
Abstract
Arachis hypogaea is an allotetraploid species with low genetic variability. Its closest relatives, all of the genus Arachis, are important sources of alleles for peanut breeding. However, a better understanding of the genome constitution of the species and of the relationships among taxa is needed for the effective use of the secondary gene pool of Arachis. In the present work, we focused on all 11 non-A genome (or B genome sensu lato) species of Arachis recognized so far. Detailed karyotypes were developed by heterochromatin detection and mapping of the 5S and the 18S–25S rRNA using FISH. On the basis of outstanding differences observed in the karyotype structures, we propose segregating the non-A genome taxa into three genomes: B sensu stricto (s.s.), F and K. The B genome s.s. is deprived of centromeric heterochromatin and is homologous to one of the A. hypogaea complements. The other two genomes have centromeric bands on most of the chromosomes, but differ in the amount and distribution of heterochromatin. This organization is supported by previously published data on molecular markers, cross compatibility assays and bivalent formation at meiosis in interspecific hybrids. The geographic structure of the karyotype variability observed also reflects that each genome group may constitute lineages that have evolved through independent evolutionary pathways. In the present study, we confirmed that Arachis ipaensis was the most probable B genome donor for A. hypogaea, and we identified a group of other closely related species. The data provided here will facilitate the identification of the most suitable species for the development of prebreeding materials for further improvement of cultivated peanut.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Members of the genus Arachis are native to South America and consist of a diverse group of autogamous and geocarpic taxa. Among them, species included within section Arachis are considered to be the most derived, diverse, and numerous of the genus (Krapovickas and Gregory 1994). A total of 26 diploids with 2n = 2x = 20, three diploids with 2n = 2x = 18, and two allopolyploid entities (2n = 4x = 40), the cultigen A. hypogaea and its presumed wild progenitor A. monticola, are recognized within the group (Fernández and Krapovickas 1994; Lavia 1996, 1998, 2000; Peñaloza and Valls 2005; Valls and Simpson 2005).
Geographically, section Arachis has the largest range of the genus (Krapovickas and Gregory 1994). It extends from the foothills of the Andes Mountains in Bolivia and northern Argentina to the Atlantic coast in Brazil and from the headwaters of the Mamoré and Guaporé rivers in northern Bolivia and the Tocantins rivers in Central Brazil, to the northern coast of La Plata River in Uruguay. Within this area, the species may either have extended ranges or limited to only one collection site. The distribution areas of the species can overlap, but sympatric populations are rarely observed. Some of the species are composed of populations scattered throughout the entire species range, but others occur in a few small populations often separated by long distances (which can be hundreds of kilometers). Each population usually has tens to hundreds of individuals, arranged in patches of different sizes or in a regular distribution.
Peanut (Arachis hypogaea) is an ancient crop of the New World, which is widely cultivated around the world in tropical, subtropical, and warm temperate climates. However, the restricted natural resistance to biotic and abiotic agents, mainly due to the limited genetic base superimposed by its polyploid nature (Halward et al. 1991; Kochert et al. 1991, 1996; Paik-Ro et al. 1992; Hilu and Stalker 1995; He and Prakash 1997; Subramanian et al. 2000; Herselman 2003), has driven the attention of breeders and geneticists toward the diploid species of section Arachis. Several desirable agronomic traits such as resistance to various pests and diseases, drought resistance and different life-cycle spans have been detected in these wild species (Singh 1986; Burow et al. 2001; Simpson 2001; Mallikarjuna 2002; Mallikarjuna et al. 2004). Some of these species have been used in introgression programs, and some breeding lines and varieties possessing alleles from wild species have been released (Johnson et al. 1977; Foster et al. 1981; Singh 1986; Burow et al. 2001; Isleib et al. 2001; Simpson and Starr 2001). Although these trials have shown the feasibility of using wild germplasm to improve traditional cultivars, the efficient implementation of these programs requires more detailed knowledge of the genomic affinities of the species. For this reason, great efforts have been directed toward understanding the genetic compatibility among the wild taxa, as well as between them and the cultivated peanut.
According to their morphological and chromosome features added to cross-compatibility assays, three different genomes (A, B and D) have been traditionally proposed for diploid species with x = 10 within the section Arachis (Smartt et al. 1978; Gregory and Gregory 1979; Singh and Moss 1982, 1984; Singh 1986; Stalker 1991; Fernández and Krapovickas 1994). Excluding A. glandulifera (D genome), which has an asymmetric karyotype, all the species have symmetric karyotypes largely composed of metacentric chromosomes (Stalker 1991; Fernández and Krapovickas 1994; Lavia 1996, 2000; Peñaloza and Valls 2005), and they have been assigned to the A or to the B genomes. The A genome is characterized by a small chromosome pair, the “A chromosomes” (Smartt et al. 1978, after Husted 1936). The “A chromosomes” have a differential condensation pattern during prometaphase (Fernández and Krapovickas 1994) and have a large heterochromatic band in the centromeric region (Seijo et al. 2004). The remaining species with symmetric karyotypes, but without “A chromosomes”, have been assigned to one single genome group named B genome (Smartt et al. 1978) or non-A genome (Seijo et al. 2004). Diploid species with x = 9 are not well characterized, and their genome constitution is still to be determined. Tetraploid species have an AABB genome constitution, and they may have originated either by hybridization of two wild diploid species followed by chromosome doubling (Gregory and Gregory 1976, 1979) or by means of bilateral sexual polyploidization (Seijo et al. 2007).
