resistance gene product of the nucleotide binding site-leucine rich repeats class can form a complex with bacterial avirulence proteins in vivo. Plant J 22:345–354
Librado P, Rozas J (2009) DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Luck JE, Lawrence GL, Dodds PN, Shepherd KW, Ellis JG (2000) Regions outside of the leucine-rich repeats of flax rust resistance proteins play a role in specificity determination. Plant Cell 12:1367–1377
Meyers BC, Dickerman AW, Michelmore RW, Sivaramakrishnan S, Cobral BW, Young ND (1999) Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J 20:317–332
Meyers BC, Kozik A, Griego A, Kuang H, Michelmore RW (2003) Genome-wide analysis of NBS-LRR-encoding genes in arabidopsis. Plant Cell 15:809–834
Michelmore RW, Meyers BC (1998) Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res 8:1113–1130
Mondragón-Palomino M, Meyers BC, Michelmore RW, Gaut BS (2002) Patterns of positive selection in the complete NBS-LRR gene family of Arabidopsis thaliana. Genome Res 12:1305–1315
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Nagy ED, Bennetzen JL (2008) Pathogen corruption and site-directed recombination at a plant disease resistance gene cluster. Genome Res 18:1918–1923
Nei M (1987) Molecular Evolutionary Genetics. Columbia University Press, New York
Parniske M, Hammond-Kosack KE, Golstein C, Thomas CM, Jones DA et al (1997) Novel disease resistance specificities result from sequence exchange between tandemly repeated genes at the Cf-4/9 locus of tomato. Cell 91:821–832
Pryor T (1987) The orgin and structure of fungal disease resistance genes in maize. Trends Genet 3:157–161
Ramakrishna W, Emberton J, Ogden M, SanMiguel P, Bennetzen JL (2002) Structural analysis of the maize Rp1 complex reveals numerous sites and unexpected mechanisms of local rearrangement. Plant Cell 14:3213–3223
Richter TE, Pryor TJ, Bennetzen JL, Hulbert SH (1995) New rust resistance specificities associated with recombination in the rp1 complex in maize. Genetics 141:373–381
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Saxena KMS, Hooker AL (1968) On the structure of a gene for disease resistance in maize. Proc Natl Acad Sci USA 61:1300
Sela H, Cheng J, Jun Y, Nevo E, Fahima T (2009) Divergent diversity patterns of NBS and LRR domains of resistance gene analogs in wild emmer wheat populations. Genome 52:557–565
Smith SM, Hulbert SH (2003) Evolution of disease resistance genes. In: Tsuyuma S, Leach J, Wolpert T (eds) Genomics and genetic analysis of plant parasitism and defense. APS Press
Smith S, Hulbert SH (2005) Recombination events generating a novel Rp1 race specificity. Mol Plant Microbe Interact 18:220–228
Smith S, Pryor A, Hulbert SH (2004) Allelic and haplotypic diversity at the Rp1 rust resistance locus of maize. Genetics 167:1939–1947
Smith SM, Steinau M, Trick H, Hulbert SH (2010) Recombinant Rp1 genes confer necrotic or nonspecific resistance phenotypes. Mol Genet Genomics 283:591–602
Song WY, Pi LY, Wang GL, Gardner J, Holsten T et al (1997) Evolution of the rice Xa21 disease resistance gene family. Plant Cell 9:1279–1287
Stirnweis D, Milani SD, Jordan T, Keller B, Brunner S (2014) Substututions of two amino acids in the nucletide-binding site domain of a resistance protein enhance the hypersensitive response and enlarge the PM3F resistance spectrum in wheat. Mol Plant Microbe Interact 27:265–276
Sudupak MA, Bennetzen JL, Hulbert SH (1993) Unequal exchange and meiotic instability of Rp1 region disease resistance genes. Genetics 133:119–125
Sun Q, Collins NC, Ayliffe M, Smith SM, Drake J et al (2001) Recombination between paralogues at the rp1 rust resistance locus in maize. Genetics 158:423–438
Tajima F (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tameling WIL, Elzinga SD, Darmin PS, Vossen JH, Takken FLW et al (2002) The tomato R gene products I-2 and Mi-1 are functional ATP binding proteins with ATPase activity. Plant Cell 14:2929–2939
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Tamura K, Peterson D, Peterson N, Stecher G, Nei M et al (2011) MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7:256–276
Wei F, Gobelman-Werner K, Morroll SM, Kurth J, Mao L et al (1999) The Mla (powdery mildew) resistance cluster is associated with three NBS-LRR gene families and suppressed recombination within a 240-kb DNA interval on chromosome 5S (1HS) of barley. Genetics 153:1929–1948
Wilkinson DR, Hooker AL (1968) Genetics of reaction to Puccinia sorghi in ten corn inbreds from Africa and Europe. Phytopathology 58:720–721
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556
Yang Z, Nielsen R, Goldman N, Pedersen AMK (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Zhang Y, Lubberstedt T, Xu M (2013) The genetic and molecular basis of plant resistance to pathogens. J Genet Genomics 40:23–35
Acknowledgments
We thank Scot Hulbert for reviewing this manuscript and his helpful discussions regarding the Rp1 locus. This research was supported by the Georgia Commodity Commission for Corn (047461-01).
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
The experiments comply with the current laws of the country (US) in which they were performed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Y. Xu.
Rights and permissions
About this article

Cite this article
Chavan, S., Gray, J. & Smith, S.M. Diversity and evolution of Rp1 rust resistance genes in four maize lines. Theor Appl Genet 128, 985–998 (2015). https://doi.org/10.1007/s00122-015-2484-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-015-2484-2