
Abstract
Evolutionary studies in plant and animal breeding are aimed at understanding the structure and organization of genetic variations of species. We have identified and characterized a genomic sequence in Phaseolus vulgaris of 1,200 bp (PvSHP1) that is homologous to SHATTERPROOF-1 (SHP1), a gene involved in control of fruit shattering in Arabidopsis thaliana. The PvSHP1 fragment was mapped to chromosome Pv06 in P. vulgaris and is linked to the flower and seed color gene V. Amplification of the PvSHP1 sequence from the most agronomically important legume species showed a high degree of interspecies diversity in the introns within the Phaseoleae, while the coding region was conserved across distant taxa. Sequencing of the PvSHP1 sequence in a sample of 91 wild and domesticated genotypes that span the geographic distribution of this species in the centers of origin showed that PvSHP1 is highly polymorphic and, therefore, particularly useful to further investigate the origin and domestication history of P. vulgaris. Our data confirm the gene pool structure seen in P. vulgaris along with independent domestication processes in the Andes and Mesoamerica; they provide additional evidence for a single domestication event in Mesoamerica. Moreover, our results support the Mesoamerican origin of this species. Finally, we have developed three indel-spanning markers that will be very useful for bean germplasm characterization, and particularly to trace the distribution of the domesticated Andean and Mesoamerican gene pools.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Phaseolus genus comprises about 70 species, and according to their geographic distribution, it is considered to be of Mesoamerican origin (Delgado-Salinas et al. 1999, 2006; Freytag and Debouck 1996, 2002). Five Phaseolus species have been domesticated, and among these, P. vulgaris is the most extensively distributed and consumed grain legume for direct human consumption (Freytag and Debouck 2002). Wild common bean, P. vulgaris, is widely distributed from northern Mexico to northern Argentina (Toro et al. 1990). P. vulgaris is characterized by two major geographic gene pools, one in Mesoamerica and the second in the southern Andes, with parallel geographic variation in the wild and domesticated beans. A third wild gene pool from Ecuador and northern Peru has been described (Debouck et al. 1993); phaseolin seed protein diversity suggests that the Ecuador and northern Peru populations are the ancestral population of P. vulgaris and consequently that this species may have originated in this region (Kami et al. 1995). Recently, Rossi et al. (2009) used amplified fragment length polymorphism (AFLP) of wild and domesticated populations of P. vulgaris to present new data arguing in favor of a Mesoamerican origin of P. vulgaris.
Two independent domestication events have been documented for the common bean in the Americas (Gepts et al. 1986): one in Mesoamerica and one in the Andes, giving origin to two major gene pools that show partial reproductive barriers and marked phenotypic differences, according to morphology, seed-storage protein variation, and differences in nuclear and chloroplast markers (see Acosta-Gallegos et al. 2007; Angioi et al. 2009; Papa et al. 2006, for reviews). One topic of discussion is whether multiple domestications have occurred within each gene pool. Multilocus molecular marker analyses have provided evidence for the single domestication in Mesoamerica (Gepts et al. 1986; Kwak and Gepts 2009; Kwak et al. 2009; Papa and Gepts 2003; Rossi et al. 2009). However, cpDNA restriction fragment length polymorphism (RFLP) data also suggest the occurrence of multiple domestications in Mesoamerica (Chacón et al. 2005). In any case, as suggested by Allaby et al. (2008), the interpretation of results from multilocus nuclear molecular markers can be problematic when distinguishing between single versus multiple domestication. Indeed, in general, models of phylogenetic reconstruction assume no recombination, which can hardly be assumed for multilocus data, although this is more realistic within a single locus (Allaby et al. 2008). In the Andes, the situation is less clear, because of the lack of geographic structure of the genetic diversity, which reduces the resolution power of molecular studies. However, both single and multiple domestications have been suggested within the Andean gene pool (Beebe et al. 2001; Chacón et al. 2005; Rossi et al. 2009; Santalla et al. 2004).
Nearly, all of the evidence regarding genetic diversity in the common bean is based on multilocus molecular markers (see Acosta-Gallegos et al. 2007; Papa et al. 2006, for reviews). Only a few studies have investigated nucleotide diversity in this important crop species, particularly for wild populations; these have included sequence diversity in wild accessions at the phaseolin locus (Kami et al. 1995) and in landraces and modern cultivars in three noncoding regions of the dihydroflavonol 4-reductase and chalcone isomerase genes (McClean et al. 2004; McClean and Lee 2007). A larger amount of sequence data (over 500 genes) was obtained in a study by McConnell et al. (2010) for the two parents of one of the major mapping populations of P. vulgaris, ‘BAT93’ × ‘JaloEPP558’ (Freyre et al. 1998), in order to enrich this genetic map and to investigate macrosynteny between the common bean and the A. thaliana, M. truncatula and L. japonicus species.
In the present study, nucleotide polymorphism in a gene fragment was analyzed for the first time in a representative panel containing both wild and domesticated P. vulgaris. We have characterized a fragment of a gene homologous to SHATTERPROOF-1 of A. thaliana (PvSHP1). SHATTERPROOF (SHP) belongs to the family of MADS-box genes, which have a wide range of functions in flowering plants, such as in the formation of flowers (including development of reproductive structures) and control of flowering time and vegetative development. SHP is a member of the AGAMOUS clade [AG/SHP1/SHP2/SEEDSTICK (STK)], which are expressed mainly in carpels/ovules and contribute to aspects of carpel and ovule identity and development in Arabidopsis (Pinyopich et al. 2003). SHP is involved in fruit shattering in Arabidopsis (Liljegren et al. 2000). The loss of fruit shattering has been under selection in most seed crops, to facilitate seed harvesting (Purugganan and Fuller 2009), while in wild plants, fruit shattering is a fundamental trait that helps to assure seed dispersal. Thus, we have selected SHP as a candidate gene of one of the key traits of the domestication syndrome. Expressed sequences corresponding to SHP1 have also been identified in other species. Vrebalov et al. (2009) showed that Tomato AGAMOUS-LIKE1 (TAGL1), which is the tomato ortholog of the duplicated SHP MADS-box genes of Arabidopsis, has an important role in the regulation of both fleshy fruit expansion and the ripening process, which are together necessary to promote seed dispersal of fleshy fruit. In legumes, orthologous sequences to Arabidopsis SHP1 have been identified in Medicago, pea and soybean (Hecht et al. 2005), but there has been only a limited number of molecular analyses of SHP1-related sequences, despite the importance of flowering time and shattering in legume domestication and crop improvement.
We analyzed the nucleotide polymorphism of the PvSHP1 gene fragment in a sample of the most agronomically important Phaseoleae species and of 91 wild and domesticated genotypes spanning the geographic distribution of the centers of origin of this species, and we compared these results with those of AFLP markers. The main objectives of this study were: (1) to analyze the variation of PvSHP1 in different Phaseoleae species; (2) to investigate the possible implications of PvSHP1 in fruit shattering; and (3) to use nucleotide data for the first time to better define the domestication and evolution of P. vulgaris.
Materials and methods
Identification and amplification of a genomic sequence of Phaseolus spp. similar to SHATTERPROOF (SHP1) of A. thaliana
We first identified two EST sequences of the runner bean (Phaseolus coccineus cv. Hammond’s Dwarf Scarlet) in GenBank that are similar to the SHATTERPROOF-1 (SHP1; At3g58780) sequence of A. thaliana. These P. coccineus sequences have sizes of 445 and 558 bp (CA902468, CA902467, respectively). To perform a similarity search of the most conserved regions for primer design for the amplification of the SHP1 region in different species of legume, we performed BLAST searches for both the Arabidopsis SHP1 gene (tBLASTx) and protein (tBLASTn) against EST databases for Medicago, Glycine, Lotus, and Phaseolus. A pair of primers, 2SHP2fw and 2SHP2rev (Table 1) was then designed based on the homology of an EST sequence of the scarlet runner bean (CA902468) and an EST sequence of P. vulgaris (CV538324) with SHP1. A single PCR product of about 1,200 bp was obtained (PvSHP1), which was sequenced in a preliminary sample of ten individuals: two accessions of P. coccineus and a sample of P. vulgaris (two accessions each of domesticated and wild beans from both Mesoamerica and South America). The parents of the core mapping population of P. vulgaris, BAT93 and Jalo EEP558 (Freyre et al. 1998), and the parents of the mapping population for the domestication syndrome traits, Midas and G12873 (Koinange et al. 1996), were added to the samples. All of the sequences were checked manually and edited according to chromatograms. The sequences were aligned using a combination of methods implemented in BioEdit, version 7.0 (Hall 1999), and ClustalW (Thompson et al. 1997), with further manual refinements. Translation of the DNA sequences obtained for all six of the reading frames was performed on-line, using the Translate tool on the ExPASy server (http://www.expasy.org; accessed on 27 November 2009). After translation of the amplified DNA sequences, the deduced 62-amino-acid sequence was compared with those in GenBank. This deduced sequence has a K-box domain that was less well conserved than the MADS domain, but which is present in all of the MIKC-type MADS-box transcription factors. It also has 80% identity with the Lotus corniculatus var. japonicus MADS-box protein AGL1 and 70% identity with the Pisum sativum MADS-box protein M8, which was identified by Hecht et al. (2005) as the product of the Pisum SHATTERPROOF-like gene (PsSHP).