Detailed cytogenetic analysis by FISH mapping of rDNAs performed on the Arachis species included within the A genome has revealed that all the entities have a common karyotype structure and supports all belonging to the same genome (Robledo et al. 2009). However, three different karyotype subgroups could be established on the basis of the number of rDNA loci and the chromosomes with centromeric heterochromatin. This arrangement is supported to some extent by cross-compatibility assays. In general, all the A genome species can produce interspecific hybrids with stained pollen (i.e., viable) usually higher than 25%, thus supporting their belonging to the same genome. Nevertheless, the available published data revealed that species within each karyotype subgroup cross more readily and produce a higher number of fertile hybrids than crosses between species from different subgroups (Krapovickas and Gregory 1994; Milla et al. 2005; Tallury et al. 2005).
The species included within the B genome remain poorly characterized, and the relationships among these taxa are largely unknown. Classical cytogenetic studies have determined that 11 wild diploid species may belong to this group (Smartt et al. 1978; Fernández and Krapovickas 1994; Lavia 1996; Peñaloza and Valls 2005; Robledo G, unpublished), and at least three of them have been proposed as possible progenitors of A. hypogaea (Smartt et al. 1978; Fernández and Krapovickas 1994; Lavia 1996; Seijo et al. 2004, 2007; Milla et al. 2005). All these species are annuals, morphologically diverse, and inhabit different ecological regions (Krapovickas and Gregory 1994; Valls and Simpson 2005). Unlike species with the A genome, species with the B genome are more diverse in their karyotype formulas (Fernández and Krapovickas 1994) and karyotype structure (Seijo et al. 2004), and the degree of fertility of their interspecific hybrids vary, from completely sterile (with less than 10% of pollen stainability) to almost completely fertile (Krapovickas and Gregory 1994; Tallury et al. 2005; Burow et al. 2009). For these reasons, this group has been suspected as being composed of different lineages (Seijo et al. 2004).
A variety of analyses using molecular markers have been carried out aiming to establish the genetic relationships among species of Arachis (Kochert et al. 1991; Raina et al. 2001; Gimenes et al. 2002; Moretzsohn et al. 2004; Milla et al. 2005; Tallury et al. 2005; Burow et al. 2009). Most of these analyses have shown that although species with the B genome are usually clearly segregated from those with the A genome, a set of two or more clusters can be easily recognized within the former. In spite of this clustering of species, the relationships between them, within each cluster and among the different clusters, remain unclear (Milla et al. 2005; Tallury et al. 2005; Burow et al. 2009).
Taking these antecedents into consideration, we constructed detailed cytogenetic maps by detecting rDNA loci using FISH and analyzing heterochromatin distribution using DAPI+ staining in 11 species currently assigned to the B or non-A genome, in order to (1) investigate the species relationships and comprehensively reevaluate their genomic status and (2) provide further information on the B genome donor of peanut by comparing the karyotypes established for diploid species with those published for the allotetraploids of the section (Seijo et al. 2004).
Materials and methods
Plant material
The original provenances and the collection numbers of the accession analyzed are cited in Table 1. Arachis hoehnei was excluded from this study according to the suggestions of previous authors (Tallury et al. 2005) and because a recent karyotype analysis made in our laboratory which revealed that the available accessions Nº K,G 30006 and V9094-2 have a karyotype constitution that corresponds to the A genome species (Robledo G, unpublished). Figure 1 shows the geographic distribution of the wild species analyzed and the center of diversity of A. hypogaea subsp. hypogaea, which is considered the most ancestral peanut subspecies.

Geographic distribution of Arachis species. 1 A. batizocoi; 2 A. cruziana; 3 A. krapovickasii; 4 A. valida; 5 A. gregoryi; 6 A. magna; 7 Arachis sp.; 8 A. ipaënsis; 9 A. williamsii; 10 A. benesis; 11 A. trinitensis; 12 Major center of landraces with ancestral characters of A. hypogaea var. hypogaea. (striped) K genome; (light grey shaded) B genome and (dark grey shaded) F genome. Dashed line indicates the geographic distribution of section Arachis
Chromosome preparations
Healthy root tips (5–10 mm long) of germinating seeds were pretreated with 2 mM 8-hydroxyquinoline for 3 h at room temperature (Fernández and Krapovickas 1994) and fixed in 3:1 absolute ethanol:glacial acetic acid for at least 12 h at 4°C. Root apices were enzymatically digested (Schwarzacher et al. 1980) and squashed in 45% acetic acid. Coverslips were removed with CO2, and the slides were air dried, aged for 1–2 days at room temperature, and kept at −20°C until use.
For some accessions chromosomes preparation were obtained from ovaries of matured flowers pretreated and fixed in the field according to Robledo and Seijo (2008).
