
Abstract
Increasing seed yield is an important breeding goal of soybean [Glycine max (L.) Merr.] improvement efforts. Due to the small number of ancestors and subsequent breeding and selection, the genetic base of current soybean cultivars in North America is narrow. The objective of this study was to map quantitative trait loci (QTL) in two backcross populations developed using soybean plant introductions as donor parents. The first population included 116 BC2F3-derived lines developed using “Elgin” as the recurrent parent and PI 436684 as the donor parent (E population). The second population included 93 BC3F3-derived lines developed with “Williams 82” as the recurrent parent and PI 90566-1 as the donor parent (W population). The two populations were evaluated with 1,536 SNP markers and during 2 years for seed yield and other agronomic traits. Genotypic and phenotypic data were analyzed using the programs MapQTL and QTLNetwork to identify major QTL and epistatic QTL. In the E population, two yield QTL were identified by both MapQTL and QTLNetwork, and the PI 436684 alleles were associated with yield increases. In the W population, a QTL allele from PI 90566-1 accounted for 30 % of the yield variation; however, the PI region was also associated with later maturity and shorter plant height. No epistasis for seed yield was identified in either population. No yield QTL was previously reported at the regions where these QTL map indicating that exotic germplasm can be a source of new alleles that can improve soybean yield.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genetic base of modern soybean [Glycine max (L.) Merr.] cultivars in North America is narrow due at least in part to the small number of ancestors that formed the base of this germplasm and subsequent breeding and selection during cultivar development. Over 80 % of the genes present in modern North American soybean cultivars could be traced to 17 plant introduction (PI) ancestors and their first progeny (Gizlice et al. 1994). Since the establishment of the North American germplasm base, most soybean yield improvements have been made using crosses among elite germplasm rather than crosses with exotic germplasm or wild relatives (Carter et al. 2004); however, soybean breeders have introduced new germplasm into the North America soybean gene pool to improve resistance to diseases and pests and to attempt to increase yield (Carter et al. 2004). Exotic germplasm has proven to be an important source of genes especially for disease and pest resistance (Carter et al. 2004); however, it has been difficult to improve yield using PIs. This difficulty stems from the lower average yield performance of PIs compared to elite breeding lines which makes it difficult for breeders to obtain similar or higher yields in selected progeny from crosses with PIs when compared with the progeny obtained using only adapted parents (Smalley et al. 2004). In addition, there is a lack of reliable methods to predict whether a PI actually carries yield increasing alleles. Despite these difficulties, there continues to be a need to identify and use genetic variability in soybean germplasm that can improve soybean yields (Diers and Kim 2008) and experimental lines derived from exotic germplasm that yield significantly more than the best public cultivars indicate that useful yield genes do exist in exotic germplasm (Nelson and Johnson 2012).
Genetic mapping with molecular markers and marker-assisted selection (MAS) are widely used in soybean breeding programs. For both soybean breeding and research, single nucleotide polymorphism (SNP) markers are becoming the marker of choice because of their high frequency, widespread distribution throughout the genome as well as their suitability for high-throughput automated genotyping (Choi et al. 2007; Hyten et al. 2010). Multiple Illumina GoldenGate assays with 384–1,536 SNP markers have been developed (Hyten et al. 2008, 2010) and used in developing genetic maps (Hyten et al. 2010), mapping genes conferring resistance to Asian soybean rust (Hyten et al. 2009; Chakraborty et al. 2009), soybean cyst nematode (Heterodera glycines Ichinohe) (Vuong et al. 2010; Kim et al. 2011), and soybean aphid (Aphis glycines Matsumura) (Jun et al. 2012), and mapping loci involved in isoflavone concentration (Gutierrez-Gonzalez et al. 2011).
Understanding the genetic architecture of complex traits is a major challenge in the post-genomic era, especially for quantitative trait loci (QTL) by QTL interactions (epistasis), QTL by environment interactions, epistasis by environment interactions and more complex higher order interactions (Yang et al. 2008). The genotypic effect of one locus on a phenotype might depend on the genotype at several or many other loci, and QTL with minor or no individual effect can also involve epistasis, a finding that is well documented for a number of physiological traits in Drosophila melanogaster (Montooth et al. 2003). Strong interactions between QTL have been detected in maize (Lukens and Doebley 1999) and soybean (Lark et al. 1995), which have implications in cultivar development programs. If alleles involved in positive epistatic interactions are not transferred together to the cultivar that is being developed, yield improvement will be unsuccessful because high yield is conditional on the presence of epistatic effects (Lark et al. 1995).
QTL alleles from exotic soybean germplasm that significantly increase seed yield have been reported previously. Kabelka et al. (2004) identified nine positive yield QTL alleles that trace to the exotic soybean germplasm accessions FC 04007B and PI 68508. Wang et al. (2004) reported four positive yield QTL alleles from G. soja PI 468916; however, the QTL were only identified when the significance threshold was reduced and the data were analyzed with simple linear regression. Li et al. (2008) reported one positive yield QTL allele from G. soja and the QTL mapped to the same region on chromosome 5 where Kabelka et al. (2004) also reported a yield QTL. Guzman et al. (2007) identified eight positive yield QTL alleles from PIs but all of them mapped to the same regions where yield QTL were reported previously. Although these results suggest that it may be difficult to identify new positive yield QTL from exotic germplasm, there is a need to identify these positive alleles to help increase the rate of yield improvement of future cultivars. The objective of this study was to identify QTL and epistatic interactions associated with important agronomic traits in soybean using two backcross populations that each has a different PI as the donor parent.
Materials and methods
Plant material
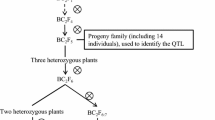
Two populations of lines developed through backcrossing were used in the study. The first population (E population) included 116 BC2F3-derived lines developed using Elgin (PI 548557) as the recurrent parent and PI 436684 as the donor parent. Elgin was developed by the Iowa Agriculture and Home Economics Experiment Station and was released in 1984 because of its superior yield compared to public cultivars of similar maturity (Fehr and Bahrenfus 1984). Elgin is a maturity group (MG) II cultivar and it has resistance to bacterial pustule (Xanthomonas axonopodis pv. glycines). PI 436684 (MG III) is the Chinese cultivar Tie feng No. 8, which was released in 1970 by the Liaoning Academy of Agricultural Sciences, Shenyang, Liaoning, China (Cui et al. 1999) and introduced in the US in 1979 (http://www.ars-grin.gov/npgs/acc/acc_queries.html/; accessed 30 April 2012). It was selected as a parent based on its yield potential in germplasm evaluations conducted at Urbana, IL, USA, in 1983 and 1984 (Nelson et al. 1988). The cross of PI 436684 by Elgin was made in 1985. Progeny from this cross were advanced through early generation testing for yield that included testing F2-derived lines in the F3 and F4 generations and F5-derived lines. LG90-2847 was selected from the original cross and used as a parent in 1992 to develop a BC1 population. Early generation testing for yield was again employed and LG98-1351 was selected as a F4-derived line and crossed to Elgin to develop the BC2 population in 2001. This population was advanced by single seed descent and BC2 F3-derived lines were harvested in Chile in the spring of 2005.