Another pair of primers was designed based on the EST sequence CA902467 (SHP4fw, SHP3rev). This primer pair also led to the amplification of a single PCR product, which was sequenced in the preliminary sample of ten individuals described above; no polymorphism was detected. After translation of the amplified DNA sequence, the comparison of this deduced amino-acid sequence with those in GenBank identified the Arabidopsis AGL1 protein (Shatterproof-1), with 90% identity across the 75-amino-acid region of the fragment obtained. The deduced amino-acid sequence also contained the conserved MADS-MEF2-like domain, which is highly conserved among MADS-box proteins.
Plant materials
The PvSHP1 regions from several species of legumes were amplified and sequenced using the primers described above. The legume species included P. coccineus, P. dumosus (formerly P. polyanthus, P. coccineus subsp. polyanthus, or P. coccineus subsp. darwinianus), P. vulgaris, P. acutifolius, P. lunatus, Vigna unguiculata and Glycine max. No amplification products were obtained in Pisum, Anthyllis, Lotus and Cicer, consistent with their more distant phylogenetic position within the Papilionoideae (Wojciechowski et al. 2004).
The sequences obtained included five wild accessions of P. coccineus, two domesticated accessions of P. dumosus, and 91 accessions representative of the geographic distribution of P. vulgaris from Mesoamerica and South America. A complete list of the accessions studied is available as Supplementary Materials (Table A1). The collection of 91 common bean accessions consisted of 39 domesticated (mostly landraces) and 52 wild (including two weedy genotypes) individuals. Based on their geographic distributions, the accessions were chosen to be representative of the different gene pools of P. vulgaris: Mesoamerican (Mexico, central America and Colombia) and Andean (South America) for both wild and domesticated forms, and the wild Ecuador and northern Peru population characterized by phaseolin type I (Debouck et al. 1993; Kami et al. 1995). Most of the accessions had already been characterized using different types of molecular markers in previous studies (Angioi et al. 2009; Rossi et al. 2009). Moreover, passport information, (i.e. race type, phaseolin type, seed color and pattern, seed weight, longitude and latitude) was available for the entire sample of genotypes.
For the domesticated genotypes, the geographic origin of the individual accessions is not a reliable indicator of gene-pool membership, because of exchange between the Andean and Mesoamerican gene pools after domestication. Thus, the gene-pool designation was based on passport data, which included race type, phaseolin type, seed color and pattern, and seed weight (following the convention of Singh et al. 1991), previous studies of the molecular diversity of the common bean (Beebe et al. 2000, 2001; Blair et al. 2006; Dìaz and Blair 2006; Duarte et al. 1999; McClean et al. 2004; McClean and Lee 2007; Pedrosa et al. 2006; Rosales-Serna et al. 2005; Singh et al. 2001), and recent molecular analyses (Angioi et al. 2009; Rossi et al. 2009). According to this information, we classified 28 domesticated accessions as Mesoamerican and 11 as Andean.
Most of the seeds were obtained from the International Centre for Tropical Agriculture (CIAT) in Cali, Colombia, and the US Department of Agriculture Western Plant Introduction Station in Pullman, WA, USA. The two recombinant inbred line (RIL) populations used to map PvSHP1 have been described before: ‘BAT93’ × ‘Jalo EPP558’ (BJ; n = 80; Freyre et al. 1998) and ‘Midas’ × ‘G12873’ (MG; n = 58; Koinange et al. 1996). The former population is the core linkage mapping population of the common bean (Freyre et al. 1998), whereas the latter was developed by Koinange et al. (1996) to map genes and quantitative trait loci (QTLs) that are involved in the domestication process of the common bean. All of the seeds were planted in a greenhouse, and each accession was represented by a randomly extracted single individual.
DNA isolation, PCR amplification and sequencing
Total genomic DNA was isolated from young expanding leaf tissues of single individuals for each sample, using the Dneasy Plant kit (QIAGEN GmbH, Hilden, Germany), following the protocols provided by the manufacturer. DNA fragments were amplified using 25 ng DNA and the following reagent concentrations: 0.25 μM each forward and reverse primer; 200 μM each dNTP, 2.5 mM MgCl2, 1× Taq polymerase buffer, 1 unit AmpliTaq DNA polymerase, and sterile double-distilled H2O, to a final volume of 100 μl. The sequencing was carried out on purified PCR-amplified templates using BigDye cycle sequencing (Applied Biosystems, Foster City, CA, USA) (performed by Qiagen Sequencing Service, Hilden, Germany).
Sequence analysis
BLASTn and BLASTp (Altschul et al. 1997) analyses were carried out against the NCBI/GenBank database (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi, as accessed on 27 November, 2009), to identify homologies with the nucleotide and peptide sequences, respectively. The searches for open reading frames (ORFs) in the PvSHP1 sequences were performed using the ORF finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html, as accessed on 27 November 2009). The identification of putative splicing sites and introns was carried out by comparing our sequences with the ESTs of Phaseolus similar to SHP1 that were characterized and identified during the BLAST searches. Alignment for the PvSHP1 sequences of the different legume species considered was performed with the webPRANK tool (http://www.ebi.ac.uk/goldman-srv/webPRANK/) (Löytynoja and Goldman 2005). Indeed, aligning orthologous sequences from different species using traditional multiple sequence alignment methods disregards the phylogenetic implications of gap patterns that they create and infers systematically biased alignments with excess deletions and substitutions, too few insertions, and implausible insertion–deletion event histories. The PRANK tool is based on an algorithm that prevents these systematic errors by recognizing insertions and deletions as distinct evolutionary events, improving the quality of sequence alignments and downstream analyses over a wide range of realistic alignment problems (Löytynoja and Goldman 2008).
An analysis of the variation of the PvSHP1 sequence in the sequences of the seven different legume species considered was carried out using DnaSP, version 5.00 software (Librado and Rozas 2009): for variable sites, parsimony informative sites and singleton sites for both coding and noncoding regions; for variable residues; and for synonymous and nonsynonymous changes only for coding regions. Furthermore, the number of nonsynonymous nucleotide substitutions per nonsynonymous site (d N ), and that of synonymous nucleotide substitutions per synonymous site (d S ), were computed using the modified Nei–Gojobori method (Nei and Gojobori 1986), implemented in the MEGA software, version 4 (Tamura et al. 2007; http://www.megasoftware.net/). The significance of the difference between d N and d S was estimated using the Fisher’s exact test of neutrality for sequence pairs. Sequence alignment and editing of the PvSHP1 sequences of P. vulgaris were performed with ClustalW (Thompson et al. 1997) and BioEdit, version 7.0.5.3 (Hall 1999). Insertions/deletions (indels) were not included in the analysis. The sequences are accessible as GenBank accession numbers JF411844–JF411934 for P. vulgaris, JF411945–JF411949 for P. coccineus; JF411943 and JF411950 for P. dumosus, JF411935, JF411951, JF411952, and JF411953 for P. acutifolius, G. max, V. unguiculata, and P. lunatus, respectively.
Linkage analysis and molecular mapping of PvSHP1
The preliminary analysis of the PvSHP1 sequences of the initial sample of P. vulgaris showed a polymorphism in introns that was useful for the development of three indel-spanning markers: SHP1-A, SHP1-B, and SHP1-C. These spanning markers were amplified by three pairs of primers that were designed based on sequences in exons I, III, and IV, and on conserved motifs of the first intron (Table 1).
The SHP1-A, SHP1-B and SHP1-C amplification products were located on the common bean linkage map using the BJ and MG RIL populations. The map locations were evaluated using the markers already mapped on the two RIL populations as a framework (Freyre et al. 1998; Koinange et al. 1996; Rossi et al. 2009), and Mapmaker/EXP software, version 3.0 (Lander et al. 1987), following standard procedures (Freyre et al. 1998; Papa et al. 2005). The markers were also placed on the MG linkage map using the same procedure.
