
Abstract
Recent advances in whole genome sequencing (WGS) have allowed identification of genes for disease susceptibility in humans. The objective of our research was to exploit whole genome sequences of 13 rice (Oryza sativa L.) inbred lines to identify non-synonymous SNPs (nsSNPs) and candidate genes for resistance to sheath blight, a disease of worldwide significance. WGS by the Illumina GA IIx platform produced an average 5× coverage with ~700 K variants detected per line when compared to the Nipponbare reference genome. Two filtering strategies were developed to identify nsSNPs between two groups of known resistant and susceptible lines. A total of 333 nsSNPs detected in the resistant lines were absent in the susceptible group. Selected variants associated with resistance were found in 11 of 12 chromosomes. More than 200 genes with selected nsSNPs were assigned to 42 categories based on gene family/gene ontology. Several candidate genes belonged to families reported in previous studies, and three new regions with novel candidates were also identified. A subset of 24 nsSNPs detected in 23 genes was selected for further study. Individual alleles of the 24 nsSNPs were evaluated by PCR whose presence or absence corresponded to known resistant or susceptible phenotypes of nine additional lines. Sanger sequencing confirmed presence of 12 selected nsSNPs in two lines. “Resistant” nsSNP alleles were detected in two accessions of O. nivara that suggests sources for resistance occur in additional Oryza sp. Results from this study provide a foundation for future basic research and marker-assisted breeding of rice for sheath blight resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sheath blight (SB), caused by the fungal pathogen Rhizoctonia solani Kuhn, causes significant yield loss and reduction in grain quality for rice (Oryza sativa L.) in the southern U.S. and other regions of the world (Lee and Rush 1983; Rush and Lindberg 1996). All current U.S. rice cultivars are susceptible to R. solani with costly fungicide applications as the primary means of control. Various studies have shown that response of different rice lines to infection by R. solani is expressed as partial resistance (Liu et al. 2009), also referred to as incomplete, quantitative, field, or horizontal resistance (Wang et al. 2010). Numerous genetic and QTL mapping studies have reported partial resistance, hereafter referred to simply as “resistance”, to R. solani is controlled by multiple regions in the genome each with small or moderate effect (http://www.gramene.org). As part of the RiceCAP research efforts (http://www.uark.edu/ua/ricecap), a recombinant inbred line mapping population was used to identify a “major” QTL for SB resistance on chromosome 9 (Liu et al. 2009). This same region was also reported in previous studies to impact SB resistance (Pinson et al. 2005; Tan et al. 2005; Zuo et al. 2008), including the RiceCAP SB2 mapping population evaluated in Louisiana and Arkansas (Nelson et al. unpublished data). A recent study reported a QTL of large effect for resistance on chromosome 11 containing 154 genes of which 11 were tandem repeats of xylanase inhibitor (chitinase) genes (Channamallikarjuna et al. 2010). Zhao et al. (2008) found 50 genes of diverse function that were transcritpionally activated in rice after challenge by R. solani. Venu et al. (2007) detected numerous up- and down-regulated rice genes after infection by R. solani using SAGE and microarray analysis. Increased resistance was observed in transgenic rice containing an engineered ribosome inactivating protein (Kim et al. 2003), thaumatin and chitinase genes from rice (Maruthasalam et al. 2007), and chitinase genes from Trichoderma atroviride and T. virens (Liu et al. 2004; Shah et al. 2009). O. nivara accessions IRGC 104443 and IRGC 100898 were shown recently to exhibit SB resistance under greenhouse, growth chamber, or laboratory conditions (Prasad and Eizenga 2008). In spite of the research efforts described above, the routine use of marker-assisted selection to enhance SB resistance in commercial rice cultivars has not been reported.
The advent of next-generation sequencing has been proposed as a rapid, cost effective alternative to Sanger sequencing for identification of candidate genes and variants underlying simple and even complex traits (Hobert 2010; Teer and Mullikin 2010). Whole genome sequencing (WGS) of one or a few individuals has recently identified single or multiple variants associated with different Mendelian disorders in humans (Rios et al. 2010; Roach et al. 2010; Sobreira et al. 2010; Tong et al. 2010; Lupski et al. 2010). Similar progress has been made with whole-exome sequencing to uncover rare or recessive variants in humans causing different diseases or adaptations to different environments (Bilgüvar et al. 2010; Krawitz et al. 2010; Ng et al. 2010a, b; Walsh et al. 2010; Yi et al. 2010). Xie et al. (2010) recently used WGS of recombinant inbred lines of rice (Oryza sativa L.) at low coverage to construct a linkage map of ~209 K SNPs that successfully identified a known QTL associated with grain width. A similar WGS strategy for chromosome segment substitution lines allowed identification of a QTL containing the sd1 locus for plant height (Xu et al. 2010). A genomic DNA library enriched for genic sequences in rice was recently constructed followed by deep sequencing that revealed ~2,600 SNPs between an indica and a tropical japonica line (Deschamps et al. 2010).
In addition to QTL mapping for SB resistance by Liu et al. (2009), RiceCAP completed WGS of 13 rice lines using the Illumina GA IIx platform in cooperation with the National Center for Genome Resources (Scheffler et al., unpublished data). The objective of our research was to use sequence data of 13 lines to identify nsSNPs and corresponding candidate genes for SB resistance. We chose to focus on nsSNPs in our study because this class of variants was reported to play a role in the function and evolution of plant resistance (Fu et al. 2010; Ling et al. 2009; McNally et al. 2009; Song et al. 1995) that may complement microarray or other gene expression studies.
Materials and methods
Plant material, sheath blight ratings, DNA isolation, and variant selection strategies
All lines tested in this study were evaluated in inoculated field/greenhouse trials from 2008 to 2010 and scored for sheath blight visual ratings based on a 0–9 scale where 0 = no disease and 9 = dead plants (J.H. Oard, M.C. Rush, D.E. Groth, F. Correra, unpublished results). Plants considered resistant in this study produced ratings of 3–5, moderately resistant/susceptible plants with rating of 6, and susceptible plants showed ratings of 7–8.
To identify nsSNPs and candidate genes by the “common variant” (CV) selection strategy (see below), three SB-resistant lines [Jasmine 85 (PI 595927), MCR010277 (GSOR 200327), TeQing (PI 536047)] and three susceptible lines [Cocodrie (PI 606331), Cypress (PI 561734), Lemont (PI 475833)] were used. To further evaluate the initial CV-selected nsSNPs, the following 11 highly/moderately resistant lines were used: Shu Feng 121-1655 (mutant of PI 615015), Rondo (mutant of PI 615022), Taducan (PI 280681), Oryzica Llanos 5 (GSOR 301111), 09DN/Rush072, Araure 3 (F. Correra, unpublished), IR64 (GSOR 301401), Jhona 349 (GSOR 301071), Jouiku 393G (GSOR 301072), O. nivara (IRGC 100898), and O. nivara (IRGC 10443). In addition, the following nine highly/moderately susceptible lines were used: Azucena (GSOR 301665), Bengal (PI 561735), Bowman (RU0404191), Francis (PI 632447), L-201 (CIor 9971), LaGrue (PI 568891), Leah (GSOR 310045), Nipponbare (GSOR 301164), and Wells (PI 612439).