The second population (W population) has 93 BC3F3-derived lines developed using Williams 82 (PI 518671) as the recurrent parent and PI 90566-1 as the donor parent. Williams 82 was developed by the USDA-ARS and the Illinois Agricultural Experiment Station through backcrossing the phytophthora rot (Phytophthora megasperma Drechs. f. sp. glycinea Kuan and Erwin) resistance gene Rps1k from Kingwa into the cultivar Williams (Bernard and Cremeens 1988). PI 90566-1 is a MG III soybean accession originating from Liaoning, China and introduced in the United States in 1930 (http://www.ars-grin.gov/npgs/acc/acc_queries.html/; accessed 30 April 2012). PI 90566-1 was tested in 1978 in a cooperative project involving both public and private soybean breeders to evaluate the yield potential of accessions from the USDA Soybean Germplasm Collection. This project was organized by Dr. Clark Jennings of Pioneer Hi-Bred International and used the existing yield data from previous general germplasm evaluations as the initial selection criterion (Bernard et al. 1998). PI 90566-1 was one of 26 MG III accessions advanced for testing at four locations in 1979 and was one of 21 accessions used as parents in 1979 when PI 90566-1 was crossed with L77-1779, which was later released as Williams 82. Progeny from this cross were advanced through early generation testing for yield potential as described for the E population. An F4-derived line, LG84-1022, was selected for first backcross which was made in 1986. The early generation testing procedure was repeated and an F5-derived MG III line, LG91-7654, was selected and crossed in 1995 to Williams 82 to develop the BC2 population. The early generation testing procedure was again repeated and an F5-derived MG III line, LG98-2080, was selected for use as a parent in the third backcross, which was made in the greenhouse during the spring of 2002.
Field trials
The E population was field tested at DeKalb and Bellflower, IL and Wooster, OH in 2005. In 2006, the field trials were conducted at Fisher and Bellflower, IL and Wooster, OH. The recurrent parent Elgin, experimental line LG98-1351, which was the donor parent used to develop the BC2 population, and the high yielding cultivar IA2065 were included as checks in the E population tests. The W population was tested at Hume and Ivesdale, IL and Wooster, OH in 2005. In 2006, the population was tested at Fisher and Hume, IL, Portageville, MO and Wooster, OH. The recurrent parent Williams 82, experimental line LG98-2080, which was the donor parent used to develop the BC3 population, and the high yielding cultivar IA3023 were included as checks at all locations and years of the W population tests. When two populations were evaluated at the same location, they were evaluated in separate tests.
All field trials of the two populations were arranged in randomized complete-block designs (RCBD) with two replications at each location. In the field tests at Wooster, OH, during 2005 and 2006, each plot consisted of eight rows. The middle six rows were spaced 19 cm apart and were harvested for seed yield. The two border rows were 0.76 m from the outside harvest rows. The plots were planted to a length of 6.4 m and were end trimmed to 4.88 m at maturity. The seeding rate was 10 seeds m−1 of row. The plots in both Missouri and Illinois were four rows wide with a 76-cm row spacing and the middle two rows were harvested to estimate seed yield. At Portageville, MO, the plots were 4.42 m long and the planting rate was 33 seeds m−1 of row. At the DeKalb, Bellflower, Fisher, Ivesdale, and Hume locations, the plots were 3.6 m long. Thirty seeds per meter were planted in the Illinois locations. Conventional tillage and herbicide practices were followed at all locations to maintain weed-free environments and recommended fertilization levels were applied. The plots were rated for maturity date, plant height, and lodging. Maturity date was recorded as the day when approximately 95 % of the pods had reached mature pod color (R8; Fehr et al. 1971). Plant height (cm) was measured at maturity as the average distance from the soil surface to the apex of the main stem. Lodging was scored at maturity on a scale of 1–5 with 1 designated as all plants standing erect and 5 as all plants prostrate. Plots were harvested to measure seed yield (kg ha−1) and yield values were adjusted to 130 g kg−1 moisture.
GoldenGate assay
A bulked leaf sample from at least 30 greenhouse grown plants of each line and parent from the two populations was used to extract DNA with the CTAB (hexadecylatri methylammonium bromide) method described by Saghai Maroof et al. (1984) with slight modifications in speed and time of centrifugation. DNA concentrations were quantified with a ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE) and diluted to 100 ng μl−1. DNA quantity and quality were confirmed by electrophoresis in 3 % agarose gels and staining with a 1 μg/ml ethidium bromide staining solution (BMA, Rockland, ME, USA). The DNA samples from the E and W populations together with the parents were tested with SNP markers using the Golden Gate 1,536 Universal Soy Linage Panel 1.0 according to methods described in Hyten et al. (2010). The GoldenGate assay data were scored with the Illumina software BeadStudio v.3.2 and visually inspected to ensure that homozygous and heterozygous clusters were properly assigned.
Statistical analysis
Agronomic traits were analyzed by the PROC GLM functions of SAS 9.2 (SAS Institute, 2002). Lines, locations, replication within locations, and the line by location interaction were analyzed as random effects in each population. Each location by year combination was considered a separate environment in the analysis (Kim and Diers 2009). Broad-sense heritabilities of additive effects for yield and other agronomic traits were calculated based on the results from PROC GLM in SAS 9.2 according to Hill et al. (1998). Pearson correlation coefficients among all traits were calculated from the mean of lines across the environments using PROC CORR function in SAS 9.2.
A genetic linkage map was constructed for each population with JOINMAP 3.0 (Van Ooijen and Voorrips 2001) using the Kosambi mapping function. A logarithm (base 10) of the odds (LOD) score of 3.0 was used as the threshold to group markers into linkage groups. QTL analysis in the two populations was conducted using both the composite interval mapping (CIM) function in MapQTL 4.0 (Van Ooijen et al. 2002) and the mixed-model-based composite interval mapping (MCIM) function in QTLNetwork v2.1 (Yang et al. 2008). Average trait values across the environments were used for CIM while raw data including all individual observations from each location were used for MCIM. For each trait and environment, LOD thresholds in CIM and critical F values in MCIM corresponding to an experiment-wide threshold of P = 0.05 were determined by 1,000 permutations. For MCIM, QTL effects were estimated using Markov chain Monte Carlo method and the genome scan was performed using a 10 cM window size and a 1 cM walk speed. QTLNetwork calculates additive and dominance effects and epistatic interactions that include both of these effects. Because BC2F3 or BC3F3-derived lines that had undergone further inbreeding after derivation were evaluated in the field tests, there was very little heterozygosity in the plants grown in the yield trials that could have contributed to dominance estimates. Therefore, although dominance effects and dominance interactions are reported in the text, only additive effects and additive by additive (AA) interactions are reported in tables.