Phylogeography and population structure
The relationships among the 91 P. vulgaris, the two P. dumosus, and the five P. coccineus sequences were analyzed by developing an unrooted neighbor-joining (NJ) tree using the MEGA software, version 4 (Tamura et al. 2007). For this NJ tree, the Kimura two-parameter model for nucleotide substitutions was used. A bootstrap analysis of the inferred NJ tree was performed with 1,000 re-samplings. A synonymous mutation rate of 6.1 × 10−9 substitutions per synonymous site per year was reported for the Solanaceae and Fabaceae (Lynch and Conery 2000; Schlueter et al. 2004). Thus, divergence times of the main clades were estimated with the assumption of a synonymous mutation rate of 6.1 × 10−9 substitutions per synonymous site per year.
To clarify the relationships among individuals, a consensus NJ tree based on genetic distances (Nei 1978) was also obtained using the AFLP data (available from Rossi et al. 2009) for all of the samples of P. vulgaris considered; this analysis was carried out with the PHYLIP package (Felsenstein 1993).
A Bayesian model-based approach, implemented in the BAPs software, version 5.3 (Corander et al. 2003, 2008; Corander and Marttinen 2006; Corander and Tang 2007), was used to infer the hidden genetic population structure of our sample (91 accessions of P. vulgaris), and thus to assign the genotypes into genetically structured groups/populations (K). The software incorporates the possibility to account for the dependence due to linkage between the sites within aligned sequences. We carried out a genetic mixture analysis to determine the most probable number of populations (K) given the data (Corander and Tang 2007; Corander et al. 2008). The ‘clustering with linked loci’ analysis was chosen. Ten iterations of K (from 1 to 10) were conducted to determine the optimal number of genetically homogeneous groups. The admixture analysis was then applied, to estimate individual admixture proportions with regards to the most likely number of K clusters identified (Corander and Marttinen 2006; Corander et al. 2008). Admixture inference was based on 100 realizations from the posterior of the allele frequencies. We repeated the admixture five times, to confirm the consistency of the results.
Diversity analyses
Diversity analyses were performed for the 91 P. vulgaris genotypes. The data were subdivided according to the different regions (coding vs noncoding) of PvSHP1, the status (wild or domesticated), and the gene pool (Andean, Mesoamerican and phaseolin type I groups) of the different genotypes. The dataset was also subdivided according to the phylogenetic reconstruction provided by the NJ tree, to take into consideration the population structure. Population genetics statistics were calculated for each partition of the data described above.
Estimations were made for the number of polymorphic sites (S), the number of parsimony informative sites, the number of singleton variable sites, the number of haplotypes, and the haplotype diversity (Hd, Nei 1987). Two measures of diversity were computed: π (Tajima 1983), the direct estimation of per-site heterozygosity derived from the average number of pairwise sequence differences in the sample, and the Watterson’s theta estimator (θ W ) (Watterson 1975), based on the number of segregating sites in the sample, as an estimation of the expected per site nucleotide heterozygosity, which is theoretically equal to the neutral mutation parameter 4Neμ.
To determine whether the sequences deviated from the mutation–drift model, estimations were made for Tajima’s D (Tajima 1989) and Fu and Li D* and F* (Fu and Li 1993) parameters. Tajima’s D test compares the differences between π and θ W (Tajima 1989). Under neutrality, Tajima’s D value is assumed to be zero; under positive selection, there is an excess of rare polymorphisms and Tajima’s D value is negative. Negative D values can also be due to population expansion. If there is balancing selection, intermediate frequency genetic variants are kept and Tajima’s D value is positive. The same holds for the Fu and Li’s D* and F* tests. The minimum number of recombination events was estimated using the four-gamete test (Hudson and Kaplan 1985).
The F ST statistic (Hudson et al. 1992) permutation test with 1,000 replicates (Hudson 2000) was computed to investigate the degree of differentiation between the wild and domesticated populations of the different gene pools. All of the calculations were carried out using DnaSP, version 5.00 (Librado and Rozas 2009).
Results
In the present study, a large collection of wild and domesticated P. vulgaris genotypes that represents all of the gene pools of the species was analyzed through DNA sequences of the PvSHP1 gene. This allowed us to identify a number of indels and single nucleotide polymorphisms (SNPs) that might be useful for further phylogenetic and population genetics analyses. The sequence diversity data were also used to develop allele-specific primers to locate the position of the PvSHP1 gene in the common bean linkage map, using the BJ and MG RIL populations.
The PCR amplifications using the primers designed in the present study were successful for the identification of a sequence homologous to SHP1 of A. thaliana in different species of Phaseoleae. A single SHP1-homologous fragment was amplified from all of the genotypes of G. max, V. unguiculata, P. lunatus, and from the four species of the “vulgaris group” (Delgado-Salinas et al. 2006), i.e., P. acutifolius, P. coccineus, P. dumosus and P. vulgaris. In this last species, the SHP1-homologous sequence obtained presumably belonged to a single-copy gene, as the three markers SHP1-A, SHP1-B and SHP1-C co-segregated as a single locus in the two RIL populations. These data are consistent with the observation that a number of paralogous gene pairs in Arabidopsis are only represented by a single gene in other legumes, such as Medicago and pea. This is true for several MADS-box genes, including SHP1 and SHP2 (Hecht et al. 2005).
Molecular mapping and diversity of the indel-spanning markers
Based on the alignments among the different genotypes of P. vulgaris, three PCR-based markers spanning indels were developed and used to map the PvSHP1 sequence. The size ranges of the three indel-spanning amplicons in the whole sample of the 91 accessions of P. vulgaris were: SHP1-A (198–206 bp); SHP1-B (211–221 bp) and SHP1-C (119–127 bp). Combining the information obtained from each of these three indel-spanning markers, nine indel-based (Ib)-haplotypes were found in P. vulgaris (Supplementary Materials, Table A1). The wild accessions that belonged to the Mesoamerican gene pool had five Ib-haplotypes, one of which was shared with the phaseolin type I genotypes and one of which was shared with wild P. coccineus. The Mesoamerican domesticated accessions had three Ib-haplotypes, all in common with the wild accessions from Mesoamerica. The Andean group of genotypes had four Ib-haplotypes, two of which were shared between the domesticated and wild accessions, with one specific of the domesticated genotypes and one specific of the wild genotypes. Remarkably, no Ib-haplotypes were shared between the Mesoamerican and Andean gene pools.
The two RIL populations ‘BAT93’ × ‘Jalo EPP558’ (BJ; Freyre et al. 1998) and ‘Midas’ × ‘G12873’ (MG; Koinange et al. 1996) were used to integrate the PvSHP1 sequence into the common bean molecular linkage map. As expected, all of the three markers co-segregated as a single locus in these two RIL populations. As shown in Fig. 1, PvSHP1 is located on chromosome Pv06, between the D0096 and DROJ9c markers that are common to both maps, and at 12 cM from two AFLP markers shared by both maps (Rossi et al. 2009). Thus, PvSHP1 is located close to the V locus for flower color (mapped by Nodari et al. 1993; McClean et al. 2002) and in the same region as QTLs for days to flowering in the Darién, Colombia, environment (DF6.2 and DF6.1) (Blair et al. 2006) and for resistance to common bacterial blight (Miklas et al. 2006).

Nucleotide variation of PvSHP1 in different Phaseoleae species
The topology of the NJ dendrogram (data not shown) obtained with the sequence data is consistent with the accepted phylogeny of Phaseoleae (Delgado-Salinas et al. 1999, 2006). As described above, the structure of the sequences obtained (coding and noncoding regions) were deduced by comparison with the EST sequences of common bean and scarlet runner bean, and with SHATTERPROOF-1 (SHP1) of A. thaliana. From these comparisons, we determined that the fragment of PvSHP1 consists of four exons and three introns of different lengths.
The sizes of the PvSHP1 fragment differed widely among the species: from 676 bp in V. unguiculata, to 1,481 bp in P. lunatus (Table 2). Among the accessions belonging to the “vulgaris group” of the Phaseolus genus, the sizes of the fragment ranged from 1,024 bp for P. coccineus, to 1,054 bp for P. dumosus and P. vulgaris (Table 2). The differences in the sizes of the fragments were mainly due to differences in intron lengths. Indeed, the coding regions of the Phaseolus species and V. unguiculata all had a length of 186 bp. The only exception was G. max, with a length of 180 bp, due to a single indel event corresponding to the loss of two codons (without impacting the reading frame) (Table 2). There were no substitutions at the nucleotide level in the coding regions in the four species that belong to the “vulgaris group”, namely P. coccineus, P. dumosus, P. acutifolis and P. vulgaris, while there was one nonsynonymous substitution in P. lunatus (a one-amino-acid replacement: Gln, Q, for Leu, L). Considering all the species together, the translated coding region contained nine mutations, due to one synonymous and eight nonsynonymous substitutions (Table 3). When more genotypes of P. coccineus and P. dumosus were included, no additional substitutions were found. However, when the whole sample of P. vulgaris was considered (91 accessions), there was one synonymous substitution in exon II in four Mexican wild accessions of P. vulgaris, and a single indel, corresponding to the loss of a codon (without impacting on the reading frame) in exon IV for 10 wild and four domesticated Andean accessions; the same codon in another 13 Andean accessions (seven domesticated and six wild accessions) showed a synonymous substitution.