For the “Principal Component-Biplot” (PB) variant selection strategy (see below), the following 13 lines were used: Bengal, Bowman, Cocodrie, Cypress, Francis, Jasmine 85, LaGrue, Lemont, L-201, MCR010277, TeQing, Shu Feng 121-1655, and Wells. Seedlings of these lines were grown in the dark for ~14 days to minimize presence of chloroplasts in the leaves collected for total DNA isolation using the DNeasy 96 Plant kit (Qiagen, Inc., Hilden, Germany). Genomic DNA from each of the 13 lines was used for WGS described below. For the remaining lines, DNA was isolated from leaves grown in light using the method described by Li et al. (2010).
WGS and SNP calling
Genomic DNA isolated from each line as described above was sheared by a Covaris S2 sonicator, and Illumina paired-end genomic libraries were built according to standard protocols. Cluster generation was performed on an Illumina cluster station using a version 2 cluster generation kit and 54-bp paired-end sequencing was carried out on an Illumina Genome Analyzer IIx. Base calling and quality filtering were performed with Illumina Pipeline version 1.4.0 with default parameters. Paired reads were aligned to version 6.0 of the MSU rice genome assembly using GSNAP (Wu and Nacu 2010) with trimming enabled and allowing up to six mismatches with indels scored as equivalent to three mismatches. Alignments were filtered and variants called and characterized for changes to coding potential via the Alpheus pipeline (Miller et al. 2008). Alignments were required to have at least 50 bp matched for a read aligned singly or 100 bp matched for a paired alignment. Reads mapping equivalently to more than five locations were discarded. Variants were called from alignments meeting these criteria where in at least one of the sequenced lines, the variant allele was detected in at least two uniquely aligning reads, with the bases calling the variant having an phred-equivalent average quality ≥20, and that ≥20% of the reads aligned to the site in that variety called the variant allele. For each variant meeting these criteria, evidence for the genotype at that site was reported for each of the lines.
Identification of non-synonymous SNPs in candidate resistance genes
The following steps were carried out for the CV filtering strategy using the six lines described above: (1) select all variants, except those identified in transposable elements, from output of Alpheus analysis pipeline with quality score ≥25; (2) select variants from Step 1 with coverage ≥5; (3) select common variants from step 2 with ≥3 reads in susceptible Cocodrie, Cypress, and Lemont; (4) select common variants from step 2 with ≥3 reads in resistant Jasmine 85, TeQing, and MCR010277; (5) given that the reference Nipponbare is SB susceptible, select variants that have 100% frequency in the resistant lines and 0% frequency in the susceptible lines; (6) select nsSNPs from Step 5 and identify corresponding candidate genes.
The PB variant selection strategy was carried out using the 13 lines described above in the following steps: (1) select all variants, except those in transposable elements, from output of Alpheus analysis pipeline with quality scores ≥25; (2) complete remaining steps using SAS software (Release 9.1.3; SAS Institute, Cary, NC); (3) select variants from Step 1 with coverage ≥5; (4) remove common variants selected in Step 3 across all 13 lines with reads ≥3; (5) perform principal component analysis (PCA) using standardized variant frequencies of the 13 lines; (6) perform Ward’s minimum variance clustering (Everitt et al. 2001) using PC1 and PC2 scores obtained in Step 5; (7) for each cluster identified in Step 6, compute average variant frequencies for the 13 lines. Given that the reference Nipponbare is SB susceptible, identify a single cluster with highest average variant frequency in resistant lines and lowest average variant frequency in susceptible lines; (8) select nsSNPs from single cluster identified in Step 7 and identify corresponding candidate genes; (9) create GGE biplot display (Yan and Tinker 2006) using PC scores from Step 5.
nsSNP-specific PCR
Primers ~25-nt long were designed to amplify ~350 bases flanking each nsSNP using the SNAP Program (http://ausubellab.mgh.harvard.edu/) based on sequences of Cocodrie and MCR010277 generated by the Illumina GA IIx platform and the reference Nipponbare sequence posted at the Gramene website (http://www.gramene.org). A 10 μL PCR reaction consisted of the following: 0.5 μL 10 ng DNA template, 1 μL 10× buffer solution (containing 1 mM MgCl2) (Applied Biosystems, Life Technologies, Carlsbad, CA, USA), 7.22 μL of dH2O, 0.8 μL of 10 mM dNTPs mix (Applied Biosystems), 0.2 μL each of 20 μM forward and reverse primers, and 0.08 μL of 5 U μL−1 of TAQ polymerase (Applied Biosystems). PCR reactions were carried out on the BioRad ICycler consisting of the following steps: 95°C, 3 min; 95°C, 20 s; 60°C, 20 s; 72°C, 20 s; repeat 30× previous three steps; 72°C, 5 min. Amplified PCR products were visualized by running on a 2% agarose gel and stained with ethidium bromide.
Sanger sequencing and SNP validation
Using the Nipponbare reference sequence from Gramene (http://www.gramene.org), 24 primer pairs were designed using the software Primer 3 (http://frodo.wi.mit.edu/primer3) to flank putative nsSNPs located within 23 CV-selected genes identified with Locus ID in Table S3a. Sequences of the primers designed and evaluated are shown in Table S3b. The 24 primer pairs were used to amplify PCR fragments of ~500 bp from the SB susceptible line Cocodrie and resistant line Araure 3. Both strands of amplified fragments were directly sequenced at the Pennington Biomedical Research Institute, Louisiana State University, Baton Rouge, LA. The nsSNPs were identified from sequence data using the ClustalW software (http://www.ebi.ac.uk/Tools/msa/clustalw2/). To detect predicted amino acid changes in “resistant” and “susceptible” alleles, nsSNP-containing codons from Sanger sequence data were compared manually with corresponding codons posted at the Gramene website.
To detect candidate nsSNPs in O. nivara accessions IRGC 104443 and IRGC 100898, primers were designed and evaluated as described above for 12 CV-selected nsSNPs in genes identified with locus ID given in Supplementary Table S4a (Online Resource 4). Sequences of the primers designed for amplification of PCR fragments containing the nsSNPs are shown in Supplementary Table S4b (Online Resource 4).