Probe labeling and fluorescent in situ hybridization
The 5S rDNA loci were localized using the pA5S probe labeled by nick translation with digoxigenin-11-dUTP (Boehringer Mannheim, Mannheim, Germany) and the 45S rDNA loci using an equimolar mixture of the probes pA18S and pA26S labeled with biotin-11-dUTP (Sigma-Aldrich, St. Louis, Missouri, USA), according to Robledo and Seijo (2008). Pretreatment of slides, chromosome and probe denaturation, conditions for the in situ hybridization, posthybridization washing, blocking, and indirect detection with fluorochrome-conjugated antibodies were performed according to Moscone et al. (1996). Except for the first set of antibodies, which consisted of anti-biotin produced in goat and monoclonal anti-digoxigenin conjugated to fluorescein isothiocyanate (FITC) produced in mouse, and the second set, which consisted of anti-goat conjugated to tetramethyl-rodamine isothiocyanate (TRITC) produced in rabbit and anti-mouse conjugated to FITC produced in sheep (all from Sigma-Aldrich). Preparations were counterstained and mounted with Vectashield medium (Vector Laboratories, Burlingame, California, USA) containing 2 mg/mL of 4′,6-diamidino-2-phenylindole (DAPI). The counterstaining with DAPI revealed a C-banding-like pattern, with major heterochromatic bands fluorescing more intensely (Seijo et al. 2004).
Fluorescence microscopy and image acquisition
Chromosomes were photographed with a Leica DMRX epifluorescence microscope (Leica, Heerbrugg, Switzerland) equipped with a computer-assisted Leica DC 350 digital camera system. Red, green, and blue images were captured in black and white, then combined using IM 1000 Leica software. The merged images were then imported into Photoshop version 7.0 (Adobe, San Jose, California, USA) for final processing using the brightness, contrast, and intensity tools, which affect the whole image equally.
Karyotype analysis and loci mapping
Chromosome measurements were carried out on at least five metaphases per individual and three individuals per species using the free version of the MicroMeasure 3.3 program http://www.colostate.edu/Depts/Biology/MicroMeasure/. The centromeric index (i = short arm length × 100/chromosome length) was used to classify the chromosomes according to Levan et al. (1964) as metacentric (m, i = 50–37.51), submetacentric (sm, i = 37.50–25.10) and subtelocentric (sm, i = 25–12.51). Data from homologous chromosomes were combined to obtain mean values—first between chromosomes in the same metaphase and subsequently among different metaphases of the same species. Mean karyotype values for each species were represented as haploid complements in the idiograms. Chromosomes were ordered primarily by morphology and then by decreasing length. Some chromosomes within each idiogram were reordered according to tentative homeologies with other diploid species and on the basis of the nomenclature proposed for A. hypogaea/A. monticola (Seijo et al. 2004). The intrachromosomal and interchromosomal asymmetry index was determined using the A1 and A2 indices (Romero Zarco 1986).
Results
General karyotype features
Morphological analysis of the karyotypes showed that all the studied species have their complements completely or mainly composed of metacentric chromosomes. However, one to four pairs of submetacentric chromosomes were observed in different species. The mean chromosome length varied between 2.60 μm (A. benensis) and 3.64 μm (A. batizocoi), and the total length of the haploid karyotype ranged from 25.18 μm (A. benensis) to 36.15 μm (A. batizocoi) (Table 2). The intrachromosomal asymmetry index (A1) ranged from 0.15 to 0.30, while the interchromosomal index (A2) ranged from 0.07 to 0.11. As a whole, the karyotypes of A. cruziana, A. batizocoi, and A. krapovickasii were the most asymmetric, while those of Arachis sp., A. magna, and A. williamsii were the least (Table 2).
Almost all species have only one pair of secondary constrictions localized on the long arms of pair 10 (Fig. 2). The exceptions are A. cruziana, with the constrictions on pair 2 (m), and A. krapovickasii, and A. gregoryi, which have two pairs of secondary constrictions. In general, the constrictions were extended at early metaphase, and the satellites remained far from the corresponding proximal segments of the chromosome arms. The constrictions on pair 9 of A. gregoryi were the only ones that never extended.

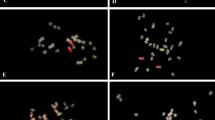
Somatic metaphases of Arachis species after double-fluorescent in situ hybridization (FISH), showing yellow-green FITC signals for the 5S rDNA loci (pA5S), and red TRITC signals for the 45S rDNA loci (pA18S and pA26S). DAPI counterstaining (light blue) subsequent to the FISH procedure was used to highlight the heterochromatin bands and to stain euchromatin. a A. batizocoi; b A. cruziana; c A. krapovickasii; d A. trinitensis; e A. benesis; f Arachis sp.; g A. ipaënsis; h A. magna; i A. williamsii; j A. gregoryi; k A. valida. Scale bar 3 μm
Heterochromatin distribution
The pattern of C-DAPI+ heterochromatin distribution varied among species, but with some commonalities among taxa. The karyotypes of A. batizocoi, A. cruziana, and A. krapovickasii were characterized by conspicuous centromeric bands in nine chromosome pairs (Table 2; Fig. 2). The last two species also have one small band in the telomeric position on the long arms of pair 8. The total amount of centromeric heterochromatin per haploid complement was conserved within this group, varying from 11.36% of the total karyotype length in A. krapovickasii to 12.55% in A. cruziana. Heterochromatin distribution among the chromosomes of any of these karyotypes was also conserved. Pair 9 bore the largest centromeric bands (0.85 μm in A. batizocoi, 0.86 μm in A. cruziana and 0.78 μm in A. krapovickasii), pair 10 had medium-sized bands (0.62 μm in A. batizocoi, 0.43 μm in A. cruziana and 0.57 μm in A. krapovickasii), and the remaining chromosomes had the smallest (0.20–0.52 μm in A. batizocoi, 0.21–0.37 μm in A. cruziana and 0.24–0.51 μm in A. krapovickasii).