Compared to exons, there was a sharp contrast in the levels of variation found in the introns. Many indels were found in the first intron (I). Intron I was 889 bp in P. lunatus, 289 bp in soybean, and only 80 bp in Vigna (Table 2). Among the species of the “vulgaris group”, intron I was around 430 bp in P. coccineus and P. acutifolius, and around 460 bp in P. dumosus and P. vulgaris. In contrast, intron II was almost the same size among Phaseolus spp. and V. unguiculata (295–322 bp), while it was more than twice this size in G. max (792 bp). Intron III had almost the same size in the different species, from 88 bp in Vigna to 148 bp in Glycine.
Most of the variation for nucleotide substitutions and indels was also found in introns. As expected, the variation in the noncoding regions of PvSHP1 decreased among the more closely related species, with the number of variable sites, ranging from 136 (considering all seven species) to 41 for the five species of Phaseolus, and 35 for the species of the “vulgaris group” (Table 4).
No significantly higher d N than d S estimates were found in the 21 pairwise comparisons of the seven legume species sequences, supporting neutral evolution for the PvSHP1 gene.
Phylogeography and population structure
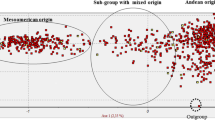
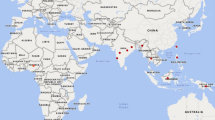
The relationships among the accessions of P. vulgaris, P. coccineus and P. dumosus were reconstructed in a NJ dendrogram inferred from the PvSHP1 sequence (Fig. 2). The five accessions of wild P. coccineus and two of P. dumosus grouped together and appeared as an outgroup of the P. vulgaris clade. The P. vulgaris clade itself was divided into four main, well-supported, groups (bootstrap values ≥70%, according to Hillis and Bull 1993): Group 1 (G1), Group 2 (G2), Group 3 (G3) and Group 4 (G4). Figure 3 shows the geographic positions of the wild genotypes of P. vulgaris used for the analysis and their membership in the four groups identified.

Unrooted neighbor-joining bootstrap linearized tree inferred from the PvSHP1 sequence data of the 91 genotypes of P. vulgaris, five accessions of wild P. coccineus, and two accessions of P. dumosus. The four main sub-clusters of the P. vulgaris groups are indicated as G1, G2, G3, G4. Pd, P. dumosus; Pc, P. coccineus; WA, wild Andean; DA, domesticated Andean; WM, wild Mesoamerican; DM, domesticated Mesoamerican; Ph_I, Phaseolin type I. For the wild accessions, the codes indicating the country and the Department (region) are given; country codes: ar Argentina, bl Bolivia, co Colombia, cr Costa Rica, ec Ecuador, gt Guatemala, mx Mexico, pr Peru. Department codes: ap Apurimac, az Azuay, ch Chiapas, ci Cihuahua, cj Cajamarca, cm Chimborazo, cn Cundinamarca, cq Chuquisaca, cu Cuzco, du Durango, hi Hidalgo, ja Jalisco, jn Junin, ju Jujuy, mo Morelos, ox Oaxaca, pu Puebla, sa Salta, sc Sacatepequez, sj San Jose, tj Tarija, tu Tucuman

Map of central and South America, showing the geographic position of the wild genotypes of P. vulgaris belonging to each of the four groups identified by the NJ tree
-
Group 1 (G1) (94% bootstrap value) is divided into two main parts, one comprising only wild genotypes from Mexico (97% bootstrap value), the other comprising three wild genotypes from Mexico that outgroup from 25 domesticated and 4 wild genotypes from Mexico (88% bootstrap value). For the three accessions that were the most closely related to the domesticated accessions, one from Tecalitlán, in the state of Jalisco, Mexico, showed a clearly wild seed phenotype (a small seed of an agouti color). The other two accessions were classified by the donor as either a weedy type from Durango or a cream-seeded type with an intermediate seed weight (0.07 g).
-
Group 2 (G2) (97% bootstrap value) consists of all of the wild accessions from Ecuador and northern Peru that were characterized by the ancestral phaseolin type I; these accessions are closely related to the Mesoamerican domesticated and wild accessions of G1, with which they form a clade with a bootstrap value of 59%.
-
Group 3 (G3) (80% bootstrap value) comprises a set of wild accessions from Colombia, Guatemala, Mexico and Costa Rica, and three domesticated accessions, two from Mexico and one from El Salvador. Furthermore, G3 is divided into three subgroups (bootstrap values > 70%). The first of these groups comprises the three wild accessions from Colombia, together with two wild accessions from Costa Rica and Guatemala, and a wild accession from Jalisco. The second G3 subgroup comprises two wild and one weedy Mexican accessions, one wild accession from Guatemala, and a domesticated accession from El Salvador with the Andean phaseolin type T (Gepts et al. 1988). The third subgroup consists of three wild accession from different states of Mexico (Chihuahua, Jalisco, Oaxaca), and two Mexican domesticated accessions.
-
Group 4 (G4) (100% bootstrap value) comprises wild genotypes from Argentina, Peru and Bolivia, along with all of the Andean domesticated genotypes from South America and one genotype from Mexico belonging to the Andean Nueva Granada race.
Age estimates were obtained assuming a molecular clock (mutation rate μ = 6.1 × 10−9; Lynch and Conery 2000; Schlueter et al. 2004), with the divergence of the P. vulgaris–P. coccineus–P. dumosus crown placed at ca. 3.7 Ma.
The genealogy tree within P. vulgaris obtained here was based on DNA sequence data. It was compared with that from a NJ tree based on the AFLP markers of Rossi et al. (2009), using a set of 73 accessions shared between their study and the present study. Based on an analyses at a single individual level, the NJ phylogram obtained from the AFLP data (Fig. 4) showed that all of the wild Mesoamerican genotypes belong to a single clade that forms an outgroup to all of the Mesoamerican domesticated accessions. A very similar situation was seen in the Andes. The phaseolin type I accessions were intermediate between the Mesoamerican and Andean gene pools, as was observed previously in an allozyme marker analysis (Debouck et al. 1993).

Analysis of the relationships among the different PvSHP1 haplogroups investigated in the NJ trees (Phylip, v 3.67) based on the AFLP data. The NJ analyses were conducted at the single-individual level. Violet and blue lines genotypes from the Mesoamerican gene pool, red lines phaseolin type I genotypes, green lines genotypes from the Andean gene pool. For the wild genotypes, the labels also indicate the country of origin (see legend of Fig. 2 for country codes)
Population structure was also investigated using the Bayesian model-based clustering method implemented in the BAPS software, version 5.3 (Corander et al. 2003; Corander and Marttinen 2006; Corander and Tang 2007; Corander et al. 2008). This BAPS analysis indicated that the best partitioning of our sample is represented by six different genetic clusters (C1, C2, C3, C4, C5, and C6). Comparing the BAPS assignment results to the NJ groups, we observed that the accessions belonging to the NJ group G1_D (represented by most of the Mesoamerican domesticated accessions) were assigned with very high percentages of membership to cluster C1 (qC1 ≥ 0.96) (Fig. 5); similarly, the accessions characterized by phaseolin I type (NJ group G2) were assigned to cluster C3 (qC3 ≥ 0.99), and the Andean accessions, of both wild (NJ group G4_W) and domesticated (NJ group G4_D), to cluster C6 (qC6 = 1.0) (Fig. 5). We observed a further subdivision for the Mesoamerican wild accessions not found by the NJ analysis; indeed, each of the previously described two Mesoamerican wild groups was once more split into two subgroups. In more detail, the G1_W group, which includes only accessions of Mexican origin, was characterized by two clusters: cluster C1 (seven accessions, one of which was from Durango, and two each from the Jalisco, Oaxaca and Puebla states, qC1 ≥ 0.98) and cluster C2 (nine accessions, six of which were from Morelos, two from Jalisco, and one from Puebla; qC2 ≥ 0.91); G3_W was split into two clusters too: cluster C4 (seven accessions, six of which were from Mexico, and one from Guatemala, qC4 ≥ 0.80) and cluster C5 (six accessions, three of which were from Colombia, and one each from Costa Rica, Guatemala and Mexico, qC5 ≥ 0.98). The domesticated Mesoamerican genotypes belonging to the G3_D NJ group were completely assigned to cluster C4 (qC4 ≥ 0.99) (Fig. 5).