Results
Genomic variants, reads, and coverage
The total number of sequenced read counts, % aligned reads, total number of reads generated, filtered reads, high quality (HQ) reads, and variants detected from HQ reads are shown in Table 1. The total number of sequence reads produced across chromosomes before filtering was ~520,000,000 with a range from ~21,000,000 for Bengal to ~92, 000,000 for Cocodrie. The average percentage of reads generated across chromosomes that aligned to the Nipponbare reference genome was ~72% with a range ~60% for LaGrue to 77% for Bengal. Moderate variation in the total number of reads and filtered reads was observed for the 13 lines except for the relatively high values of Cocodrie and the low values for Bengal. Variation in the number of HQ reads and variants detected with HQ reads for this study was found to be consistent with next-generation sequencing of whole genomes in other plant species (Farmer and Woodward, unpublished results). The average coverage across lines of 5.3× generated in this study was nearly identical to that reported for WGS of six maize (Zea mays L.) inbred lines (Lai et al. 2010).
The number of variants with minimum quality scores of 25 and coverage of 5 for each chromosome, and the number of selected nsSNPs and genes are shown in Table 2. Across all chromosomes, the total number of unselected variants before filtering relative to the reference Nipponbare varied from ~300 to 500 K that represented ~10% of the total unfiltered reads produced by the Illumina procedure. Only a small reduction in variants with minimum quality was observed, but a approximately tenfold reduction in those with minimum coverage relative to the original variants was also found. Wide variation in the number of selected nsSNPs across chromosomes was observed with 1.3 nsSNPs detected on average for each gene.
Figure 1 shows the percentage of reads and coverage in different combinations for the 13 lines. Variation was observed across lines for depth of coverage and reads where Cocodrie produced the highest percentage of reads ≥3 and coverage ≥5 while Bengal generated the lowest percentage of all lines. The combination of reads ≥3 and coverage ≥5 comprised the highest percentage for all lines except for Bengal.

From left to right, frequency of reads <3, coverage <5 (tan bar); reads <3, coverage ≥5 (purple); reads ≥3, coverage <5 (light green); reads ≥3, coverage ≥5 (dark green) from whole genome sequencing of 13 rice lines (color figure online)
The percentage of all variants within intergenic, untranslated 5′, untranslated 3′, coding sequence, and intron regions for each chromosome across all 13 lines is shown in Fig. 2. A large majority of variants (~60%) were detected within intergenic regions across lines that consisted of both indica and tropical japonica sub populations. Coding sequences and introns shared similar proportions across all lines (~15–20%) while untranslated 5′and untranslated 3′ regions comprised a small percentage of the total variants (~1, ~2%, respectively).

From left to right, frequency of all variants detected by Alpheus pipeline analysis for intergenic (light green bar), untranslated 5′(UTR 5) (yellow), untranslated 3′(UTR 3) (purple), coding sequencing (CDS) (orange), and intron (blue) regions within and across 13 rice lines (color figure online)
Figure 3 shows the percentage of variants identified as insertions, deletions, non-synonymous SNPs, and synonymous SNPs for each chromosome across all 13 lines. The most striking result was the large percentage (~80%) of variants that consisted of synonymous SNPs for both indica and tropical japonica lines. The second largest class was comprised of nsSNPs, although at a much smaller percentage at ~12%, while the remaining variants were made up of insertions and deletions at very low frequencies (~3, ~4%, respectively).

From left to right, frequency of all insertions (yellow bar), deletions (red), non-synonymous SNPs (nsSNPs) (blue), and synonymous SNPs (sSNPs) (orange) detected by Alpheus pipeline analysis for each chromosome across all 13 lines (color figure online)
Detection of nsSNPs and candidate genes within SB QTL qShB9-2 on chromosome 9
qShB9-2, a QTL for sheath blight, was mapped to a region at the bottom of chromosome 9 consisting of ~1.2 M bp flanked by SSR markers RM215 and RM245 (Liu et al. 2009). Before the CV selection procedure was carried out, a total of 155 variants were detected within qShB9-2 with reads ≥3 and coverage ≥5 from resistant Jasmine 85, TeQing, and MCR010277. The majority of variants in qShB9-2 were classified as sSNPs (73%), a substantially smaller percentage as nsSNPs (26%), and the smallest fractions identified were insertions (1.0%) or deletions (0%). When the CV selection procedure was carried out to identify candidate nsSNPs for SB resistance within qShB9-2, relatively few selected nsSNPs (10) were found that mapped throughout most (~1.1 M bp) of the QTL. The nsSNPs were detected in a total of 10 genes that were placed into seven groups based on gene ontology/gene function. The physical location of selected nsSNPs within qShB9-2 along with corresponding genes are shown in Supplementary Table S1 (Online Resource 1).
Detection of nsSNPs, candidate genes, and new regions outside of qShB9-2
QTL qShB9-2 explained ~25% of the observed variation for SB resistance when Jasmine 85 was used as the resistant parent (Liu et al. 2009). Because the majority of variation was detected outside of qShB9-2, we scanned all remaining regions of the genome other than this QTL using the CV selection strategy. The selected regions also showed that sSNPs were the most common variant at 78% while insertions and deletions were rare at 0.35%. As shown in Supplementary Table S2 (Online Resource 2), the distribution of selected nsSNPs and corresponding genes across chromosomes was not uniform. For example, a maximum of 70 nsSNPs and 49 genes were found on chromosome 2 whereas 0, 7, and 9 nsSNPs and 0, 7, and 2 genes were detected on chromosomes 10, 5, and 7, respectively.
Supplementary Table S2 (Online Resource 2) includes three new candidate regions for SB resistance that have not been reported in the literature. These regions include the top of chromosome 2 (975,892–6,210,412 bp), the bottom of chromosome 3 (30,523,344–35,667,086 bp), and the bottom of chromosome 5 (21,585,027–28,979,361 bp). The homologues of certain selected genes within these regions have been implicated in stress and disease response of plants and humans. Examples include phytosulfokine receptors (LOC_Os02g06200, LOC_Os02g06210) (Motose et al. 2009), cytokinin-O-glucosyltransferase (LOC_Os02g11130) (Havlova et al. 2008), U5 small nuclear ribonucleoprotein helicase (LOC_Os03g53220) (Hahn and Beggs 2010), and CCR4-NOT transcription factor (LOC_Os05g40790) (Sarowar et al. 2007). The following three genes on chromosome 5 are reported here for the first time as candidates associated with SB resistance in plants: VHS and GAT domain containing protein (LOC_Os05g39760), kri1 protein (LOC_Os05g41100), and PX domain containing protein (LOC_Os05g50660).