The karyotypes of A. trinitensis and A. benensis characteristically had small and faint centromeric bands in only seven and eight chromosome pairs, respectively (Table 2; Figs. 2, 3). Additionally, all the bands had a similar size (0.16–0.26 μm in A. benensis and 0.17–0.31 μm in A. trinitensis). The total amount of heterochromatin per complement was almost half (5.89% in A. trinitensis and 7.52% in A. benensis) of that observed in the karyotypes of the aforementioned species (Table 2).

Idiograms of Arachis species showing the distribution of 5S (striped) and 18S-26S rDNA loci (black shaded), DAPI-enhanced heterochromatic bands (white shaded), DAPI-pale low condensed euchromatic regions (light grey shaded), and normally condensed euchromatin (dark grey shaded). Genes were mapped by FISH and, heterochromatic and differentially condensed euchromatic regions were distinguished by DAPI counterstaining. The idiogram of A. hypogaea/A. monticola was redrawn from Seijo et al. 2004. In the diploid species, the chromosomes were ordered by morphology, then according to decreasing size. Some chromosomes within each idiogram were re-ordered according to tentative homeologies with other diploid species and on the basis of the nomenclature proposed for A. hypogaea/A. monticola (Seijo et al. 2004). Chromosomes with similar morphology that lack any characteristic landmark were arranged in groups. Scale bar 3 μm
The remaining species had karyotypes completely devoid of centromeric bands. However, while A. ipaënsis, Arachis sp. and A. magna were completely devoid of detectable heterochromatin, A. gregoryi, A. valida, and A. williamsii had one small interstitial or distal band in the short arms of pair 3 (Figs. 2, 3), which covered from 0.20 μm in A. williamsii to 0.40 μm in A. valida.
Chromosome mapping of the 5S and 45S rRNA genes by FISH
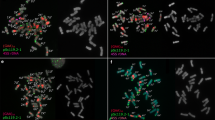
All the species had one pair of 5S rDNA loci localized in proximal position on the short arms of the metacentric pair 3 (Figs. 2, 3). Exceptions to this generality were A. batizocoi, in which these loci were interstitial, and A. gregoryi and A. valida, in which the loci were proximally located but on the long arms. In most species, this is the only pair of 5S rDNA clusters in the karyotypes, and each signal covered approximately one-fourth of the arm length (Fig. 3). However, A. cruziana, A. batizocoi, and A. krapovickasii had two additional pairs of loci (Figs. 2, 3), one of them proximally localized in the long arms of pair 10 and the other interstitially located in the long arms of pair 8 (Fig. 3). In A. batizocoi, the size and intensity of the three loci were similar, while in A. krapovickasii and A. cruziana they differed greatly. In A. krapovickasii, the signals in pair 8 were the largest and brightest, while the others were small and faint. In contrast, A. cruziana had conspicuous signals in pairs 3 and 8, but those of pair 10 were faint (Fig. 2).
The number of 45S rDNA loci ranged from two in A. gregoryi and A. trinitensis to four pairs in A. magna and A. valida (Fig. 2). Most of them were located in pericentromeric or interstitial position on the long arms. However, A. magna and A. benensis had one pair of 45S rDNA loci on the short arms, and A. ipaënsis had one pair in the subterminal position. Arachis valida and A. williamsii were characterized by two pairs of 45S rDNA loci on the long arms of pair 10, one of them close to the centromere and the other in a more distal position (Fig. 3).
In most species, the largest and brightest 45S rDNA loci corresponded to the clusters located in the secondary constrictions of the satellited chromosomes (pair 10), while the remaining ones were small and pale. In the cases of A. valida and A. williamsii, the largest clusters were located distally on pair 10 (Fig. 2). The secondary constrictions located on pair 9 of A. gregoryi were the only ones that did not hybridize with the 45S rDNA probe.
One 45S rDNA and one 5S rDNA loci were localized on the same chromosome in six species (Figs. 2, 3). In A. batizocoi, A. cruziana, and A. krapovickasii, these loci colocalized on the long arms of pair 10, while in A. benensis and A. magna they colocalized on the short arms of pair 3. In A. ipaënsis, both rDNA loci mapped to pair 3, but on different arms.
Karyotype pattern
In this study, we exploited the high number of chromosome markers detected to establish homologies with a high degree of accuracy for most of the pairs of each species complement. A further analysis of the conservative number, size, and distribution of the rDNA sites and the features of the heterochromatic bands between species was useful to determine tentative chromosome homeologies. According to the homeologies established, the species were arranged into three different groups (Fig. 3).
One of them includes A. batizocoi, A. cruziana, and A. krapovickasii and is characterized by having conspicuous heterochromatic bands in nine chromosome pairs and three 5S rDNA loci. A second one, integrated by A. benensis and A. trinitensis, has karyotypes with small and faint bands in seven or eight chromosome pairs and only one 5S rDNA locus. The last group is composed of all the species without pericentromeric heterochromatic bands in their karyotypes, namely, A. ipaënsis, Arachis sp., A. gregoryi, A. valida, A. williamsii, and A. magna.
Discussion
In the present work, in situ hybridization of rDNA and detection of heterochromatin were used to identify homologous chromosomes in 11 Arachis species. This information was used to suggest interspecific relationships among the species previously assigned to the B or non-A genome. The basic premise for this was that closely related species will have more similar karyotypes than will distantly related ones.