Percentages of membership (q) for each of the six clusters identified for the 91 accessions of P. vulgaris by the BAPS analysis at K = 6
PvSHP1 diversity and recombination in P. vulgaris
The nucleotide diversity was analyzed considering the major population subdivisions that have already been described for P. vulgaris, including the three wild gene pools (Mesoamerican, Andean and phaseolin type I groups) and the two groups of domesticated genotypes (Mesoamerican and Andean). These subdivisions are based on the geographic distributions of the samples. Based on nucleotide variations, the sequences of the 91 genotypes of P. vulgaris defined 36 different haplotypes; none of these were shared among the three gene pools (Mesoamerican and Andean for both wild and domesticated genotypes, and the phaseolin type I gene pool).
The Mesoamerican gene pool had the greatest number of haplotypes (24): 17 in the wild accessions, four in the domesticated accessions, and three shared among the wild and domesticated accessions; the wild accessions from Colombia had a unique haplotype that was shared with one accession from Costa Rica. There were two PvSHP1 haplotypes in the phaseolin type I accessions from Ecuador and northern Peru. In the Andean gene pool, 10 haplotypes were found; seven were specific for the wild forms, and one for the domesticated form, while two haplotypes were shared between these forms. For the entire PvSHP1 sequence, for both of the estimators used, as π (Tajima 1983) and θ (Watterson 1975), the nucleotide variation among all of the accessions was 15 × 10−3 (Table 5).
For the PvSHP1 locus, the Tajima’s D statistic was negative (D = −0.32), but not significant, as well as for the other tests.
Among all of the groups, the highest diversity was seen in the Mesoamerican wild genotypes (π = 17.34 × 10−3). This was more than one order of magnitude higher than for the Andean wild pool (π = 1.33 × 10−3), and there was an even lower diversity in the wild genotypes with phaseolin type I (π = 0.28 × 10−3). The Mesoamerican domesticated genotypes showed a lower diversity (π = 5.43 × 10−3) compared to the Mesoamerican wild genotypes, but higher than the Andean wild populations and domesticated types (π = 1.33 × 10−3 and 0.61 × 10−3, respectively). Comparing the diversity in the wild and domesticated accessions, there was an overall loss of diversity due to domestication of 34%. Considering the two gene pools separately, the Andean domesticated gene pool showed a loss of diversity of 54%. The loss of diversity estimated for the Mesoamerican domesticated gene pool was slightly higher, at 69% when compared with all of the Mesoamerican wild populations, and 65% when considering only the wild genotypes from Mexico.
The differences between the diversities measured as π or θ can be summarized by the analysis of the Tajima’s D. The wild forms from the Mesoamerican gene pool showed a positive Tajima’s D (D = 0.98), while in the phaseolin type I wild accessions, the Tajima’s D was negative (D = −1.01). However, these values were not statistically significant in either of these cases. In contrast, the wild form of the Andean gene pool deviated significantly from the mutation–drift model, with a statistically significant negative Tajima’s D value (D = −2.05; P < 0.05).
The domesticated forms of the two gene pools showed negative Tajima’s D values. For the Andean gene pool, the value was not significant (D = −0.29), but it was significant for the Mesoamerican gene pool (D = −1.94; P < 0.05). The estimation of the minimum number of recombination events tested within regions showed that there was no evidence of recombination for the four coding regions and for intron III of the PvSHP1 sequence, while for introns I and II, recombination events were detected only for the wild genotypes from the Mesoamerican gene pool.
The F ST pairwise estimates between the different P. vulgaris groups showed that there was no differentiation between Andean wild and domesticated genotypes (Table 6). All of the other F ST estimates were significantly different from 0 (Permutation test; Hudson 2000). The F ST estimate between the Mesoamerican wild and domesticated genotypes was the lowest significant value (F ST = 0.23). Interestingly, the Mesoamerican wild group was the least differentiated with regard to the other groups (F ST = 0.48, 0.51 and 0.52 with Phaseolin I type, Andean wild, and Andean domesticated groups, respectively) (Table 6).
On the basis of the relationships among P. vulgaris accessions reconstructed by the NJ analysis, six different subgroups were identified: wild accessions of G1, G1_W; domesticated accessions of G1, G1_D; phaseolin type I accessions, G2; wild accessions of G3, G3_W; wild accessions of G4, G4_W; domesticated accessions of G4, G4_D. As the G2 group corresponds to the Phaseolin wild-type genotypes, and the G4_W and G4_D to the wild and domesticated Andean genotypes analyzed previously, only the Mesoamerican NJ groups were used to recalculate the genetic statistics (Table 7). This analysis was performed to investigate differences in the levels of genetic diversity of these subgroups that belong to the same gene pool. The subgroup that included the domesticated accessions of G3 (G3_D) was not included in this analysis because of the low sample size (3 genotypes).
The wild genotypes that belong to G3 (from different origins: Mexico, Colombia, Costa Rica and Guatemala) showed the highest level of diversity (G3_W, π × 10−3 = 11.60). This was almost double that for the nucleotide variation of the Mesoamerican wild genotypes of G1 (all from Mexico) (G1_W, π × 10−3 = 5.66) The domesticated genotypes from G1 showed a similar level of diversity (G1_D, π × 10−3 = 1.32) as the Andean wild, while their nucleotide diversity was greater compared with the Andean domesticated genotypes (G4_D/AD, π × 10−3 = 0.61).
We detected a significant departure from the neutral equilibrium model of the Tajima’s D test in the domesticated genotypes of G1 (G1_D, D = −2.31; P < 0.01). For this group, the other tests of neutrality, including the Fu and Li D* and F* values, were also significant (G1_D, D* = −3.28 and F* = −3.49, P < 0.02 for both). The Tajima’s D values were positive, although without significance, only in the wild genotypes of G1 (G1_W, D = 0.50) and G3 (G3_W, D = 0.97), which were all of Mesoamerican origin.
Three recombination events (Rm = 3, Hudson and Kaplan 1985) were estimated among the Mexican wild genotypes of G1, with recombination detected between sites (388, 393) of intron I, and (404, 464), (513, 553) and (553, 698) of intron II.
Discussion
Locus characterization
The polymorphisms of the PvSHP1 sequence seen in P. vulgaris were useful for the development of PCR-based markers spanning the indels that we used to map the PvSHP1 locus using two different segregating populations. The PvSHP1 locus is located on linkage group Pv06, in proximity to the V gene for flower color, and in a genomic region where QTLs for days to flowering and common bacterial blight resistance have been located (Blair et al. 2006; Miklas et al. 2006). The sequence tagged site (STS) markers developed, as demonstrated by the analysis of the indel-based haplotypes, should be very useful in diversity studies, and particularly to trace the distribution of the domesticated Mesoamerican and Andean gene pools, as also showed by Angioi et al. (2010).
Since SHP in Arabidopsis is involved in fruit shattering (Liljegren et al. 2000), we hypothesized that the homologous PvSHP1 in Phaseolus should co-map/be the same gene as St, which is responsible for the presence or absence of pod string, and was mapped phenotypically on linkage group Pv02 using a domesticated (Midas) × wild (G12873) RIL population (Koinange et al. 1996). The PvSHP1 fragment did not map to the gene St, thus our data imply that the phenotypic variation for the presence/absence of pod string seen in the segregating population is not due to a mutation at or near the PvSHP1 locus. However, we cannot totally exclude that PvSHP1 is somewhat involved in genetic control of fruit shattering in Phaseolus. Our results indicate that similar phenotypic effects may not be produced by orthologous loci, even in closely related species, in agreement to what was found by Doust et al. (2004) from the analysis in foxtail millet of the teosinte branched1 (tb1) ortholog, which is the major gene controlling the branching phenotype in maize. Indeed, they found that the orthologous gene in foxtail millet has only a minor and variable effect on the control of branching.