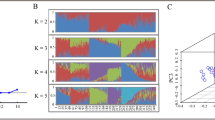
Principal component-biplot (PB) display of variants on chromosome 9
The PB selection strategy was conducted across all chromosomes for the 13 lines described above. An important step in this procedure was the construction of a biplot that simultaneously displayed the relationships among variants, relationships among lines, and the underlying interactions between variants and lines (Yan and Tinker 2006). For ease of visualization, Fig. 4 shows the biplot of variants on chromosome 9 from the PB selection among four SB resistant (MCR010277, Jasmine 85, TeQing, Shu-Feng 121-1655), one moderately susceptible (Bengal), and eight highly susceptible lines (Cocodrie, Cypress, Lemont, Bowman, LaGrue, Francis, L-201, and Wells). Vectors (solid lines) in the biplot showed a clear separation between the four resistant lines and the nine remaining lines. TeQing (TQNG) and MCR010277 (MCR) were found in the same region, which was reasonable given that TeQing was one SB-resistant parent of MCR010277. Resistant Shu-Feng 121-1655 was found in the same region as TeQing and MCR010277, but its pedigree was not known because the parental line is an undescribed accession from China (http://www.ars-grin.gov). All nine remaining lines occurred in one large region including Bengal that generated a relatively short vector length compared to the susceptible lines in that region.

Biplot display of all variants on chromosome 9 in four sheath blight resistant lines (MCR010277 (MCR), Jasmine 85 (J85), TeQing (TQNG), Shu Feng 121-1655 (SHUF) and nine highly/moderately susceptible lines [Cocodrie (CCDR), Cypress (CPRS), Lemont (LMNT), Bengal (BNGL), Bowman (BWMN), LaGrue (LGRU), Francis (FRCS), L-201, and Wells (WLLS)]
Grouping of CV-selected candidate genes based on gene family/gene ontology
Figure 5 shows the groupings of CV-selected candidate genes across all lines and chromosomes based on gene family/gene ontology. A total of 240 genes were assigned to 42 diverse groups with kinase, nucleotide binding, and peptide repeat as the top three with the greatest number of candidate genes. One-half (22/42) of the groups contained only one or two candidate genes.

Grouping of CV-selected candidate genes based on gene family/gene ontology
Genotypes of selected candidate nsSNPs evaluated in different resistant and susceptible lines
Based on selected nsSNPs from the six lines used in the CV selection strategy, we examined nsSNP profiles of the remaining seven lines sequenced by the Illumina method. The susceptible lines Bowman, Francis, L-201, LaGrue, Leah, and Wells were found with “susceptible” alleles at all loci consistent with susceptible Cocodrie, Cypress, and Lemont. The moderately susceptible Bengal displayed a combination of “susceptible” and “resistant” nsSNPs (results not shown).
A subset of 24 nsSNPs found in 23 randomly selected candidate genes was selected for further study (Supplementary Table S3a, Online Resource 3). All nsSNPs in this subset were found in dbSNP (posted on Gramene website). All PCR-generated SNP-specific alleles for susceptible Nipponbare were consistent with those from the Illumina WGS results and the published Nipponbare reference genome sequence. Susceptible Azucena and Leah produced the same allele profiles as those of Nipponbare. “Resistant” SNP genotypes generated from PCR amplification in resistant MCR010277 and TeQing were in complete agreement for all 23 genes and were consistent with all corresponding genotypes produced by the Illumina GA IIx platform. Profiles for the remaining seven moderately resistant lines varied when compared with MCR010277 and TeQing, ranging from one allele difference in IR64 and Shu Feng 121-1655 to five in Oryzica Llanos 5 and Jhona 349.
The two O. nivara accessions, IRGC 104443 and IRGC 100898, along with resistant Araure 3 and susceptible Catahoula, were screened with 12 random CV-selected nsSNPs (Supplementary Table S4a, Online Resource 4). Araure 3 produced “resistant” alleles from nine genes (LOC_Os02g19200, LOC_Os02g54330, LOC_Os02g54500, LOC_Os03g37720, LOC_04g59540, LOC_Os06g28124, LOC_Os06g29700, LOC_Os06g32350, LOC_Os09g37880). Susceptible Catahoula carried only “susceptible” alleles. IRGC 104443 produced “resistant” and “susceptible” alleles at heterozygous loci from two genes on chromosome 2 (LOC_Os02g54330, LOC_Os02g54500) while IRGC 100898 produced one resistant allele on chromosome 4 (LOC_Os04g59540).
We also genotyped eight individuals derived from the moderately resistant Louisiana inbred (F6) line 09DN/Rush072 with 11 CV-selected nsSNPs chosen at random (data not shown). No individual possessed all 11 “resistant” alleles, although five individuals contained seven resistant alleles from LOC_Os01g52880, LOC_Os02g56380, LOC_Os04g20680, LOC_Os04g55760, LOC_12g06740, LOC_Os12g09710, and LOC_Os12g10180. All eight individuals carried “susceptible” alleles from four genes on chromosome 9 (LOC_Os09g36900, LOC_Os09g37590, LOC_Os09g37800, LOC_Os09g37880).
Selection of variants using the CV vs. the PB selection strategies
The CV strategy for selection of variants in this study was developed as a modification of the approaches used to identify variants for rare human disorders. As shown here, the CV method appears to successfully select candidates associated with SB resistance, but the procedure is somewhat tedious. We therefore developed the PB approach that does involve more steps, but is actually less time consuming and more systematic than the CV method. As part of the PB strategy, the biplot display allows rapid and informative inspection of variant information not possible by other statistical methods. After the CV and PB procedures were completed for QTL qShB9-2 and the remaining portions of the genome, we found that if a low number of clusters was identified for an individual chromosome, the PB approach selected slightly greater numbers of variants than the CV method. With high numbers of clusters, both methods were virtually indistinguishable in terms of selected variants.
Sanger sequencing of fragments containing candidate nsSNPs
Both strands of 12 putative nsSNP-containing fragments from Cocodrie and Araure 3 were sequenced by the Sanger method for the following CV-selected genes: NBS-LRR type disease resistance protein Rps1-k-2 (LOC_Os12g10180), receptor-like protein kinase 2 (LOC_Os09g17630), resistance protein (LOC_Os02g35210), OsFBDUF47-F box and DUF domain containing protein (LOC_Os09g37590), receptor protein kinase TMK1 precursor (LOC_Os04g58910), OsFBDUF14-F-box and DUF domain containing protein (LOC_Os02g54330), leucine-rich repeat family protein (LOC_Os01g52880), NBS-LRR type disease resistance protein Rps1-k-1 (LOC_Os03g37720), phosphatidylinositol-4-phosphate 5-Kinase (LOC_Os04g59540), THION21-Plant thionin family protein precursor (LOC_Os02g02650), OsFBD11-F-box and FBD domain containing protein (LOC_Os06g29700), and glycosyltransferase (LOC_Os06g28124). Sanger sequencing results confirmed presence of nsSNPs within all 12 genes (results not shown). In addition, predicted amino acid changes of all nsSNPs were consistent between Sanger and GA IIx sequencing results.