Single marker analysis (SMA) and regression analysis (SRA) were conducted to detect QTL using PROC GLM in SAS 9.2 when segregating SNP markers were not joined onto linkage groups by JOINMAP 3.0. Multiple regression analysis for QTL was conducted using PROC REG function with markers linked to significant QTL and epistatic interactions identified by CIM, MCIM, and SMA to determine the total phenotypic variance explained (R 2) by QTL and epistatic interactions. The proportion of the genotypic variance for yield explained by all significant QTL in the multivariate model was estimated from the ratio R 2/H 2 (Schön et al. 1994).
To test the impact of maturity on yield QTL, yield estimates of lines in both populations were adjusted using maturity as a covariate with PROC MIXED in SAS 9.2. The adjusted yield values were used to map yield QTL with CIM and SMA.
Results
Field data analysis
E population
There were significant (P < 0.0001) differences for seed yield, maturity date, plant height, and plant lodging (P = 0.004) among the three check genotypes in the E population tests across the six environments. The yield of LG98-1351, the BC1 parent of the BC2 population, was significantly greater (P = 0.05) than the recurrent parent, Elgin. The yields of the check and parental genotypes were 4,435 kg ha−1 for IA2065, 4,212 kg ha−1 for LG98-1351, and 3,620 kg ha−1 for Elgin. LG98-1351 matured significantly (P = 0.05) later than Elgin (3 days) and IA2065 (4 days).
There were significant (P < 0.0001) effects of lines, environments, and the interaction of lines by environments for seed yield, days to maturity, plant height, and lodging score in the population across the six environments. Seed yield was positively correlated with plant maturity (r = 0.55, P < 0.0001) and plant height (r = 0.69, P < 0.0001) but not significantly (P = 0.05) correlated with lodging. The average yield of the lines in the population across the six environments was 3,915 kg ha−1 (Table 1) and the average yields for environments ranged from 4,671 kg ha−1 at DeKalb in 2005 to 3,012 kg ha−1 in Wooster in 2006. The average yield of the population in 2005 was significantly greater than that in 2006 (Table 1). The broad-sense heritability for yield was 0.66 across environments in 2005, 0.54 in 2006, and 0.60 over the 2 years (Table 1). Across environments, 81 lines in the population yielded significantly (P = 0.05) more than the recurrent parent Elgin while no line yielded significantly greater than LG98-1351.
W population
Across the seven environments, there were significant differences (P < 0.0001) in seed yield, maturity date, and plant height, and lodging score (P = 0.03) among the three check genotypes in the W population tests. The BC2 parent of the population, LG98-2080, yielded significantly more than the recurrent parent Williams 82. The average yield across environments for IA3023 was 4,083, 3,520 kg ha−1 for LG98-2080, and 3,275 kg ha−1 for Williams 82.
Across environments, there were significant (P < 0.0001) effects of lines and environment for seed yield, days to maturity, plant height, and lodging score. The line by environment interaction was significant for seed yield, days to maturity but not for plant height or lodging score. Seed yield was positively correlated with plant maturity (r = 0.45, P < 0.0001) while negatively correlated with plant height (r = −0.37, P = 0.0002) and there was no significant correlation with lodging score. The average yield of the W population was 3,442 kg ha−1 across environments (Table 1) and the average for environments ranged from 3,986 kg ha−1 for Fisher in 2006 to 2,864 kg ha−1 for Hume in 2006. The lines at Fisher in 2006 also had the highest lodging score (2.3). The average yield of the population in 2005 was 3,520 and 3,384 kg ha−1 in 2006 (Table 1). The board-sense heritability for yield was 0.64 across the 2005 environments, 0.37 for the 2006 environments and 0.66 over the 2 years. Like the E population, there were lines in the W population that yielded greater than both parents with 21 lines yielding significantly (P = 0.05) more than Williams 82 and one line yielding significantly more than LG98-2080.
Genetic map construction
E population
Of the 1,536 SNP markers in the GoldenGate assay, 513 were polymorphic between Elgin and PI 436684, the original donor parent of the population. Of these polymorphic SNP markers, 106 (21 % of the polymorphic markers) were segregating in this BC2 population. The segregating markers mapped to 16 chromosomes while chromosomes 3, 7, 19 and 20 were fixed for the Elgin alleles. The genetic map covered a length of 469 cM out of a total map size of 2,241 cM and the relative positions of the markers were generally consistent with the G. max consensus map 4.0 (Hyten et al. 2010; http://soybase.org). Chromosome 9 had the largest number of segregating SNP markers (16) covering approximately 74 cM while chromosomes 10 and 11 had only one segregating SNP marker each.
W population
Four hundred and three out of 1,536 SNP markers in the GoldenGate assay showed polymorphisms between Williams 82 and PI 90566-1, and 83 (21 % of polymorphic markers) SNP markers segregated in this BC3 population. The genetic map covered a distance of 238 cM. Chromosome 15 had the largest number of segregating SNP markers (18) covering 40 cM, while chromosomes 1, 5, 7, 10, 11, 14 and 19 were fixed for the Williams 82 alleles. Like the E population, the positions of markers on the genetic map were generally consistent with the G. max consensus map 4.0.
QTL identified in the E population
Yield QTL
Across the six environments, three yield QTL were identified by CIM with MapQTL in the E population with an experiment-wide threshold of P = 0.05 (Table 2). For the chromosome 4 and 18 yield QTL, the alleles from PI 436684 conferred significantly greater yield than the Elgin allele while the Elgin allele conferred greater yield than the PI allele for the chromosome 14 QTL (Table 2). Across the 2 years, the QTL on chromosomes 4, 14, and 18 explained 12.2, 7.8 and 10.6 % of the phenotypic variance for yield and their additive effects were 62, 39, and 61 kg ha−1, respectively (Table 2). Out of six locations in which the population was tested, the QTL on chromosomes 4 and 14 were significant at two locations, and the chromosome 18 QTL was significant at three locations (Table 2).
The yield QTL on chromosomes 4 and 18 identified with CIM were also detected with MCIM; however, the chromosome 14 QTL was not detected with MCIM. The MCIM analysis showed that the QTL on chromosome 4 had significant additive, dominance and additive by environment interaction effects while the QTL on chromosome 18 had additive and additive by environment interaction effects (Table 2). The magnitude of the effects of yield QTL identified by MCIM was similar to what was observed by CIM with the PI 436684 alleles for the QTL on chromosome 4 and 18 having an additive effect of 67 and 62 kg ha−1 for yield, respectively (Tables 2).