Heterochromatin distribution and rDNA loci localization
Our results demonstrated that the species included in the B genome, although diverse in their karyotype organization, can be arranged into three different groups on the basis of the presence and size of the heterochromatic bands. The group composed by A. batizocoi, A. cruziana, and A. krapovickasii is the most homogeneous, and the general pattern here observed coincides with that previously described for A. batizocoi (Seijo et al. 2004). The distribution of heterochromatic bands observed in this group show many similarities with those previously reported for the A genome species, particularly with the Chiquitano group (Robledo et al. 2009). Not only is the presence of heterochromatin in the centromeric region of nine of their chromosome pairs similar, but also the relative amount of heterochromatin present in each chromosome. The only outstanding differences with the A genome karyotypes are the morphology and dynamics of chromatin condensation of pair 9. In the A genome group, this pair corresponds to the “A chromosomes”, which are characteristically metacentric have the largest heterochromatic bands of the complement, and the distal euchromatic portions of their arms condensed allocyclically. In this group of B genome species, pair 9 also has the largest heterochromatic bands of the complement, but they are submetacentric, and their condensation proceeds in a fashion similar to the rest of the complement. The similarities in the general pattern of heterochromatin distribution observed between the chiquitano group of the A genome and the B genome group of species with heterochromatic bands raise new questions about the relationships between the species within the Arachis section.
The heterochromatin distribution described for A. benensis and A. trinitensis constitutes a new pattern for Arachis species and has two obvious particularities. One of them is the quite uniform amount of centromeric heterochromatin along the seven- or eight-banded chromosomes of the complement, contrasting with the heterogeneous sizes observed in the species of the A. batizocoi group. The other is that the total amount of centromeric heterochromatin present in the karyotypes of these species is approximately one half of that observed in other groups of Arachis species (Seijo et al. 2004; Robledo and Seijo 2008; Robledo et al. 2009). This reduction of heterochromatin is due not only to the absence of bands in two or three pairs of chromosomes, but also to the smaller size of the bands present in each chromosome.
The karyotypes without centromeric bands observed in A. ipaënsis, A. gregoryi, A. magna, A. valida, A. williamsii, and Arachis sp. are very particular and may have interesting phylogenetic implications. Since centromeric bands have been observed in all other species of the genus so far analyzed (Raina and Mukai 1999), this pattern probably constitutes a derived character within Arachis. Although the lack of detectable centromeric heterochromatin makes the group uniform, the presence of one interstitial or distal band on the short arm of pair 3 in A. gregoryi, A. valida, and A. williamsii, probably reflects a closer relationship among these taxa.
Concerning the nature of the heterochromatic bands, direct CMA/DAPI staining demonstrated that all the centromeric ones are composed of AT-rich sequences, that those of the secondary constrictions are composed of GC-rich sequences, and that the interstitial ones are of unknown composition (G. Seijo et al. unpublished data). This base composition of heterochromatic bands is in accordance with the composition of the heterochromatin reported for other species of Arachis (Seijo et al. 2004; Robledo et al. 2009). The preferential localization of heterochromatin in the centromeric region of all, or almost all, the chromosomes of the karyotypes is a common pattern in Arachis, and, according to Guerra (2000), is the rule for species with small chromosomes.
Considering the model of Schweizer and Loidl (1987), the equilocality of heterochromatin distribution can be attributed to mechanisms that operate above the level of individual chromosomes. The basic molecular processes, namely, amplification, transfer, and homogenization of sequences may be constrained by the chromosomal proximity determined by the ordered disposition of chromosomes in the mitotic (Rabl’s orientation) and meiotic (the bouquet polarization) nuclei. Similar predictions can be deduced from the Bennett (1983) nucleotype model, in which heterochromatin dispersion within a karyotype is constrained by heterologous chromosome pairing.
Ribosomal RNA gene clusters provided additional chromosome markers for the characterization of the 11 species analyzed. Our results coincide with some of the previously reported data for five B genome species (Raina and Mukai 1999; Seijo et al. 2004), although we revealed new rDNA loci for most of the taxa. In the case of A. williamsii and A. valida, the number of 5S rDNA loci agrees with that reported previously, although we detected one and two additional 45S rDNA loci, respectively. For A. ipaënsis, our results add one more 45S rDNA locus to those observed by Raina and Mukai (1999) and are consistent in number and location to that previously reported by our group (Seijo et al. 2004). For A. benesis, we observed the same number of rDNA loci as did Raina and Mukai (1999). Nonetheless, the 5S rDNA mapped to a different locus. The pattern found here for A. batizocoi is different from those available in the literature (Raina and Mukai 1999; Seijo et al. 2004). This fact suggests that materials of A. batizocoi conserved at different seed banks may have fixed different chromosome rearrangements, and that this species thus deserves a more detailed analysis including populations from different localities of its geographic range.
The comparative analysis of the rDNA loci showed that the presence of three 5S rDNA loci in some species constitutes an additional diagnostic feature of the group with large heterochromatic bands. However, the number, location, and size of the 5S rDNA loci in the other two groups of species were highly conserved, like the general pattern usually found in Arachis (Raina and Mukai 1999; Seijo et al. 2004). Concerning the 45S rDNA loci, although the pattern was conserved in the species with large heterochromatic bands, it was variable and useful for characterization in the other species.