Discussion
A major rice breeding goal for the southern U.S. is the development of high-yielding cultivars that are resistant to sheath blight, a disease that causes substantial reductions in grain yield and quality in the southern U.S., South America, and Asia. There is currently no resistant U.S. commercial cultivar, primarily due to challenges in selection for quantitative resistance and inconsistencies in phenotyping across years and locations. Several QTLs of small effect have been reported over the years from different studies using Jasmine 85, TeQing, MCR010277, and other lines as sources of resistance. Liu et al. (2009) crossed Jasmine 85 with Lemont to generate a mapping population that showed a QTL at the bottom of chromosome 9 with a “large” effect (R 2 = 0.25). However, only a modest increase in resistance was observed using three markers within this region for selection in a backcross population (Zuo et al. 2008). This result highlights the quantitative nature of SB resistance and the need to identify additional markers across the entire genome to assist in development of new cultivars with high levels of resistance.
Several recent studies in humans have shown the potential of WGS to identify variants and genes responsible for rare Mendelian disorders (Rios et al. 2010; Roach et al. 2010; Sobreira et al. 2010; Tong et al. 2010; Lupski et al. 2010). Based on initial success of the human sequencing efforts, we initiated a study to evaluate WGS of rice by the Illumina GA technology to identify candidate nsSNPs that are associated with resistance to sheath blight. An important component of the RiceCAP efforts was to complete WGS of 13 inbred rice lines that have been used in applied breeding of elite U.S. southern cultivars. As shown in Table 1, the number of total and high quality variants produced by the Illumina platform differed across the 13 lines, a result that is consistent with other plant species using the Illumina GA IIx technology. The average coverage across lines in our study was nearly identical to that reported for WGS of six maize (Zea mays L.) inbred lines (Lai et al. 2010).
Figure 2 shows that the majority of variants detected by the Alpheus pipeline for the 13 lines occurred not in the coding sequences, but in the intergenic regions. Therefore, only a small portion of the rice genome from the coding sequences was actually evaluated in this study for candidate variants associated with SB resistance. It is therefore likely that more variants other than the nsSNPs detected in this study could play a role in resistance. Similar conclusions can be drawn from inspection of variant distributions as shown in Fig. 3 of insertions, deletions, nsSNPs, and sSNPs.
The display in Fig. 4 shows that Bengal produced a relatively short biplot vector length compared to the remaining eight susceptible lines. One interpretation to account for the difference is that Bengal, classified as a line of medium grain-length, possesses a different genetic makeup compared to the remaining susceptible long-grain types. A second possibility is that U.S. southern medium grain-length lines such as Bengal generally exhibit slightly higher, and therefore slightly different, levels of SB resistance than most long-grain lines. However, the most likely explanation for the short vector length of Bengal is that it represents a reduced ability to discriminate among variants compared to the remaining susceptible lines. This reduction is consistent with the number of HQ variants generated for Bengal which was the smallest for all 13 lines (Table 1). The removal of Bengal variant data should therefore be considered for initial identification of candidate nsSNPs and corresponding genes. This conclusion might not have been possible using only PCA, cluster or other similar statistical approaches, and demonstrates the potential value of biplot display during the variant selection process.
When the CV selection strategy was applied across all lines and chromosomes, a wide array of gene families was identified based on gene ontology/gene function as shown in Fig. 5. Other than the 25 families grouped together, each with less than four genes, the kinase, nucleotide binding, peptide repeat, and F-box protein categories were the top four that have been detected in several previous investigations of rice and A. thaliana resistance (Jwa et al. 2006; Venu et al. 2007; Zhao et al. 2008). Fourteen families, including calcium binding, heat shock, and polygalacturonase, consisted of a single candidate gene. Similar high levels of gene family diversity were also found in previous studies of resistance to the rice blast pathogen Magnaporthe oryzae (Vergne et al. 2010), to soybean Glycine max L. pathogen Phytophthora sojae Kaufm. and Gerd (Wang et al. 2010), and response of A. thaliana to a plant defense elicitor (Libault et al. 2007). Although many of the same gene families were shared, none of the candidate SB resistance genes isolated by our CV or PB selection strategies was identified by suppression subtractive hybridization or MPSS/SAGE methods in rice (Venu et al. 2007; Zhao et al. 2008). This discrepancy may be explained by the possibility that genes identified in the RNA-based methods contained variants other than nsSNPs.
The QTL qShB9-2 reported by Liu et al. (2009) represents a region of potential importance for SB resistance breeding because of the relatively large stable effect detected across different greenhouse and field conditions. We therefore decided to identify candidate markers and genes within this region as shown in Supplementary Table S1 (Online Resource 1). Genes homologous to four selected candidates within qShB9-2 have been implicated previously in resistance to different pathogens. For example, serine/threonine kinases such as those at LOC_Os09g37800 and LOC_Os09g37880 in the current study have been shown previously to play a role in disease resistance (Afzal et al. 2008). F Box proteins such as OsFBDUF47 at LOC_Os09g37590 were reported to improve disease resistance in tobacco (Cao et al. 2008). Zinc finger proteins like that at LOC_Os09g38970 were reported to be important in resistance signaling in barley (Shirasu et al. 1999). The wall-associated kinase OsWAK91 at LOC_Os09g38850 represents a category found to be associated with resistance to Pseudomonas syringae in A. thaliana (He et al. 1998). The following five selected candidates have not been reported in the literature to be associated with biotic stress in rice, and therefore represent potential new factors contributing to SB resistance: aspartic proteinase nepenthesin (LOC_Os09g38380), WD domain, G-beta repeat domain containing protein (LOC_Os09g36900), STRUBBELIG-RECEPTOR FAMILY 5 precursor (LOC_Os09g38700), HEAT repeat family protein (LOC_Os09g38710), and potassium transporter (LOC_Os09g38960).