Markers in the E population that were not placed in linkage groups and therefore were not included in either the CIM or MCIM analysis were tested for associations with agronomic traits with SMA. The SNP marker BARC-044481-08709 (BARC8709) on chromosome 5 was significantly (P = 0.0022) associated with yield across the environments (Table 3). The other 12 segregating SNP markers on the chromosome 5 were grouped together in one genetic linkage map by JOINMAP 3.0 while BARC8709 was not. Based on the G. max consensus map 4.0, there was a distance of at least 8.8 cM between BARC8709 and other SNP markers that were grouped together. BARC8709 had an additive effect on yield of 44 kg ha−1 and the yield increasing allele was from PI 436684. This marker was not significantly associated with any other trait and no other significant QTL was identified with the non-linked markers by single factor analysis in the E population. When the three significant yield QTL identified by CIM and the BARC8709 were placed into a multivariate model, all QTL were significant (P < 0.001) except BARC8709 and the total variance explained was 0.23 (Table 1).
Maturity, plant height, and plant lodging QTL
Five QTL controlling maturity, six QTL for plant height, and two for plant lodging were mapped in the E population with either CIM or MCIM methods. QTL for maturity, plant height and lodging were mapped within 9 cM of the yield QTL on chromosome 4 (Table 4). The allele for later maturity increased plant height and greater lodging was from the PI parent, which was the source of the yield increasing allele. QTL controlling maturity and plant height also were mapped to the same positions as the yield QTL on both chromosomes 14 and 18 (Tables 2, 4). Similar to what was observed for the QTL on chromosome 4, the source of the allele that increased yield conferred later maturity and greater plant height for both chromosomes. Additional QTL for both maturity and plant height were mapped on chromosomes 9 and 17 (Table 4). The allele for later maturity and taller plants was from Elgin for the chromosome 9 QTL, while the allele for later maturity and taller plants was from PI 436684 for the chromosome 17 QTL (Table 4). An additional QTL for plant height was mapped on chromosome 2 and a QTL for lodging was mapped on chromosome 1.
QTL identified in the W population
Yield QTL
Across environments, only one QTL, located on chromosome 3, was significant for yield with CIM (Table 5). For this yield QTL across environments, the SNP marker BARC-060031-16308 had the greatest LOD score (7.3) and the allele from PI 90566-1 had an additive effect of 80 kg ha−1 greater yield than the allele from Williams 82 (Table 5). This QTL was significant at six of the seven locations in which the population was evaluated based on the CIM (Table 5).
A similar trend was observed for the chromosome 3 yield QTL with the MCIM analysis. The MCIM analysis revealed a 77 kg ha−1 additive effect (Table 5) and it was significant in four of the seven test locations. The QTL also showed significant dominance and additive by environment interaction effects.
The only non-linked marker that was significant in the SMA was BARC-059943-16234 (BARC16234) on chromosome 16 and this marker was significantly (P = 0.0264) associated with yield (Table 3). The other 14 markers on this chromosome formed a cluster that was at least 20.2 cM from BARC16234 based on the G. max consensus map 4.0. This QTL had an additive effect of 40 kg ha−1 and the positive effect was from Williams 82. When the significant QTL identified by CIM and the second QTL identified by SMA were placed into a multivariate model, both QTL were significant and together their R 2 value was 0.34 (Table 1).
Maturity, plant height, and plant lodging QTL
Three QTL for maturity, two QTL for plant height and two QTL for plant lodging were identified with either CIM or MCIM analysis in the W population (Table 6). QTL for both maturity and plant height were mapped to the same region on chromosome 3 as the significant yield QTL. The allele from PI 90566-1 had an additive effect of 1.5 days later maturity and 2.8 cm shorter height than the allele from Williams 82. QTL controlling maturity, plant height, and lodging also were mapped to chromosome 18 with the Williams 82 allele having an additive effect of 1.1 day later maturity, 1.4 cm greater plant height, and 0.7 less lodging than the allele from PI 90566-1 (Table 6). QTL on chromosome 15 were detected for both maturity and lodging with the Williams 82 allele having an additive effect of 0.2 days later maturity, and 0.1 less lodging than the allele from PI 90566-1 (Table 6).
Epistasis identified by MCIM in QTLNetwork
E population
Across the six environments, significant epistasis (experiment wide probability of P < 0.05) for maturity, plant height and lodging was detected with MCIM, while no significant epistasis was found for seed yield (Table 7). An AA epistatic interaction for maturity was identified between loci on chromosomes 2 and 14. These two loci were not detected individually with the MCIM analysis; however, the locus on chromosome 14 was detected individually by CIM (Table 4). Epistasis for plant height and lodging was identified between loci that had no individual effect (Tables 4, 7). The epistasis for plant height was detected between loci on chromosomes 1 and 5 (Table 7). The epistasis for lodging was detected between loci on chromosomes 15 and 17 and had only AD with environmental interaction (Table 7). The proportion of the phenotypic variation for maturity explained by the three major QTL identified by both CIM and MCIM was 0.46, while total amount of the variation explained by the effects of the major QTL together with epistasis was 0.63 (Table 1). For plant height and lodging, epistasis could explain 10 and 13 % of phenotypic variation beyond what was explained by main effect QTL, respectively (Table 1).
W population
In the W population, two significant AA epistatic (P < 0.05) interactions for plant maturity were identified and neither was found to interact with the environment (Table 7). There was no significant epistasis for plant height, lodging and seed yield. The first epistatic interaction was between regions on chromosomes 13 and 18. A maturity QTL was mapped to the same region on chromosome 18 through both CIM and MCIM and no individual effect maturity QTL was mapped to the chromosome 13 region. The second epistatic interaction was detected between loci on chromosomes 16 and 20 and these had no significant individual effects for maturity based on CIM and MCIM analysis (Table 7). The total amount of phenotypic variation for maturity explained by the two major QTL identified through both the CIM and the MCIM analysis and two epistatic interactions was 0.72, whereas the amount of variation explained by only the two major QTL on chromosomes 3 and 18 was 0.56 (Table 1).
Discussion
The backcross populations used in this study were developed through selections made over 25 years. The development of these populations was begun before technology was available for large-scale QTL mapping and they were not initially intended for that use. This is not a recommended strategy for yield QTL mapping but the end result of this backcrossing was the development of lines that outperformed the recurrent parent. An advantage of using these backcross populations is that positive alleles from the exotic parents are segregating in a very limited proportion of the recurrent parent genome. Another advantage is that the donor alleles are segregating in a more elite genetic background compared to using a population developed from a standard two-way cross. Even when an exotic parent has alleles that could potentially improve agronomic traits, exotic accessions often have poor overall agronomic performance that makes the resulting populations difficult to evaluate for agronomically important traits. Negative features of QTL mapping using a backcross population are the inability to assay the entire genomes of either parent for QTL because much of the genome of the backcross population is fixed for alleles from the recurrent parent.