A bottleneck during the formation of the Andean gene pool
Recently, using a comparison between their AFLP data and the simple sequence repeat (SSR)-based data of Kwak and Gepts (2009) and Rossi et al. (2009) suggested that P. vulgaris originated in Mexico or Mesoamerica, rather than in the Ecuador–northern Peru region, as proposed by Kami et al. (1995). They indicated that this is the most likely explanation for the strong reduction in molecular diversity when comparing Andean versus Mesoamerican populations for low mutating markers (AFLPs), compared to the very small reduction in diversity seen using markers with higher mutation rates (SSRs). Indeed, for a population that experiences a bottleneck, the number of generations needed for the recovery of the original level of genetic diversity (H) is expected to be close to the reciprocal of the mutation rate (Nei et al. 1975). Thus, markers that differ substantially in their mutation rates can show very different patterns of molecular diversity, as found by Rossi et al. (2009). Nucleotide substitution has a much lower mutation rate (10−9; Lynch and Conery 2000; Schlueter et al. 2004) compared with both AFLPs (10−6 to 10−5; Gaudeul et al. 2004; Kropf et al. 2009; Mariette et al. 2001) and SSRs (10−3 to 10−4; Thuillet et al. 2005; Vigouroux et al. 2002; Weber and Wong 1993). Thus, if the hypothesis of Rossi et al. (2009) is valid, in comparison with AFLPs, for the nucleotide data we should expect an even stronger reduction in the level of diversity in the Andean compared to the Mesoamerican wild populations. Our data are consistent with this expectation, and they support the hypothesis of Rossi et al. (2009). Indeed, for nucleotide variation, there was a much higher reduction in diversity in the Andes compared to the Mesoamerican wild populations, which is about twice that measured with AFLPs (Rossi et al. 2009) and 13-fold that measured with SSRs (Kwak and Gepts 2009), when using very similar samples of accessions. Moreover, the occurrence of a bottleneck is also in agreement with the significant and negative Tajima’s D values in the Andean wild population for the PvSHP1 locus. As Rossi et al. (2009) pointed out, this occurrence of a bottleneck during the formation of the Andean wild gene pool supports the Mesoamerican origin of the species and rejects the hypothesis of an Ecuador–northern Peru origin. Our current data provide additional evidence for the occurrence of a bottleneck in the Andes prior to domestication; thus, they support the Mesoamerican origin of P. vulgaris as a species.
Additional support of the Mesoamerican origin of P. vulgaris comes from recent SSR analyses of domesticated accessions (Blair et al. 2009), which showed, on average, a pattern similar to that of Kwak and Gepts (2009). Moreover, Blair et al. (2009) compared gene-based vs genomic SSRs. Using their data, we tested for differences between gene pools and between the two marker classes for H (expected heterozygosity) and for Na (number of alleles) using a Wilcoxon Rank Sums nonparametric test (P < 0.05). In agreement with expected different roles of purifying selection, while in both gene pools gene-based SSRs showed less diversity than genomic SSRs, the two classes showed a contrasting diversity pattern across the gene pools (for both H and Na). Significantly higher diversity was seen in Mesoamerica for gene-based SSRs compared to the higher diversity of genomic SSRs in Andean accessions. Thus, although based only on domesticated samples, our analysis of the data of Blair et al. (2009) provides additional confirmation of the Rossi et al. (2009) hypothesis.
Domestication
The data from the present study offer the first estimates of the effects of domestication on nucleotide variation in the common bean. The loss of diversity in the domesticated accessions in the Andes was 54%. In Mesoamerica, the loss of diversity in the domesticated accessions ranged from 65 to 69% when compared with only the wild accessions from Mexico and with all of the Mesoamerican wild populations, respectively.
These estimations are higher than those obtained using molecular markers. Indeed, very low reductions were seen in the Andean gene pool using SSRs (10%) and AFLPs (7%), while in Mesoamerica, a greater reduction was seen using AFLPs (34%). These differences might be associated with the different mutation rates of the various molecular markers, and after the bottleneck of domestication, with greater restoration of the molecular diversity for markers with a higher mutation rate (Glémin and Bataillon 2009). The reduction in genetic diversity in domesticated species has been summarized and reviewed (Glémin and Bataillon 2009; Haudry et al. 2007), and in some outbreeding species the loss of diversity was described: for maize, as 38% (Tenaillon et al. 2004; Wright et al. 2005), for Medicago, 31% (Muller et al. 2006), and for Helianthus annus, 55% (Liu and Burke 2006). Among inbreeding species, in soybean, Hyten et al. (2006) calculated a loss of diversity of 34%, while more severe bottlenecks associated with domestication have been described for barley, as 57% (Caldwell et al. 2006; Kilian et al. 2006), for rice, with a loss of diversity of 58 and 52% for Oryza sativa japonica and indica main varietal groups, respectively (Caicedo et al. 2007), and for tetraploid wheat, 65% (Haudry et al. 2007). However, among inbreeders, no loss of diversity was found by Kilian et al. (2007) for Einkorn (Triticum monococcum) when the diversity of the domesticated form was compared with that of the wild race β from which domestication is considered to have taken place, or there was relatively low reduction when the comparison was made with the whole wild gene pool (37%).
Our results indicate that P. vulgaris underwent a severe reduction in diversity that was similar to most inbreeding species, even though more loci were needed to obtain a detailed picture of the overall effect of drift in reducing the diversity of the domesticated form of the common bean. Indeed, as only one gene was analyzed in our study, we cannot estimate the relative role of drift and selection (if any) in such a large reduction in diversity, which would resolve this question. The neutrality tests provide no real help here; even though they indicate a departure from the mutation–drift equilibrium, they do not resolve whether this departure is determined just by the occurrence of a bottleneck (i.e. domestication), or whether to some extent the departure from neutrality is due to positive selection. More genes will need to be analyzed to obtain estimates of the bottleneck effects during the domestication of the common bean, which would then allow full and correct testing for selection at specific loci.
Our data indicate the occurrence of a single domestication event in Mesoamerica. Previous studies have also suggested that a single domestication event occurred in Mesoamerica (Gepts et al. 1986; Kwak and Gepts 2009; Kwak et al. 2009; Papa and Gepts 2003; Papa et al. 2005; Rossi et al. 2009), even if multiple domestication was suggested as one possible explanation of the results of a diversity study carried out using cp-RFLP data (Chacón et al. 2005). The present study shows that almost all of the Mesoamerican domesticated accessions are included in the single, homogeneous and well-supported G1 subclade (bootstrap value, 88%), along with four wild or weedy accessions; this group corresponds to the C1 cluster identified by BAPS analysis. Thus, our study confirms the hypothesis of a single domestication event in Mesoamerica using sequence data of a fragment that had little possibility of recombination, and was thus less subject to the confounding effects of hybridization in phylogeny reconstruction (Avise 1995; Posada et al. 2000).
However, three Mesoamerican domesticated genotypes fall into G3 (C4 for BAPS). This might be the result of hybridization after domestication or of an independent minor domestication event. The AFLP data provide an answer to this question, indicating that these genotypes are probably determined by introgression from wild populations. Indeed the AFLP analysis shows that all of the domesticated G3 genotypes form a group together with all of the G1 domesticated wild accessions, rather than being closer to the wild G3, suggesting that the G3 PvSHP1 alleles were introduced in these genotypes by gene flow rather than by common ancestry. Our data strongly support the occurrence of a single Mesoamerican domestication event that occurred in Mexico. Moreover, even if our sampling does not provide the appropriate power to indicate the domestication area of the common bean, our data are compatible with those of Kwak et al. (2009) on the origin of domestication of the common bean, which they located to the Lerma–Santiago Basin of Mexico. Indeed, one of the three wild genotypes that form an outgroup of the domesticated G1 genotypes is from Jalisco, Mexico, while the other two are more probably weedy types. For the domestication in the Andes, the lack of polymorphism does not provide any clues towards an answer to the analogous questions.
Conclusions
In the present study, we have described the successful identification and mapping of PvSHP1, a sequence that is homologous to SHATTERPROOF-1 (SHP1) of A. thaliana. We amplified a SHP1-related sequence from the agronomically most important Phaseoleae species. Within Phaseoleae, there was a high degree of intraspecies and interspecies diversity in the introns, while the coding region appeared to be conserved across distant taxa. The sizes of the amplified fragments differed greatly among distant taxa (eg. Glycine vs. Phaseolus), due to the different sizes of the noncoding regions, while they were almost the same among the most closely related species of Phaseolus. Using three markers that span indels, PvSHP1 was mapped to chromosome 6 in P. vulgaris, in proximity of the flower color V gene and in the same region where QTLs for flowering time and common bacterial blight resistance are located. However, our data suggest that the phenotypic variation for the presence or absence of pod string seen in a segregating population is not due to variations at the PvSHP1 locus. A very interesting point of this study is also seen from the use of the amplification products for the development of markers that will be very useful for bean germplasm characterization, and particularly to trace the distribution of the domesticated Andean and Mesoamerican gene pools. This is shown by our analysis of the diversity of indel-based haplotypes (Ib-haplotypes) and was confirmed by Angioi et al. (2010), who used these markers to study the effects of the introduction of P. vulgaris into and across Europe.
PvSHP1 was also sequenced in a sample of 91 wild and domesticated genotypes spanning the geographic distribution of the centers of origin of this species. The analysis of the nucleotide diversity confirms the hypothesis of Rossi et al. (2009), of a Mesoamerican origin of P. vulgaris with a strong bottleneck associated with the colonization of the Andes.