Supplementary Table S2 (Online Resource 2) shows candidate nsSNPs and genes identified by the CV selection strategy outside of qShB9-2. A review of the candidates shows that many selections belong to different plant resistance pathways. For example, various kinases and corresponding receptors like those detected on chromosomes 1–4, 6, 8, 9, and 12 in this study have been reported to play a role in disease resistance. Different F-Box and ubiquitin proteins like those found on chromosomes 2–6, 9, and 12 presumably assist in regulation of the salicylic acid pathway (Llorente et al. 2008). Thionins similar to those on chromosomes 2 and 6 have been shown to accumulate after jasmonic acid induction (Andresen et al. 1992). NB-ARC and leucine-rich repeat proteins help modulate R gene-based resistance (Zhang et al. 2003) with homologues in this study detected on chromosomes 1–4, 8, and 12. The pathogenesis-related (PR) protein glucan endo-1,3-beta-glucosidase (glucanase) was found at two loci on chromosomes 8 and 9. Others of interest include a GTPase on chromosome 2, heat shock protein on chromosome 4, a MYB family transcription factor on chromosome 5, a “cell death” protein on chromosome 11, and a RING-H2 finger protein on chromosome 12. Certain selected candidates outside of qShB9-2 not reported in the literature represent potential new resistance factors. Examples include the rapid alkalinization factor protein (LOC_Os01g10470), cystathionine β-synthase (CBS) domain-containing protein (LOC_Os02g42640), multidrug resistance protein (LOC_Os02g46680), mitochondrial transcription termination factor (LOC_Os02g54200), KIP1 (LOC_Os03g43684), amidase (LOC_Os04g10460), and cadmium tolerance factor (LOC_Os06g19110).
Supplementary Table S3a (Online Resource 3) shows PCR-based SNP allele genotypes from 23 candidate genes of MCR010277 and TeQing originally used in the CV selection process and 10 additional resistant and susceptible lines. Complete agreement observed between PCR and Illumina-generated alleles for all 23 genes of MCR010277 and TeQing suggests that the Illumina platform is suitable for accurate genotyping of rice breeding material. Similarly, all genotypes found for Azucena, Leah, Nipponbare are consistent with their known susceptibility to R. solani. The remaining seven lines showed different combinations of “resistant” and “susceptible” alleles that are in accord with their moderate level of resistance compared to MCR010277 and TeQing. Similar results were obtained for the LA breeding line 09DN/Rush072 (results not shown). Taken together the PCR-based profiles of the 12 lines described here are consistent with corresponding nsSNPs identified from Illumina sequencing of Jasmine 85, TeQing, and MCR010277.
We also examined the possibility that 12 selected nsSNPs were present in two resistant accessions of O. nivara and a SB-resistant indica line from South America. Two O. nivara accessions contained three resistant nsSNPs that were common with the nine resistant nsSNPs from the indica-resistant line. These results suggest that sources of SB resistance do occur in related species other than O. sativa consistent with previous work of Prasad and Eizenga (2008). Channamallikarjuna et al. (2010) identified a stable SB QTL on chromosome 11 from the cultivar Tetep that contained 11 xylanase inhibitor genes presumably functioning as class III chitinases. No such genes were found on chromosome 11 in our study which suggests that additional candidates may occur in other SB-resistant sources. It is interesting that a xylanase inhibitor gene was identified in this study on chromosome 9.
Our study was the first to use WGS to identify candidate rice genes associated with SB resistance. The outcome from this investigation suggests that WGS may be a useful strategy to identify candidate variants associated with other rice diseases that can complement QTL mapping and microarray/transcriptome approaches. Several new candidate genes were identified in our study that warrant further investigation. Moreover, SNP profiles detected in the original three resistant lines were found to be consistent with additional resistant/tolerant material. This information may prove valuable in development of marker-assisted breeding for SB resistance. Proof that these selected genes actually play a role in resistance will require transgene over-expression and/or knock-out experiments.
The candidate markers and genes identified in this study appear promising, but it is important to state that the WGS approach used in our research very likely did not detect all genes associated with SB resistance for the following reasons: (1) the majority of variants detected in the initial screening were not nsSNPs. Additional research will be required to determine any potential role(s) of sSNPs and other variants in SB resistance; (2) the Illumina GA IIx and other similar sequencing platforms generate data from short DNA fragments that cannot readily identify large deletions, insertions, or copy number variants; (3) additional accessions, lines or Oryza sp. may contain different alleles or genes not evaluated in this study. Nevertheless, the output generated from this study should provide new information for future basic and applied research of SB resistance in rice.
Abbreviations
- PCR:
-
Polymerase chain reaction
- SB:
-
Sheath blight
- SNP:
-
Single nucleotide polymorphism
- WGS:
-
Whole genome sequencing
References
Afzal AJ, Wood AJ, Lightfoot DA (2008) Plant receptor-like serine threonine kinases: roles in signaling and plant defense. Mol Plant Microbe Interact 21:507–517
Andresen I, Becker W, Schlüter K, Burges J, Parthier B, Apel K (1992) The identification of leaf thionin as one of the main jasmonate-induced proteins of barley (Hordeum vulgare). Plant Mol Biol 19:193–204
Bilgüvar K, Oztürk AK, Louvi A, Kwan KY, Choi M, Tatli B, Yalnizoğlu D, Tüysüz B, Cağlayan AO, Gökben S, Kaymakçalan H, Barak T, Bakircioğlu M, Yasuno K, Ho W, Sanders S, Zhu Y, Yilmaz S, Dinçer A, Johnson MH, Bronen RA, Koçer N, Per H, Mane S, Pamir MN, Yalçinkaya C, Kumandaş S, Topçu M, Ozmen M, Sestan N, Lifton RP, State MW, Günel M (2010) Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467:207–210