Species groups and biogeography
One source of data that usually provides strong support for phylogenetic considerations is the biogeography of the species group. The B genome species of Arachis included within each karyotype group tend to have more similar distribution areas than do species belonging to different groups. Considering the biogeographic treatments of Bolivia (Navarro and Maldonado 2002) and South America (Cabrera and Willink 1973; Morrone 2001; Prado 1993; Spichiger et al. 2004), biogeographic and ecological segregation between species with different karyotype structures is also evident in most of the cases. In this sense, the species that have large centromeric bands grow in sand patches of the xerophytic forests characteristic of the northern and western edges of the Chaco Boreal biogeographic province. Two of these species, A. cruziana and A. krapovickasii, have overlapping distributions along the Chiquitano Meridional biogeographic district, in the Cerrado biogeographic Province of Bolivia (Navarro and Maldonado 2002). This area constitutes an ecotone between the biogeographic provinces of the Chaco Boreal and the Cerrado. The other species included in this group, A. batizocoi, grows westward of the Chaco Boreal, along the lower sub-Andean hills in the Subandino del Río Grande biogeographic district, in the Boliviano-Tucumana biogeographic province, southwest of Santa Cruz city. Its populations usually grow in patches of yellow-to-brown sand dunes, which are considered as paleochannels of the Parapetí River. Although this river now drains into the Izozog swamps, its paleochannels suggest an ancient connection with the Paraguay-Paraná river basin. During the Pleistocene, these paleochannels may have served as a dispersal pathway into the chacoan plains some hundred of kilometers away from the extant Bolivian populations of A. batizocoi. This hypothesis may explain the existence of some isolated populations of A. batizocoi in sand islands of the dry chacoan forest in northwestern Paraguay (Krapovickas and Gregory 1994).
The species included in the group without heterochromatic bands are mainly distributed over a large area of semi-deciduous forests and savannas of the cerrado associated with the Chiquitano highlands and peripheral mountain ranges of the western portion of the Brazilian Precambrian shield. This area is included within the Cerrado biogeographic province (Navarro and Maldonado 2002). Most of these Arachis species have disjunct ranges, probably as a consequence of the patchy distribution of different vegetation types in the region. Arachis magna, Arachis sp. and A. gregoryi grow in the central area of the Chiquitano plateau and extend toward the west of the Mato Grosso state in Brazil. However, they have never been observed growing in sympatry. Populations of A. valida have been collected eastward of this area on the slopes of the Precambrian Brazilian shield that surrounds the westside of the Brazilian Pantanal. Arachis williamsii grows in a disjunct small and elevated area of cerrado-like savannas in the floodplain of the Marmore River, close to Trinidad city, Bolivia. Although this area belongs to the El Beni biogeographic province, it is considered a relict of the Cerrado biogeographic province, nowadays surrounded by seasonally flooded rain forest (Navarro and Maldonado 2002). Arachis ipaënsis is the only species of the group not closely associated with the cerrado vegetation. The only known population of this species grows at the top of the sand banks of streams in an ecotone between the Tucumano-Oranence deciduous forest and the chacoan xerophytic forest, in the first slopes of the lower sub-Andean hills, some kilometers northward of Villamontes city, Bolivia.
The group of species with tiny centromeric bands includes two species with small distribution areas in the savannas of the cerrado surrounding Trinidad city (biogeographic province of El Beni). In this location, both species grow on sandy to clayey soils and share their distribution area with A. williamsii (of the formerly described group). This is the only case in which species of the B genome that have different karyotype structures have overlapping areas. Populations of A. trinitensis are known only from Trinidad city, while A. benensis has also been collected in Guayaramerín city, northern Bolivia. However, since this locality is very far from Trinidad and separated by seasonally flooded forest, anthropic dispersion is suspected (Krapovickas A, Instituto de Botánica del Nordeste, Argentina, personal communication).
As can be deduced from the above discussion, the species within each group tend to be co-distributed in different biogeographic regions, each with particular ecological characteristics. This situation brings two relevant aspects. One clearly suggests that each group of species is under different selective pressures, which may have aided, and are still probably influencing, the fixation of particular alleles, and in this way contributing to the diversification of the groups. The second is that, due to the particular reproductive characteristics of Arachis species, autogamy and geocarpy, the co-distribution of species within each group may be reflecting a common ancestry of the taxa. Further, the fact that all the species included in each of the groups here established have the smallest genetic distance estimated by molecular markers (Tallury et al. 2005; Burow et al. 2009), strongly support the common origin of the taxa included in each group.
Genome re-assignation of the non-A genome species
The A and B genomes in Arachis species were initially established by Smartt et al. (1978) on the basis of the existence of two different chromosome complements in the allotetraploid (AABB) A. hypogaea, as reported by Husted (1936). The A genome is characterized by the presence of “A chromosomes” and the B genome by their absence. Later, several authors reported the presence of “A chromosomes” in the karyotypes of some diploid species within the Arachis section, but not in others. Accordingly, a list of diploid species were assigned to the A genome, homologous to that described in A. hypogaea, whereas the remaining diploids were widely considered as belonging to the B genome or non-A group (Smartt et al. 1978; Stalker and Dalmacio 1981; Fernández and Krapovickas 1994; Lavia 1996; Seijo et al. 2004). Thus, the membership in the B or non-A genome was not defined by the presence of a specific marker common to all the species of the group, but by the mere absence of the “A chromosomes”. The analysis of several cross compatibility assays and data on molecular markers suggests a more complex genomic relationship between the species of this group (Seijo et al. 2004; Tallury et al. 2005; Burow et al. 2009).