Our study is the first report of mapping QTL controlling yield using the combination of Illumina GoldenGate assays and backcross populations in soybean. Although GoldenGate assays were previously used for genetic mapping in soybean, it has been widely used to map simply inherited traits such as disease or pest resistance controlled by a single gene or a few genes including Asian soybean rust (Hyten et al. 2009; Chakraborty et al. 2009), soybean cyst nematode (Heterodera glycines Inchinoe) (Vuong et al. 2010; Kim et al. 2011), and soybean aphid (Aphis glycines Matsumura) (Jun et al. 2012). Our study shows that the GoldenGate assays are a powerful tool to quickly map major QTL since the assay is capable of testing 192 DNA samples with 1,536 SNPs in 3 days (Hyten et al. 2008).
Some QTL were identified by both CIM and MCIM, others were identified by either CIM or MCIM, and still others by only SMA (Tables 2, 3, 4, 5 and 6). It is not surprising that different mapping algorithms may lead to different results, even when the same phenotypic and genotypic data were used (Kassem et al. 2006). Because the QTL identification is based on a statistical approach, it is also possible to identify false positive and false negative QTL (Mackay and Powell 2007; McElroy et al. 2006). However, reliability of identified QTL can be enhanced using more than one analysis method (Ravi et al. 2011). This is the reason that both MapQTL and QTLNetwork were employed to identify QTL in the present study. The yield QTL identified by both programs were mapped to almost the same genomic regions which further strengthens our confidence in the reliability of these QTL. QTL detected by only one QTL mapping method may be false positives and there is a need for validation by other approaches. MCIM in QTLNetwork, which uses most sources of variation, should be more effective in detecting both QTL with major and minor effects than the analysis done with CIM in MapQTL, which used the average trait values across environments or across replications in individual environments (Gutierrez-Gonzalez et al. 2010). In our study, the yield QTL identified by CIM and MCIM on chromosome 3 in the W population had the most consistent effects across environments (Table 5).
There is no consistent pattern in the relationship between yield and the other important agronomic traits in soybean but it has been shown that generally higher yield is associated with later maturity and taller plant height (Ablett et al. 1989; Cober and Morrison 2010; Mansur et al. 1996). For example, the regions on chromosomes 4 and 18 from PI 436684 in the E population where QTL alleles for increased yield mapped were also significant for additive effects of 0.6 and 1.1 days delay in maturity and 2.2 and 1.7 cm increase in plant height, respectively (Tables 2, 4). When yields adjusted using maturity as a covariate were analyzed with CIM and SMA, the QTL on chromosome 4, 14 and 18 were still significant for yield. The additive effects for the chromosome 4 and 14 QTL changed little from after adjustment while the effect of the chromosome 18 QTL allele from PI 436684 increased from 61 kg ha−1 before adjustment to 146 kg ha−1 after adjustment. For the W population, the yield QTL on chromosome 3 was no longer significant after analysis by CIM and SMA with the yields adjusted for maturity. These results suggest that the yield QTL on chromosome 3 in the W population could be a maturity gene that increases yield through delaying maturity. However, this is not always the case for maturity QTL as significant maturity QTL of similar magnitude on chromosomes 9 and 17 in the E population and chromosome 18 in the W population were not significantly associated with increased yield.
Identified yield QTL in the present study could explain only a portion of the total variation despite near complete SNP marker coverage of the areas of the genome segregating in the two populations. Across the environments, 23 % of phenotypic variation and 38 % of the genotypic variance for yield were explained by the three yield QTL on chromosomes 4, 14, and 18 in the E population. In the W population, 34 % of phenotypic variation and 52 % of the genotypic variance for yield were explained by the yield QTL on chromosomes 3 and 16. These results suggest that a larger number of QTL with effects too small to detect are involved in controlling the quantitative genetic variation for the traits measured in addition to other factors such as environmental interaction and epistasis. Alternatively, it might be possible that some of the remaining non-explained effects were in regions with inadequate marker coverage.
Previous work has provided evidence showing that the effects of epistasis may vary from a large to small impact on quantitative traits. Epistasis was found to be an important factor underlying the genetic basis of complex traits such as soybean seed isoflavone content (Gutierrez-Gonzalez et al. 2010), soybean seed yield (Lark et al. 1995), maize grain yield (Ma et al. 2007), and grain protein content in wheat (Kulwal et al. 2005). In contrast, linolenic acid content in soybean seed (Han et al. 2011), seed grain yield in wheat (Reif et al. 2011), and flowering time in maize (Buckler et al. 2009) were found to be controlled primarily by additive (main) effects rather than epistasis. In the present study, epistasis for plant maturity, height and lodging was detected, but their effects were minor compared to the effects of individual QTL (Tables 1, 7). A potential reason for the relatively small role of epistasis in our study is our use of backcross populations. Some epistatic interactions that could have been observed in two-way crosses may have been missed in our backcross populations because one or both interacting regions were not segregating in the backcross populations (Li et al. 1997).
E population
In the present study, two positive yield QTL alleles from an exotic source were identified on chromosomes 4 and 18 by both CIM and MCIM, a third QTL allele with a positive effect from the adapted parent Elgin was identified on chromosome 14 by only CIM, and a fourth QTL with a positive allele from the exotic source was mapped on chromosome 5 with SMA (Tables 2, 3). In Table 8, only previously reported yield QTL mapped on the same chromosomes with the positive yield QTL identified by both CIM and MCIM in the present study were listed because they are likely more reliable than other QTL detected by only one analysis method. The chromosome 4 yield QTL was mapped to approximately 14 cM on this chromosome based on the G. max consensus map 4.0 (Table 8; Hyten et al. 2010; http://soybase.org). Several yield QTL were previously reported on chromosome 4 (Smalley et al. 2004; Guzman et al. 2007; Yuan et al. 2002; Kassem et al. 2006; Sebastian et al. 2010; Table 8); however, only the QTL by Smalley et al. (2004) was mapped within 20 cM of the yield QTL mapped in our study (Table 8). This previously mapped QTL was located between SOYGPATR at 3 cM and Satt565 at 5.7 cM on the G. max consensus map 4.0 (Table 8), respectively. These two QTL are sufficiently close that they may be the same QTL.