Cao Y, Li Z, Chen T, Zhang Z, Zhang J, Chen S (2008) Overexpression of a tobacco small G protein gene NtRop1 causes salt sensitivity and hydrogen peroxide production in transgenic plants. Sci China C Life Sci 51:383–390
Channamallikarjuna V, Sonah H, Prasad M, Rao GJN, Chand S, Upreti HC, Singh NK, Sharma TR (2010) Identification of major quantitative trait loci qSBR11-1 for sheath blight resistance in rice. Mol Breed 25:155–166
Deschamps S, La Rota M, Ratashak JP, Biddle P, Thureen DA, Luck S, Beatty M, Nagasawam N, Michael L, Llaca V, Sakai H, May G, Lightner J, Campbell MA (2010) Rapid genome-wide single nucleotide polymorphism discovery in soybean and rice via deep resequencing of reduced representation libraries with the Illumina Genome Analyzer. Plant Genome 3:53–68
Everitt BS, Landau S, Leese M (2001) Cluster analysis, 4th edn. Oxford University Press, Oxford
Fu YB, Peterson GW, McCallum BD, Huang L (2010) Population-based resequencing analysis of improved wheat germplasm at wheat leaf rust resistance locus Lr21. Theor Appl Genet 121:271–281
Hahn D, Beggs JD (2010) Brr2p RNA helicase with a split personality: insights into structure and function. Biochem Soc Trans 38:1105–11509
Havlova M, Dobrev PI, Motyka V, Storchová H, Libus J, Dobrá J, Malbeck J, Gaudinová A, Vanková R (2008) The role of cytokinins in responses to water deficit in tobacco plants over-expressing trans-zeatin O-glucosyltransferase gene under 35S or SAG12 promoters. Plant Cell Environ 31:341–353
He HH, He D, Kohorn BD (1998) Requirement for the induced expression of a cell wall associated receptor kinase for survival during the pathogen response. Plant J 14:55–63
Hobert O (2010) The impact of whole genome sequencing on model system genetics: get ready for the ride. Genetics 184:317–319
Jwa NS, Agrawal GK, Tamogami S, Yonekura M, Han O, Iwahashi H, Rakwal R (2006) Role of defense/stress-related marker genes, proteins and secondary metabolites in defining rice self-defense mechanisms. Plant Physiol Biochem 44:261–273
Kim JK, Jang IC, Wu R, Zuo WN, Boston RS, Lee YH, Ahn IP, Nahm BH (2003) Co-expression of a modified maize ribosome-inactivating protein and a rice basic chitinase gene in transgenic rice plants confers enhanced resistance to sheath blight. Transgenic Res 12:475–484
Krawitz PM, Schweiger MR, Rödelsperger C, Marcelis C, Kölsch U, Meisel C, Stephani F, Kinoshita T, Murakami Y, Bauer S, Isau M, Fischer A, Dahl A, Kerick M, Hecht J, Köhler S, Jäger M, Grünhagen J, de Condor BJ, Doelken S, Brunner HG, Meinecke P, Passarge E, Thompson MD, Cole DE, Horn D, Roscioli T, Mundlos S, Robinson PN (2010) Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet 2:827–829
Lai J, Li R, Xu X, Jin W, Xu M, Zhao H, Xiang Z, Song W, Ying K, Zhang M, Jiao Y, Ni P, Zhang J, Li D, Guo X, Ye K, Jian M, Wang B, Zheng H, Liang H, Zhang X, Wang S, Chen S, Li J, Fu Y, Springer NM, Yang H, Wang J, Dai J, Schnable PS, Wang J (2010) Genome-wide patterns of genetic variation among elite maize inbred lines. Nat Genet 42:1027–1030
Lee FN, Rush MC (1983) Rice sheath blight: a major rice disease. Plant Dis 67:829–832
Li JF, Li L, Sheen J (2010) Protocol: a rapid and economical procedure for purification of plasmid or plant DNA with diverse applications in plant biology. Plant Methods 6:1. doi:10.1186/1746-4811-6-1
Libault M, Wan J, Czechowski T, Udvardi M, Stacey G (2007) Identification of 118 Arabidopsis transcription factors and 30 ubiquitin-ligase genes responding to chitin, a plant-defense elicitor. Mol Plant Microbe Interact 20:900–911
Ling KS, Harris KR, Meyer JD, Levi A, Guner N, Wehner TC, Bendahmane A, Havey MJ (2009) Non-synonymous single nucleotide polymorphisms in the watermelon eIF4E gene are closely associated with resistance to zucchini yellow mosaic virus. Theor Appl Genet 120:191–200
Liu M, Sun ZX, Zhu J, Xu T, Harman GE, Lorito M (2004) Enhancing rice resistance to fungal pathogens by transformation with cell wall degrading enzyme genes from Trichoderma atroviride. J Zhejiang Univ Sci 5:133–136
Liu G, Jia Y, Correa-Victoria FJ, Prado GA, Yeater KM, McClung A, Correll JC (2009) Mapping quantitative trait loci responsible for resistance to sheath blight in rice. Phytopathology 99:1078–1084
Llorente F, Muskett P, Sánchez-Vallet A, López G, Ramos B, Sánchez-Rodríguez C, Jordá L, Parker J, Molina A (2008) Repression of the auxin response pathway increases Arabidopsis susceptibility to necrotrophic fungi. Mol Plant 1:496–509
Lupski JR, Reid JG, Gonzaga-Jauregui C, Deiros DR, Chen DCY, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, McGuire AL, Zhang F, Stankiewicz P, Halperin JJ, Yang C, Gehman C, Guo D, Irikat RK, Tom W, Fantin NJ, Muzny DM, Gibbs RA (2010) Whole-genome sequencing in a patient with Charcot–Marie–Tooth neuropathy. N Engl J Med 362:1181–1191
Maruthasalam S, Kalpana K, Kumar KK, Loganathan M, Poovannan K, Raja JA, Kokiladevi E, Samiyappan R, Sudhakar D, Balasubramanian P (2007) Pyramiding transgenic resistance in elite indica rice cultivars against the sheath blight and bacterial blight. Plant Cell Rep 26:791–804
McNally KL, Childs KL, Bohnert R, Davidson RM, Zhao K, Ulata VJ, Zeller G, Clark RM, Hoen DR, Bureau TE, Stokowski R, Ballinger DG, Frazer KA, Cox DR, Padhukasahasram B, Bustamante CD, Weigel D, Mackill DJ, Bruskiewich RM, Ratsch G, Buell CR, Leung H, Leach JE (2009) Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc Natl Acad Sci USA 28:12273–12278
Miller NA, Kingsmore SF, Farmer A, Langley RJ, Mudge J, Crow JA, Gonzalez AJ, Schilkey FD, van Velkinburgh J, Kim RJ, May GD, Black CF, Myers MK, Utsey JP, Frost NS, Sugarbaker DJ, Bueno R, Gullans SR, Baxter SM, Day SW, Retzel EF (2008) Management of high-throughput DNA sequencing projects: Alpheus. J Comput Sci Syst Biol 1:132–148
Motose H, Iwamoto K, Endo S, Demura T, Sakagami Y, Matsubayashi Y, Moore KL, Fukuda H (2009) Involvement of phytosulfokine in the attenuation of stress response during the transdifferentiation of Zinnia mesophyll cells into tracheary elements. Plant Phys 150:437–447
Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ (2010a) Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet 42:30–35
Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, Smith JD, Rieder MJ, Yoshiura K, Matsumoto N, Ohta T, Niikawa N, Nickerson DA, Bamshad MJ, Shendure J (2010b) Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 42:790–793