Our results confirm the gene pool structure seen in P. vulgaris, along with the independent domestication process in the Andes and in Mesoamerica, and they provide additional evidence for a single domestication event in Mesoamerica.
References
Acosta-Gallegos JA, Kelly JD, Gepts P (2007) Pre-breeding in common bean and use of genetic diversity from wild germplasm. Crop Sci 47:44–59
Allaby RG, Fuller DQ, Brown TA (2008) The genetic expectations of a protracted model for the origins of domesticated crops. Proc Natl Acad Sci USA 105:13982–13986
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res 25:3389–3402
Angioi SA, Desiderio F, Rau D, Bitocchi E, Attene G, Papa R (2009) Development and use of chloroplast microsatellites in Phaseolus spp. and other legumes. Plant Biol 11:598–612
Angioi SA, Rau D, Attene G, Nanni L, Bellucci E, Logozzo G, Negri V, Spagnoletti Zeuli PL, Papa R (2010) Beans in Europe: origin and structure of the European landraces of Phaseolus vulgaris L. Theor Appl Genet 121:829–843
Avise JC (1995) Mitochondrial DNA polymorphism and a connection between genetics and demography of relevance to conservation. Conserv Biol 9:686–690
Beebe S, Skroch P, Tohme J, Duque M, Pedraza F, Nienhuis J (2000) Structure of genetic diversity among common bean landraces of middle-American origin based on correspondence analysis of RAPD. Crop Sci 40:264–273
Beebe S, Rengifo J, Gaitan E, Duque MC, Tohme J (2001) Diversity and origin of Andean landraces of common bean. Crop Sci 41:854–862
Blair MW, Iriarte G, Beebe S (2006) QTL analysis of yield traits in an advanced backcross population derived from a cultivated Andean × wild common bean (Phaseolus vulgaris L.) cross. Theor Appl Genet 112:1149–1163
Blair MW, Diaz LM, Buendia HF, Dunque MC (2009) Genetic diversity. seed size associations and population structure of a core collection of common beans (Phaseolus vulgaris L.). Theor Appl Genet 119:955–972
Caicedo AL, Williamson SH, Hernandez RD, Boyko A, Fledel-Alon A, York TL, Polato NR, Olsen KM, Nielsen R, McCouch SR, Bustamante CD, Purugganan MD (2007) Genome-wide patterns of nucleotide polymorphism in domesticated rice. PLoS Genet 3(9):e163
Caldwell KS, Russell J, Langridge P, Powell W (2006) Extreme population-dependent linkage disequilibrium detected in an inbreeding plant species, Hordeum vulgare. Genetics 172:557–567
Chacón SMI, Pickersgill B, Debouck DG (2005) Domestication patterns in common bean (Phaseolus vulgaris L.) and the origin of the Mesoamerican and Andean cultivated races. Theor Appl Genet 110:432–444
Corander J, Waldmann P, Sillanpää MJ (2003) Bayesian analysis of genetic differentiation between populations. Genetics 163:367–374
Corander J, Marttinen P (2006) Bayesian identification of admixture events using multi-locus molecular markers. Mol Ecol 15:2833–2843
Corander J, Tang J (2007) Bayesian analysis of population structure based on linked molecular information. Math Biosci 205:19–31
Corander J, Marttinen P, Sirén J, Tang J (2008) Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinf 9:539
Debouck DG, Toro O, Paredes OM, Johnson WC, Gepts P (1993) Genetic diversity and ecological distribution of Phaseolus vulgaris in northwestern South America. Econ Bot 47:408–423
Delgado-Salinas A, Turley T, Richman A, Lavin M (1999) Phylogenetic analysis of the cultivated and wild species of Phaseolus (Fabaceae). Syst Bot 24:438–460
Delgado-Salinas A, Bibler R, Lavin M (2006) Phylogeny of the Genus Phaseolus (Leguminosae): a recent diversification in an ancient landscape. Syst Bot 31:779–791
Dìaz LM, Blair MW (2006) Race structure within the Mesoamerican gene pool of common bean (Phaseolus vulgaris L.) as determined by microsatellite markers. Theor Appl Genet 114:143–154
Doust AN, Devos KM, Gadberry MD, Gale MD, Kellogg EA (2004) Genetic control of branching in foxtail millet. Proc Natl Acad Sci USA 101:9045–9050
Duarte J, Dos Santos J, Melo L (1999) Genetic divergence among common bean cultivars from different races based on RAPD markers. Genet Mol Biol 22:419–426
Felsenstein J (1993) Phylogeny Inference Package (PHYLIP). Version 3.5. Distributed by the author. Department of Genetics. University of Washington, Seattle, USA
Freyre R, Skroch P, Geffroy V, Adam-Blondon AF, Shirmohamadali A, Johnson W, Llaca V, Nodari RO, Pereira PA, Tsai SM, Tohme J, Dron M, Nienhuis J, Vallejos CE, Gepts P (1998) Towards an integrated linkage map of common bean. 4. Development of a core map and alignment of RFLP maps. Theor Appl Genet 97:847–856
Freytag GF, Debouck DG (1996) Phaseolus costaricensis, a new wild bean species (Phaseolinae, Leguminosae) from Costa Rica and Panama, central America. Novon 6:157–163
Freytag GF, Debouck DG (2002) Taxonomy, distribution, and ecology of the genus Phaseolus (Leguminosae-Papilionoideae) in North America, Mexico and central America. Botanical Research Institute of Texas. Ft. Worth, TX
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Gaudeul M, Till-Bottraud I, Barjon F, Manel S (2004) Genetic diversity and differentiation in Eryngium alpinum L. (Apiaceae): comparison of AFLP and microsatellite markers. Heredity 92:508–518
Gepts P, Bliss FA (1986) Phaseolin variability among wild and cultivated common beans (Phaseolus vulgaris) from Colombia. Econ Bot 40:469–478
Gepts P, Osborn TC, Rashka K, Bliss FA (1986) Phaseolin-protein variability in wild forms and landraces of the common bean (Phaseolus vulgaris): evidence for multiple centers of domestication. Econ Bot 40:451–468
Gepts P, Kmiecik K, Pereira P, Bliss FA (1988) Dissemination pathways of common bean (Phaseolus vulgaris, Fabaceae) deduced from phaseolin electrophoretic variability. I. The Americas. Econ Bot 42:73–85
Glémin S, Bataillon T (2009) A comparative view of the evolution of grasses under domestication. New Phytol 183:273–290
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Haudry A, Cenci A, Ravel C, Bataillon T, Brunel D, Poncet C, Hochu I, Poirier S, Santoni S, Glémin S, David J (2007) Grinding up wheat: a massive loss of nucleotide diversity since domestication. Mol Biol Evol 24:1506–1517
Hecht V, Foucher F, Ferrandiz C, Macknight R, Navarro C, Morin J, Vardy ME, Ellis N, Beltrán JP, Rameau C, Weller JL (2005) Conservation of Arabidopsis flowering genes in model legumes. Plant Physiol 137:1420–1434
Hillis DM, Bull JJ (1993) An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst Biol 42:182–192
Hudson RR, Kaplan NL (1985) Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111:147–164
Hudson RR, Slatkin M, Maddison WP (1992) Estimation of levels of gene flow from DNA sequence data. Genetics 132:583–589
Hudson RR (2000) A new statistic for detecting genetic differentiation. Genetics 155:2011–2014
Hyten DL, Song Q, Zhu Y, Choi IY, Nelson RL, Costa JM, Specht JE, Shoemaker RC, Cregan PB (2006) Impacts of genetic bottlenecks on soybean genome diversity. Proc Natl Acad Sci USA 103:16666–16671
Kami J, Becerra Velásquez V, Debouckand DG, Gepts P (1995) Identification of presumed ancestral DNA sequences of phaseolin in Phaseolus vulgaris. Proc Natl Acad Sci USA 92:1101–1104
Kilian B, Özkan H, Kohl J, von Haeseler A, Barale F, Deusch O, Brandolini A, Yucel C, Martin W, Salamini F (2006) Haplotype structure at seven barley genes: relevance to gene pool bottlenecks, phylogeny of ear type and site of barley domestication. Mol Genet Genom 276:230–241
Kilian B, Özkan H, Walther A, Kohl J, Dagan T, Salamini F, Martin W (2007) Molecular diversity at 18 loci in 321 wild and 92 domesticate lines reveals no reduction in nucleotide diversity during Triticum monococcum (Einkorn) domestication: implications for the origin of agriculture. Mol Biol Evol 24:2657–2668
Koinange EMK, Singh SP, Gepts P (1996) Genetic control of the domestication syndrome in common bean. Crop Sci 36:1037–1045
Kropf M, Comes HP, Kadereit JW (2009) An AFLP clock for the absolute dating of shallow-time evolutionary history based on the intraspecific divergence of southwestern European alpine plant species. Mol Ecol 18:697–708