The yield QTL on chromosome 18 was mapped to an interval between 97.3 and 103.1 cM on the G. max consensus map 4.0. Kabelka et al. (2004, 2006) both mapped a yield QTL within 20 cM of this QTL using two very different populations (Table 8). The allele for greater yield was from the cultivar BSR 101 in Kabelka et al. (2004) and from the Glycine soja Sieb. and Zucc. PI 468916 in Kabelka et al. (2006). The yield increasing allele from PI 468916 maps to the same position as an allele that provides resistance to soybean cyst nematode (SCN, Heterodera glycines Ichinohe) so it is likely that this yield QTL is a secondary effect of the SCN resistance. The QTL in both former studies were detected by SMA and the QTL in Kabelka et al. (2004) was also associated with plant height and the QTL in Kabelka et al. (2006) was associated with both plant height and lodging score. The yield QTL mapped on chromosome 18 in our study was associated with additive effects of 1.1 day delay in maturity and 1.7 cm increase in plant height (Table 4). This QTL is located in a 9.6-cM gene-rich interval outside the pericentromeric region on chromosome 18 based on the G. max genome (assembly version 1.01) (Schmutz et al. 2010; http://soybase.org). This interval has a relatively high rate of recombination as the 9.6 cM corresponds to an 845 kb region or 88 kb cM−1. This recombination rate is greater than the average genetic-to-physical ratio of approximately 197 kb cM−1 for soybean euchromatic chromosome arms (Table 8; http://soybase.org).
The yield QTL on chromosome 14 was identified only by CIM and was not detected by MCIM in any single environment or across environments (Table 2). The genetic position of the marker association with the yield QTL was 85.5 cM on chromosome 14 based on the G. max consensus map 4.0. The closest known yield QTL on chromosome 14 was detected by Satt066 (Smalley et al. 2004) and its genetic position was approximately 68 cM (http://soybase.org). The yield QTL on chromosome 5 that was mapped with BARC8709 by SMA were linked close to a yield QTL previously mapped by Kabelka et al. (2004) with Satt382. The genetic locations of Satt382 and BARC8709 on the G. max consensus map 4.0 are 26 and 22.5 cM and their additive effects were 50 and 44 kg ha−1, respectively. These results suggest that it is possible that the same QTL was mapped in both studies.
Eight major maturity genes (E1–E8) have been reported and six of them were mapped and placed on chromosomes 6 (E1) (Song et al. 2004), 10 (E2) (http://soybase.org), 19 (E3) (Molnar et al. 2003), 20 (E4) (Abe et al. 2003; Molnar et al. 2003), 6 (E7) (Molnar et al. 2003), and 4 (E8) (Cober and Morrison 2010). Three maturity QTL identified by both CIM and MCIM were mapped on chromosome 4, 17, and 18 in this study (Table 4). Although the QTL on chromosome 4 is on the same chromosome as E8, it was positioned at least 40 cM from E8 based on the G. max consensus map 4.0, so it is unlikely that it is E8. Six other maturity QTL were mapped on chromosome 4 by Keim et al. (1990), Orf et al. (1999), and Lee et al. (1996); however, they were all mapped at least 30 cM from the QTL we identified based on the G. max consensus map 4.0. Therefore, QTL on chromosome 4 in our study is likely a new maturity QTL.
A maturity QTL was previously mapped on chromosome 17 by Satt186 (Guzman et al. 2007) and its genetic location was 92.2 cM on the G. max consensus map 4. BARC-021991-04246, the marker closest to the chromosome 17 QTL identified in the present study, was mapped to 72.2 cM on G. max consensus map 4.0. The distance between these QTL is sufficiently great to suggest that the chromosome 17 maturity QTL mapped in our study might be new.
Three maturity QTL were previously mapped to an interval between 43.3 and 89.4 cM on chromosome 18 based on the G. max consensus map 4.0 (Guzman et al. 2007; Kabelka et al. 2004, 2006). In the present study, the genetic locations of the SNP markers flanking the QTL associated with both yield and maturity in the G. max consensus map 4.0 were 97.3 and 103.1 cM (Table 8) which indicate that the QTL from PI 436684 may be the same QTL as previously identified by Guzman et al. (2007) and Kabelka et al. (2004).
W population
Compared to the E population, fewer QTL were identified in the W population. This was expected because the W population is in the BC3 generation and therefore should have only one half as much of the genome segregating as the E population, which is in the BC2 generation. The backcross generations of these populations are consistent with the observed size of the map in each population with the W population having a map of 238 cM and the E population having a map size of 469 cM. In addition, the number of lines in the W population was smaller than the E population, which would make it less likely to identify small effect QTL in the W population.
The only yield QTL mapped by CIM and MCIM in the W population is on chromosome 3 and several yield QTL have been previously reported on this chromosome (Smalley et al. 2004; Kassem et al. 2006; Kabelka et al. 2004; Specht et al. 2001; Wang et al. 2004). The LOD peak for the yield QTL on chromosome 3 mapped in the present study is at 92.1 cM (Table 8; http://soybase.org). The closet previously mapped yield QTL was linked to Satt257 (Wang et al. 2004; Guzman et al. 2007) and its genetic location was 74.7 cM in the G. max consensus map 4.0 (Table 8). Because the QTL we mapped is 17.4 cM from these previously mapped QTL, it is likely that this is a newly identified QTL. This QTL is relatively stable under the different environments tested and is associated with additive effects of 1.5 days delay in maturity and 2.8 cm decrease in plant height (Tables 5, 6). The positive QTL allele from PI 90566-1 had a 77–80 kg ha−1 additive effect, additive by environment interaction, and dominance effect (Table 5).
A yield QTL was identified by BARC16234 on chromosome 16 in the W population by SMA (Table 3). Guzman et al. (2007) previously identified yield QTL from PI 407720 with the SSR marker Satt547, and its genetic location on the G. max consensus map 4.0 is 74.9 cM, which is 8.1 cM from BARC16234. Although the additive effects of Satt547 (90 kg ha−1) is greater than for BARC16234 (41 kg ha−1), it is possible that the same QTL was mapped in both studies.
The maturity QTL identified on chromosome 18 was mapped near a maturity QTL identified in a previous study. Guzman et al. (2007) found that Satt191 locus from Lawrence was associated with an additive effect of 2 days delay in plant maturity by SMA and the genetic location of the marker was 89.4 cM in the G. max consensus map 4.0. The maturity QTL we identified on chromosome 18 mapped to approximately 94 cM with both CIM and MCIM and the QTL had an additive effect of 1.1 days delay in the maturity (Table 6). The genetic positions and additive effects of these QTL suggest that we may have mapped the same maturity QTL that was previously mapped.
Results from this study indicate that exotic soybean germplasm can be a good resource for improving yield in North American soybean cultivars. The QTL from PI 90566-1 was significant in both CIM and MCIM across environments as well as in each year and in most of the individual environments. No previous studies have reported a yield or maturity QTL near the QTL region on the chromosome. Confirmation of the yield QTL in the E and W population is currently being done. The SNP markers flanking the QTL will be especially useful in MAS and pyramiding of positive QTL in soybean breeding programs, because of the availability of efficient SNP marker detection assays such as TaqMan and melting curve assay. In addition, the identification of the physical location of the QTL on the soybean genome will greatly facilitate discovery of candidate genes, map-based cloning and functional characterization of the QTL.