Pinson SRM, Capdevielle FM, Oard JH (2005) Confirming QTLs and finding additional loci conditioning sheath blight resistance in rice using recombinant inbred lines. Crop Sci 45:503–510
Prasad B, Eizenga GC (2008) Rice sheath blight disease identified in Oryza spp accessions. Plant Dis 92:1503–1509
Rios J, Stein E, Shendure J, Hobbs HH, Cohen JC (2010) Identification by whole genome resequencing of gene defect responsible for severe hypercholesterolemia. Hum Mol Genet 19:4313–4318
Roach JC, Glusman G, Smit AFA, Huff CD, Hubley R, Shannon RT, Rowen L, Pant KP, Goodman N, Bamshad M, Shendure J, Drmanac R, Jorde LB, Hood L, Galas DJ (2010) Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328:636–639
Rush MC, Lindberg GD (1996) Rice disease research. Rice J 77:49–52
Sarowar S, Oh HW, Cho HS, Baek KH, Seong ES, Joung YH, Choi GJ, Lee S, Choi D (2007) Capsicum annuum CCR4-associated factor CaCAF1 is necessary for plant development and defence response. Plant J 51:792–802
Shah JM, Raghupathy V, Veluthambi K (2009) Enhanced sheath blight resistance in transgenic rice expressing an endochitinase gene from Trichoderma virens. Biotech Lett 31:239–244
Shirasu K, Lahaye T, Tan MW, Zhou F, Azevedo C, Schulze-Lefert P (1999) A novel class of eukaryotic zinc-binding proteins is required for disease resistance signaling in barley and development in C. elegans. Cell 99:355–366
Sobreira NLM, Cirulli ET, Avramopoulos D, Wohler E, Oswald GL, Stevens EL, Ge D, Shianna KV, Smith JP, Maia JM, Gumbs CE, Pevsner J, Thomas G, Valle D, Hoover-Fong JE, Goldstein DB (2010) Whole-genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene. PLoS Genet 6:e1000991. doi:10.1371/journal.pgen.1000991
Song WY, Wang GL, Chen LL, Kim HS, Pi LY, Holsten T, Gardner J, Wang B, Zhai WX, Zhu LH, Fauquet C, Ronald P (1995) A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 270:1804–1806
Tan CX, Ji XM, Yang Y, Pan XY, Zuo SM, Zhang YF, Zou JH, Chen ZX, Zhu LH, Pan XB (2005) Identification and marker-assisted selection of two major quantitative genes controlling rice sheath blight resistance in backcross generations. Chin J Genet 32:399–405
Teer JK, Mullikin JC (2010) Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet 19:145–151
Tong P, Prendergast JGD, Lohan AJ, Farrington SM, Cronin SC, Friel N, Bradley DG, Hardiman O, Evans A, Wilson JF, Loftus BJ (2010) Sequencing and analysis of an Irish human genome. Genome Biol 11:R91. doi:10.1186/gb-2010-11-9-r91
Venu RC, Jia Y, Gowda M, Jia M, Jantasuriyarat C, Stahlberg E, Li H, Rhineheart A, Boddhireddy P, Singh P, Rutger N, Kudrna D, Wing R, Nelson JC, Wang GL (2007) RL-SAGE and microarray analysis of the rice transcriptome after Rhizoctonia solani infection. Mol Genet Genomics 278:421–431
Vergne E, Grand X, Ballini E, Chalvon V, Saindrenan P, Tharreau D, Nottéghem JL, Morel JB (2010) Preformed expression of defense is a hallmark of partial resistance to rice blast fungal pathogen Magnaporthe oryzae. BMC Plant Biol 10:206. doi:10.1186/1471-2229-10-206
Walsh T, Shahin H, Elkan-Miller T, Lee MK, Thornton AM, Roeb W, Abu Rayyan A, Loulus S, Avraham KB, King MC, Kanaan M (2010) Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am J Hum Genet 87:90–94
Wang H, Waller L, Tripathy S, St. Martin SK, Zhou L, Krampis K, Tucker DM, Mao Y, Hoeschele I, Maroof MAS, Tyler BM, Dorrance AE (2010) Analysis of genes underlying soybean quantitative trait loci conferring partial resistance to Phytophthora sojae. Plant Genome 3:23–40
Wu TD, Nacu S (2010) Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26:873–881
Xie W, Feng Q, Huihui Y, Xuehui H, Xing Y, Zhao Q, Yu S, Han B, Zhang Q (2010) Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing. Proc Natl Acad Sci USA 107:10578–10583
Xu J, Zhao Q, Du P, Xu C, Wang B, Feng Q, Liu Q, Tang S, Gu M, Han B, Liang G (2010) Developing high throughput genotyped chromosome segment substitution lines based on population whole-genome re-sequencing in rice (Oryza sativa L.). BMC Genomics 11:656
Yan W, Tinker NA (2006) Biplot analysis of multi-environment trial data: principles and applications. Can J Plant Sci 86:623–645
Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZX, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, Zheng H, Liu T, He W, Li K, Luo R, Nie X, Wu H, Zhao M, Cao H, Zou J, Shan Y, Li S, Yang Q, Asan NiP, Tian G, Xu J, Liu X, Jiang T, Wu R, Zhou G, Tang M, Qin J, Wang T, Feng S, Li G, Huasang, Luosang J, Wang W, Chen F, Wang Y, Zheng X, Li Z, Bianba Z, Yang G, Wang X, Tang S, Gao G, Chen Y, Luo Z, Gusang L, Cao Z, Zhang Q, Ouyang W, Ren X, Liang H, Zheng H, Huang Y, Li J, Bolund L, Kristiansen K, Li Y, Zhang Y, Zhang X, Li R, Li S, Yang H, Nielsen R, Wang J, Wang J (2010) Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329:75–78
Zhang Y, Goritschnig S, Dong X, Li X (2003) A gain-of-function mutation in a plant disease resistance gene leads to constitutive activation of downstream signal transduction pathways in suppressor of npr1-1, constitutive 1. Plant Cell 15:2636–2646
Zhao CJ, Wang AR, Shi YJ, Wang LQ, Liu WD, Wang ZH, Lu GD (2008) Identification of defense-related genes in rice responding to challenge by Rhizoctonia solani. Theor Appl Genet 116:501–516
Zuo S, Zhang L, Wang H, Yin Y, Zhang Y, Chen Z, Ma Y, Pan X (2008) Prospect of the QTL-qSB-9Tq utilized in molecular breeding program of japonica rice against sheath blight. J Genet Genomics 35:499–505
Acknowledgments
We thank the RiceCAP project (USDA/CSREES Grant 2004-35317-14867) for generation of sequence data, generation of the SB5 mapping population and associated QTL data. We recognize the Louisiana Rice Research Board for partial funding of this research. The Mississippi Rice Promotion Board funded the sequencing of the variety Bowman. We acknowledge Dr. Georgia Eizenga, USDA-ARS Dale Bumpers National Rice Research Center, for her generous gift of DNA isolated from two O. nivara accessions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Xu.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Silva, J., Scheffler, B., Sanabria, Y. et al. Identification of candidate genes in rice for resistance to sheath blight disease by whole genome sequencing. Theor Appl Genet 124, 63–74 (2012). https://doi.org/10.1007/s00122-011-1687-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-011-1687-4