Our results using molecular cytogenetics provide strong evidence for the existence of three different karyotype structures within the B group of species. Usually, when chromosome complements with differences of the magnitude observed here are put together in a hybrid nucleus, important cell processes become altered (Neves et al. 1997). In interphasic nuclei, the chromosome territories and genome dispositions (Heslop-Harrison and Bennett 1984; Leitch et al. 1991), functional domains (Jones and Hegarty 2010), epigenetic patterns (Rapp and Wendel 2005; Josefsson et al. 2006), and gene expression (Adams and Wendel 2005; Lai et al. 2006; Adams 2007; Michalak 2009) may be differently affected. In meiotic cells, homologous recombination and segregation are usually compromised (Darlington 1965; Stebbins 1971). All these mechanisms can potentially lead to hybrid inviability or sterility and, thus, to reproductive isolation.
Several works (Smartt et al. 1978; Krapovickas and Gregory 1994; Tallury et al. 2005; Burow et al. 2009) suggest that interspecific crosses between Arachis species of the same genome usually range between 25 and 70% of pollen stainability. Higher proportions of stained pollen usually have been taken into account to consider accessions as belonging to the same species (Stalker 1991; Krapovickas and Gregory 1994; Burow et al. 2009). By contrast, records of pollen stainability lower than 10% in F1 hybrids are widely accepted as indicative of intergenomic crosses. Burow et al. (2009) reported that viabilities between 12 and 25% are difficult to interpret because those values can be observed in F1 resulting from interspecific crosses (but mainly in reference to accessions of the widely distributed A. duranensis). However, values below 25% are usually observed in F1 obtained by intergenomic crosses, that is, between A genome and B genome species or between A. glandulifera (D genome) and any species of the A or B genomes (Stalker 1991; Krapovickas and Gregory 1994; Gregory and Gregory 1979).
In crosses between A. batizocoi and A. ipaënsis or A. williamsii, the pollen viability is always less than 4%, whereas between A. batizocoi and A. benesis all the seeds in F1 pods are aborted (Burow et al. 2009; Tallury et al. 2005). Also, crosses between A. gregoryi and A. benensis result in F1 with pollen viability less than 1% (Custodio 2009). All the analyses of pollen viabilities published for F1 resulting from crosses between B genome species with different karyotype structures have always revealed values lower than 10%. By contrast, hybrids between B genome species that share the same karyotype structure always have viabilities higher than 25%, except when A. gregoryi is involved in the crosses. These facts may support the hypothesis that the B genome groups of species established on the basis of karyotype structure may belong to different genomes.
The available data on meiotic chromosome pairing support this hypothesis. Bivalent formation at metaphases of hybrids obtained by crosses between species with the same genome (A genome) is usually higher than 9.5 out of the 10 expected. However, in intergenomic hybrids (between B genome and A genome species), the number of bivalents formed is lower than 7.5 (Tallury et al. 2005). For example, hybrids between A. batizocoi and A. herzogii or A. kempff-mercadoi (the latter of which are A genome species) showed 5.5 and 6.2 bivalents, whereas hybrids between A. duranensis (A genome) and A. batizocoi showed bivalents between 6.17 and 7.35 (Stalker et al. 1991). Similarly, values from 5.1 to 6.5 were obtained in crosses between A genome species and A. glandulifera of the D genome (Stalker 1991). Considering the crosses between the B genome species, a mean bivalent formation higher than 9.5 has been reported for the F1 obtained by crossing A. batizocoi × A. cruziana and for the F1 between A. ipaënsis and A. magna, both of which are crosses between species that share the same karyotype structure. However, F1 hybrids resulting from crosses between B genome species with different karyotype structures, like crosses between A. batizocoi and A. williamsii or between A. batizocoi and A. ipaënsis, formed 5.8 and 6.9–7.9 bivalents, respectively (Smartt et al. 1978; Stalker and Wynne 1979; Krapovickas and Gregory 1994; Tallury et al. 2005). Therefore, the number of bivalents formed in F1 obtained by crosses between species with large heterochromatic bands (A. batizocoi) and those without them (A. ipaënsis and A. williamsii) is in the range expected for intergenomic hybrids. On the other hand, homologous pairing in the hybrids between two species with similar karyotypes (A. cruziana and A. batizocoi) clearly reflects the values expected for an intragenomic F1.
Both sets of data, pollen stainability and chromosome pairing at meiosis of interspecific hybrids, indicate that B species with different karyotype structure have a high reproductive isolation, stronger than that usually expected for species belonging to the same genome. On these bases, we propose to segregate the species so far included within the non-A genome (widely considered as B genome) into three different genomes considering their karyotype structure. First, we propose that the group devoid of heterochromatic bands, which includes A. ipaënsis, A. magna, A. gregoryi, A. sp., A. valida and A. williamsii, should retain the B genome designation. This is based on the fact that the B genome was originally assigned to one of the chromosome complements of the cultigen (Smartt et al. 1978); thus the wild donor of this genome, A. ipaënsis, and all the relatives that share the same karyotype structure should belong to same genome.
Second, we propose to designate the group of species characterized by karyotypes with large centromeric bands and that includes A. batizocoi, A. cruziana A. krapovickasii as the K genome group. Similarly, we propose the F genome designation for the species that have karyotypes with tiny centromeric bands in seven or eight chromosome pairs, i.e., A. trinitensis and A. benesis.