Abbreviations
- PI:
-
Plant introduction
- MAS:
-
Marker-assisted selection
- SNP:
-
Single nucleotide polymorphism
- QTL:
-
Quantitative trait loci
- CIM:
-
Composite interval mapping
- MCIM:
-
Mixed-model-based composite interval mapping
References
Abe J, Xu D, Miyano A, Komatsu K, Kanazawa A, Shimamoto Y (2003) Photoperiod-insensitive Japanese soybean landraces differ at two maturity loci. Crop Sci 43:1300–1304
Ablett GR, Beversdorf WD, Dirks VA (1989) Performance and stability of indeterminate and determinate soybean in short-season environments. Crop Sci 29:1428–1433
Bernard RL, Cremeens CR (1988) Registration of Williams 82 soybean. Crop Sci 28:1027–1028
Bernard RL, Cremeens CR, Cooper RL, Collins FI, Krober OA, Athow KL, Laviolette FA, Coble CJ, and Nelson RL (1998) Evaluation of the USDA Soybean Germplasm Collection: Maturity Groups 000 to IV (FC 01.547 to PI 266.807). US Department of Agriculture Technical Bulletin No. 1844
Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S, Garcia A, Glaubitz JC, Goodman MM, Harjes C, Guill K, Kroon DE, Larsson S, Lepak NK, Li HH, Mitchell SE, Pressoir G, Peiffer JA, Rosas MO, Rocheford TR, Romay MC, Romero S, Salvo S, Villeda HS, da Silva HS, Sun Q, Tian F, Upadyayula N, Ware D, Yates H, Yu JM, Zhang ZW, Kresovich S, McMullen MD (2009) The genetic architecture of maize flowering time. Science 325:714–718
Carter TE Jr, Nelson RL, Sneller CH, Zhanglin C (2004) Genetic diversity in soybean. In: Boerma HR, Specht JE (eds) Soybeans: improvement, production, and uses, 3rd edn. American Society of Agronomy, Madison, pp 303–416
Chakraborty N, Curley J, Frederick RD, Hyten DL, Nelson RL, Hartman GL, Diers BW (2009) Mapping and confirmation of a new allele at Rpp 1 from soybean PI 594538A conferring RB lesion type resistance to soybean rust. Crop Sci 49:783–790
Choi IY, Hyten DL, Matukumalli LK, Song Q, Chaky JM, Quigley CV, Chase K, Lark KG, Reiter RS, Yoon MS, Hwang EY, Yi SI, Young ND, Shoemaker RC, van Tassell CP, Specht JE, Cregan PB (2007) A soybean transcript map: gene distribution, haplotype and single-nucleotide polymorphism analysis. Genetics 176:685–696
Cober ER, Morrison MJ (2010) Regulation of seed yield and agronomic characters by photoperiod sensitivity and growth habit genes in soybean. Theor Appl Genet 120:1005–1012
Cui, Z, Carter TE Jr, Gai J, Qui J, Nelson RL (1999) Origin, description, and pedigree of Chinese soybean cultivars released from 1923 to 1995. US Department of Agriculture Technical Bulletin No. 1871
Diers BW, Kim KS (2008) Improving soybean using exotic germplasm. Proceedings of the 12th International Lupin Conference. pp 222–225
Fehr WR, Bahrenfus JB (1984) Registration of Elgin soybean. Crop Sci 24:385–386
Fehr WR, Caviness CE, Burmood DT, Pennington JS (1971) Stage of development descriptions for soybeans, Glycine max (L.) Merrill. Crop Sci 11:929–931
Gizlice Z, Carter TE Jr, Burton JW (1994) Genetic base for North American public soybean cultivars released between 1947 and 1988. Crop Sci 34:1143–1151
Gutierrez-Gonzalez JJ, Guttikonda SK, Tran LS, Aldrich DL, Zhong R, Yu O, Nguyen HT, Sleper DA (2010) Differential expression of isoflavone biosynthetic genes in soybean during water deficits. Plant Cell Physiol 51:936–948
Gutierrez-Gonzalez JJ, Vuong TD, Zhong R, Yu O, Lee JD, Shannon G, Ellersieck M, Nguyen HT, Sleper DA (2011) Major locus and other novel additive and epistatic loci involved in modulation of isoflavone concentration in soybean seeds. Theor Appl Genet 123:1375–1385
Guzman PS, Diers BW, Neece DJ, Martin SKS, Leroy AR, Grau CR, Hughes TJ, Nelson RL (2007) QTL associated with yield in three backcross-derived populations of soybean. Crop Sci 47:111–122
Han Y, Xie D, Teng W, Zhang S, Chang S, Li W (2011) Dynamic QTL analysis of linolenic acid content in different developmental stages of soybean seed. Theor Appl Genet 122:1481–1488
Hill J, Becker HC, Tigerstedt PMA (1998) Quantitative and ecological aspects of plant breeding. Chapman & Hall, London
Hyten D, Song Q, Choi I, Yoon M, Specht J, Matukumalli L, Nelson R, Shoemaker R, Young N, Cregan P (2008) High-throughput genotyping with the GoldenGate assay in the complex genome of soybean. Theor Appl Genet 116:945–952
Hyten DL, Smith J, Frederick RD, Tucker ML, Song Q, Cregan PB (2009) Bulked segregant analysis using the GoldenGate assay to locate the Rpp 3 locus that confers resistance to soybean rust in soybean. Crop Sci 49:265–271
Hyten DL, Choi IY, Song Q, Specht JE, Carter TE, Shoemaker RC, Hwang EY, Matukumalli LK, Cregan PB (2010) A high density integrated genetic linkage map of soybean and the development of a 1,536 universal soy linkage panel for QTL mapping. Crop Sci 50:960–968
Jun TH, Mian MAR, Michel AP (2012) Genetic mapping revealed two loci for soybean aphid resistance in PI 567301B. Theor Appl Genet 124:13–22
Kabelka EA, Diers BW, Fehr WR, LeRoy AR, Baianu IC, You T, Neece DJ, Nelson RL (2004) Putative alleles for increased yield from soybean plant introductions. Crop Sci 44:784–791
Kabelka EA, Carlson SR, Diers BW (2006) Glycine soja PI 468916 SCN resistance loci associated effects on soybean yield and other agronomic traits. Crop Sci 46:622–629
Kassem MA, Shultz J, Meksem K, Wood AJ, Iqbal MJ, Lightfoot DA (2006) An updated ‘Essex’ by ‘Forrest’ linkage map and first composite interval map of QTL underlying six soybean traits. Theor Appl Genet 113:1015–1026
Keim P, Diers BW, Olson TC, Shoemaker RC (1990) RFLP mapping in soybean: association between marker loci and variation in quantitative traits. Genetics 126:735–742
Kim KS, Diers BW (2009) The associated effects of the soybean aphid resistance locus Rag1 on soybean yield and other agronomic traits. Crop Sci 49:1726–1732
Kim M, Hyten DL, Niblack TL, Diers BW (2011) Stacking resistance alleles from wild and domestic soybean sources improves soybean cyst nematode resistance. Crop Sci 51:934–943