Kwak M, Gepts P (2009) Structure of genetic diversity in the two major gene pools of common bean (Phaseolus vulgaris L., Fabaceae). Theor Appl Genet 118:979–992
Kwak M, Kami J, Gepts P (2009) The putative Mesoamerican domestication center of Phaseolus vulgaris is located in the Lerma-Santiago Basin of Mexico. Crop Sci 49:554–563
Lander P, Green J, Abrahamson A, Barlow M, Lincoln DS, Newberg L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Liljegren S, Ditta G, Eshed Y, Savidge B, Bowmanand J, Yanofsky M (2000) Control of fruit dehiscence in Arabidopsis by the SHATTERPROOF MADS-box genes. Nature 404:766–769
Liu A, Burke JM (2006) Patterns of nucleotide diversity in wild and cultivated sunflower. Genetics 173:321–330
Löytynoja A, Goldman N (2005) An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci USA 102:10557–10562
Löytynoja A, Goldman N (2008) A model of evolution and structure for multiple sequence alignment. Philos Trans R Soc Lond B Biol Sci 363:3913–3919
Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155
Mariette S, Chagne D, Lezier C, Pastuszka P, Raffin A, Plomion C, Kremer A (2001) Genetic diversity within and among Pinus pinaster populations: comparison between AFLP and microsatellite markers. Heredity 86:469–479
McConnell M, Mamidi S, Lee R, Chikara S, Rossi M, Papa R, McClean P (2010) Syntenic relationships among legumes revealed using a gene-based genetic linkage map of common bean (Phaseolus vulgaris L.). Theor Appl Genet 121:1103–1116
McClean PE, Lee RK, Otto C, Gepts P, Bassett M (2002) Molecular and phenotypic mapping of genes controlling seed coat pattern and color in common bean (Phaseolus vulgaris L.). J Hered 93:148–152
McClean PE, Lee RK, Miklas PN (2004) Sequence diversity analysis of dihydrofl avonol 4-reductase intron1 in common bean. Genome 47:266–280
McClean PE, Lee RK (2007) Genetic architecture of chalcone isomerase non-coding regions in common bean (Phaseolus vulgaris L.). Genome 50:203–214
Miklas PN, Kelly JD, Beebe SE, Blair MW (2006) Common bean breeding for resistance against biotic and abiotic stresses: from classical to MAS breeding. Euphytica 147:106–131
Muller MH, Poncet C, Prosperi JM, Santoniand S, Ronfort J (2006) Domestication history in the Medicago sativa species complex: inferences from nuclear sequence polymorphism. Mol Ecol 15:1589–1602
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability of populations. Evolution 29:1–10
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Nei M, Gojobori T (1986) Simple methods for estimating the number of synonymous and non-synonymous nucleotide substitutions. Mol Biol Evol 3:418–426
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nodari RO, Tsai SM, Gilbertson RL, Gepts P (1993) Towards an integrated linkage map of common bean. II. Development of an RFLP-based linkage map. Theor Appl Genet 85:513–520
Papa R, Gepts P (2003) Asymmetry of gene flow and differential geographical structure of molecular diversity in wild and domesticated common bean (Phaseolus vulgaris L.) from Mesoamerica. Theor Appl Genet 106:239–250
Papa R, Acosta J, Delgado-Salinas A, Gepts P (2005) A genome-wide analysis of differentiation between wild and domesticated Phaseolus vulgaris from Mesoamerica. Theor Appl Genet 111:1147–1158
Papa R, Nanni L, Sicard D, Rau D, Attene G (2006) The evolution of genetic diversity in Phaseolus vulgaris L. In: Motley TJ, Zerega N, Cross H (eds) New approaches to the origins. Evolution and conservation of crops. Darwin’s Harvest. Columbia University Press, New York, pp 121–142
Pedrosa HA, Souza de Almeida CC, Mosiolek M, Blair MW, Schweizer D, Guerra M (2006) Extensive ribosomal DNA amplification during Andean common bean (Phaseolus vulgaris L.) evolution. Theor Appl Genet 112:924–933
Pinyopich A, Ditta GS, Baumann E, Wisman E, Yanofsky MF (2003) Unraveling the redundant roles of MADS-box genes during carpel and fruit development. Nature 424:85–88
Posada D, Crandall KA, Templeton AR (2000) GeoDis: a program for the cladistic nested analysis of the geographical distribution of genetic haplotypes. Mol Ecol 9:487–488
Purugganan MD, Fuller D (2009) The nature of selection during plant domestication. Nature 457:843–848
Rosales-Serna R, Hernandez-Delgado S, Gonzalez-Paz M, Acosta-Gallegos JA, Mayek-Perez N (2005) Genetic relationships and diversity revealed by AFLP markers in Mexican common bean bred cultivars. Crop Sci 45:1951–1957
Rossi M, Bitocchi E, Bellucci E, Nanni L, Rau D, Attene G, Papa R (2009) Linkage disequilibrium and population structure in wild and domesticated populations of Phaseolus vulgaris L. Evol Appl 2:504–522
Santalla M, Menéndez-Sevillano MC, Monteagudo AB, De Ron AM (2004) Genetic diversity of Argentinean common bean and its evolution during domestication. Euphytica 135:75–87
Schlueter JA, Dixon P, Granger C, Grant D, Clark L, Doyle JJ, Shoemaker RC (2004) Mining EST databases to resolve evolutionary events in major crop species. Genome 47:868–876
Singh SP, Gepts P, Debouck DG (1991) Races of common bean (Phaseolus vulgaris L., Fabaceae). Econ Bot 45:379–396
Singh RJ, Kim HH, Hymowitz T (2001) Distribution of rDNA loci in the genus Glycine Willd. Theor Appl Genet 103:212–218
Tajima F (1983) Evolution relationship of DNA sequences in finite populations. Genetics 105:437–460
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software, version 4.0. Mol Biol Evol 24:1596–1599
Tenaillon MI, U’Ren J, Tenaillon O, Gaut BS (2004) Selection versus demography: a multilocus investigation of the domestication process in maize. Mol Biol Evol 21:1214–1225
Thompson JD, Gibson TJ, Plewniak F, Jeanmouginand F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res 25:4876–4882
Thuillet AC, Bataillon T, Poirier S, Santoni S, David JL (2005) Estimation of long-term effective population sizes through the history of durum wheat using microsatellite data. Genetics 169:1589–1599
Toro O, Tohme J, Debouck DG (1990) Wild Bean (Phaseolus vulgaris L): description and distribution. Centro Internacional de Agricultura Tropical, Cali, Colombia
Vigouroux Y, McMullen M, Hittinger CT, Houchins K, Schulz L, Kresovich S, Matsuoka Y, Doebley J (2002) Identifying genes of agronomic importance in maize by screening microsatellites for evidence of selection during domestication. Proc Natl Acad Sci USA 99:9650–9655
Vrebalov J, Panb IL, Arroyo AJ, McQuinn R, Chung M, Poole M, Rose J, Seymoure G, Grandillo S, Giovannoni J, Irish VF (2009) Fleshy fruit expansion and ripening are regulated by the tomato SHATTERPROOF gene TAGL1. Plant Cell 21:3041–3062
Watterson GA (1975) On the number of segregating sites in genetic models without recombination. Theor Popul Biol 7:256–276
Weber J, Wong C (1993) Mutation of human short tandem repeats. Hum Mol Genet 2:1123–1128
Wojciechowski MF, Lavin M, Sanderson MJ (2004) A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am J Bot 91:1846–1862
Wright SI, Bi IV, Schroeder SG, Yamasaki M, Doebley JF, McMullen MD, Gaut BS (2005) The effects of artificial selection on the maize genome. Science 308:1310–1314
Acknowledgments
Nanni L. and Bitocchi E. made equal contributions to this study, and so should be considered joint first authors. This study was supported by the Italian Government (MIUR) grant no. # 2005071310, Project PRIN 2005, and by the Università Politecnica delle Marche (years 2005–2009).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by B. Diers.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2011_1671_MOESM1_ESM.xls
Supplementary material descriptive captions Table A1. Genotypes of Phaseolus spp. and other legumes used in this study. (XLS 54 kb)
Rights and permissions
About this article
Cite this article
Nanni, L., Bitocchi, E., Bellucci, E. et al. Nucleotide diversity of a genomic sequence similar to SHATTERPROOF (PvSHP1) in domesticated and wild common bean (Phaseolus vulgaris L.). Theor Appl Genet 123, 1341–1357 (2011). https://doi.org/10.1007/s00122-011-1671-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-011-1671-z