Kulwal PL, Kumar N, Kumar A, Gupta RK, Balyan HS, Gupta PK (2005) Gene networks in hexaploid wheat: interacting quantitative trait loci for grain protein content. Funct Integr Genomics 5:254–259
Lark KG, Chase K, Adler F, Mansur LM, Orf JH (1995) Interactions between quantitative trait loci in soybean in which trait variation at one locus is conditional upon a specific allele at another. Proc Natl Acad Sci USA 92:4656–4660
Lee SH, Bailey MA, Mian MAR, Carter TE, Ashley DA Jr, Hussey RS, Parrott WA, Boerma HR (1996) Molecular markers associated with soybean plant height, lodging, and maturity across locations. Crop Sci 36:728–735
Li Z, Pinson SRM, Park WD, Paterson AH, Stansel JW (1997) Epistasis for three grain yield components in rice (Oryza sativa L.). Genetics 145:453–465
Li D, Pfeiffer TW, Cornelius PL (2008) Soybean QTL for yield and yield components associated with Glycine soja alleles. Crop Sci 48:571–581
Lukens LN, Doebley J (1999) Epistatic and environmental interactions for quantitative trait loci involved in maize evolution. Genet Res 74:291–302
Ma XQ, Tang JH, Teng WT, Yan JB, Meng YJ, Li JS (2007) Epistatic interaction is an important genetic basis of grain yield and its components in maize. Mol Breed 20:41–51
Mackay I, Powell W (2007) Methods for linkage disequilibrium mapping in crops. Trends Plant Sci 12:57–63
Mansur LM, Orf JH, Chase K, Jarvik T, Cregan PB, Lark KG (1996) Genetic mapping of agronomic traits using recombinant inbred lines of soybean. Crop Sci 36:1327–1336
McElroy JP, Zhang W, Koehler KJ, Lamont SJ, Dekkers JC (2006) Comparison of methods for analysis of selective genotyping survival data. Genet Sel Evol 38:637–655
Molnar SJ, Rai S, Charette M, Cober ER (2003) Simple sequence repeat (SSR) markers linked to E1, E3, E4, and E7 maturity genes in soybean. Genome 46:1024–1036
Montooth KL, Marden GH, Clark AG (2003) Mapping determinants of variation in energy metabolism, respiration and flight in Drosophila. Genetics 165:623–635
Nelson RL, Johnson EOC (2012) Registration of the High-Yielding Soybean Germplasm Line LG04-6000. J Plant Reg 6:1–4
Nelson RL, Amdor PJ, Orf JH, Cavins JF (1988) Evaluation of the USDA Soybean Germplasm Collection: Maturity Groups 000 to IV (PI 427.136 to PI 445.845). US Department of Agriculture Technical Bulletin No. 1726
Orf JH, Chase K, Jarvik T, Mansur LM, Cregan PB, Adler FR, Lark KG (1999) Genetics of soybean agronomic traits. I. Comparison of three related recombinant inbred populations. Crop Sci 39:1642–1651
Ravi K, Vadez V, Isobe S, Mir RR, Guo Y, Nigam SN, Gowda MVC, Radhakrishnan T, Bertioli DJ, Knapp SJ, Varshney RK (2011) Identification of several small main-effect QTLs and a large number of epistatic QTLs for drought tolerance related traits in groundnut (Arachis hypogaea L.). Theor Appl Genet 122:1119–1132
Reif JC, Maurer HP, Korzun V, Ebmeyer E, Miedaner T, Würschum T (2011) Mapping QTLs with main and epistatic effects underlying grain yield and heading time in soft winter wheat. Theor Appl Genet 123:283–292
Saghai Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
SAS Institute (2002) The SAS system for Windows. Release 9.2. SAS Institute, Cary
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183
Schön CC, Melchinger AE, Boppenmaier J, Brunklaus-Jung E, Herrmann RG, Seitzer JF (1994) RFLP mapping in maize: quantitative trait loci affecting testcross performance of elite European flint lines. Crop Sci 34:378–389
Sebastian S, Streit L, Stephens P, Thompson J, Hedges B, Fabrizius M, Soper J, Schmidt D, Kallem R, Hinds M (2010) Context-specific marker-assisted selection for improved grain yield in elite soybean populations. Crop Sci 50:1196–1206
Smalley MD, Fehr WR, Cianzio SR, Han F, Sebastian SA, Streit LG (2004) Quantitative trait loci for soybean seed yield in elite and plant introduction germplasm. Crop Sci 44:436–442
Song QJ, Marek LF, Shoemaker RC, Lark KG, Concibido VC, Delannay X, Specht JE, Cregan PB (2004) A new integrated genetic linkage map of the soybean. Theor Appl Genet 109:122–128
Specht JE, Chase K, Macrander M, Graef GL, Chung JU, Markwell JP, Germann M, Orf JH, Lark KG (2001) Soybean response to water: a QTL analysis of drought tolerance. Crop Sci 41:493–509
Van Ooijen JW, Voorrips RW (2001) Joinmap 3.0. Software for the calculation of genetic linkage maps. Plant Research International, Wageningen
Van Ooijen JW, Boer MP, Jansen RC, Maliepaard C (2002) MapQTL 4.0. Software for the calculation of QTL positions on genetic maps. Plant Research International, Wageningen
Vuong TD, Sleper DA, Shannon JG, Nguyen HT (2010) Novel quantitative trait loci for broad-based resistance to soybean cyst nematode (Heterodera glycines Ichinohe) in soybean PI 567516C. Theor Appl Genet 121:1253–1266
Wang D, Graef GL, Procopiuk AM, Diers BW (2004) Identification of putative QTL that underline yield in interspecific soybean backcross populations. Theor Appl Genet 108:458–467
Yang J, Hu CC, Hu H, Yu RD, Xia Z, Ye XZ, Zhu J (2008) QTLNetwork: mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 24:721–723
Yuan J, Njiti VN, Meksem K, Iqbal MJ, Triwitayakorn K, Kassem MA, Davis GT, Schmidt ME, Lightfoot DA (2002) Quantitative trait loci in two soybean recombinant inbred line populations segregating for yield and disease resistance. Crop Sci 42:271–277
Acknowledgments
This research was funded through support from the Illinois Soybean Association.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by I. Rajcan.
Rights and permissions
About this article
Cite this article
Kim, KS., Diers, B.W., Hyten, D.L. et al. Identification of positive yield QTL alleles from exotic soybean germplasm in two backcross populations. Theor Appl Genet 125, 1353–1369 (2012). https://doi.org/10.1007/s00122-012-1944-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-012-1944-1