Molecular analyses performed in section Arachis have usually included incomplete sets of non-A genome species (Milla et al. 2005; Tallury et al. 2005; Burow et al. 2009). However, in all of them, the groups of species without bands (i.e., the B genome as defined here) always formed a cluster separated from that formed by A. batizocoi, A. cruziana, and A. krapovickasii (K genome). Similarly, whenever A. benensis and A. trinitensis (F genome) were included in those analyses, they always grouped together and in a separate cluster (Milla et al. 2005). Therefore, the clustering of species observed in those dendrograms highly supports the genome arrangement defined here on the basis of karyotype features.
Genome donors of A. hypogaea
Traditionally, A. batizocoi was considered the most probable donor of the B genome to A. hypogaea, mainly because of its chromosome morphology and its ability to produce fertile hybrids with the cultigen (Singh and Smartt 1998). More comprehensive cytogenetic analysis has argued this hypothesis and proposed A. ipaënsis as the best B genome donor (Fernández and Krapovickas 1994). Further analyses using molecular markers, in situ hybridization of rDNA and GISH were concordant with the candidacy of A. ipaënsis as the best B genome donor (Kochert et al. 1996; Seijo et al. 2004; Burow et al. 2009). However, other species have been considered to be closely related to the B genome of peanut in the past two decades (Krapovickas and Lavia 2000; Milla et al. 2005), and a set of non-A species recently collected still remains poorly characterized.
In this sense, this is the first comprehensive analysis of the whole set of non-A genome species. Our detailed karyotype analysis supports A. ipaënsis as the most probable B genome donor because (1) its chromosomes are completely deprived of heterochromatic bands and (2) its pattern of rDNA loci is the most similar (among the wild diploids) to that observed in the complement without bands of the cultigen. Geographically, A. ipaënsis grows close to the most probable A genome donor (A. duranensis) and to the wild allotetrapoid A. monticola (Fig. 1), a fact that supports its candidacy as the progenitor of the cultigen.
Long-standing ideas about the center of origin of the cultivated peanut, which were based on the morphological variability of the landraces and biogeography (Gregory et al. 1980; Krapovickas and Gregory 1994), place the origin of the domesticated peanut in northern Argentina and southern Bolivia, in a transition area between the Tucumano-Boliviano forest and the Chaco lowlands (Fig. 1). The distributions of most of the putative A and B genome donors for A. hypogaea and the location of A. monticola in this area provide additional support for this hypothesis.
In summary, through the analysis of the heterochromatic bands and 45S rDNA loci patterns the present work provides the detailed karyotypes for the entire set of the non-A genome species known so far. On the basis of the karyotype structure of the species, we propose that they be grouped into three different genomes, B, F, and K. This arrangement is supported by data from molecular markers, cross compatibility, and bivalent formation at meiosis. Chromosome variability was geographically structured and provided additional support to the genome arrangement proposed. The results strongly hold A. ipaënsis as the most probable B genome donor of A. hypogaea and establish the group of species more closely related to the cultigen. We envision that the data will be of use to peanut breeders for the management of germplasm collections, selection of parents for hybridization, and the comprehensive use of the range of genetic variability present in wild species.
References
Adams KL (2007) Evolution of duplicate gene expression in polyploid and hybrid plants. J Hered 98:136–141
Adams KL, Wendel JF (2005) Allele-specific, bi-directional silencing of an alcohol dehydrogenase gene in different organs of interspecific diploid cotton hybrids. Genetics 171:2139–2142
Bennett MD (1983) The spatial distribution of chromosomes. In: Brandham PE, Bennett MD (eds) Kew chromosome conference II. Allen and Unwin, London, pp 71–79
Burow MD, Simpson CE, Starr JL, Paterson A (2001) Transmission genetics of chromatin from a synthetic amphidiploid to cultivated peanut (Arachis hypogaea L.): broadening the gene pool of a monophyletic polyploid species. Genetics 159:823–837
Burow MD, Simpson CE, Faries MW, Starr JL, Paterson A (2009) Molecular biogeographic study of recently described B- and A-genome Arachis species, also providing new insights into the origins of cultivated peanut. Genome 52:107–119
Cabrera AL, Willink A (1973) Biogeografía de América Latina. Organización de Estados Americanos, Serie Biología, Monografía Nº 13, pp 117
Custodio RC (2009) Relações de cruzabilidade entre espécies e acessos de germoplasma do gênero Arachis associados ao genoma B do amendoim (Arachis hypogaea L.). Universidade Federal de Santa Catarina, Florianópolis, Brazil, p 137
Darlington CD (1965) Cytology. J & A Churchill LTD, London
Fernández A, Krapovickas A (1994) Cromosomas y evolución en Arachis (Leguminosae). Bonplandia 8:187–220
Foster DJ, Stalker HT, Wynne JC, Beute MK (1981) Resistance of Arachis hypogaea L. and wild relatives to Cercospora arachidicola Hori. Oleagineux 36:139–143
Gimenes MA, Lopes CR, Galgaro ML, Valls JFM, Kochert G (2002) RFLP analysis of genetic variation in species of section Arachis, genus Arachis (Leguminosae). Euphytica 123:421–429
Gregory WC, Gregory MP (1976) Groundnut. Arachis hypogaea (Leguminosae-Papilionatae). In: Simmonds NW (ed) Evolution of crop plants. Longman Group Ltd, London, pp 151–154
Gregory MP, Gregory WC (1979) Exotic germoplasm of Arachis L. interspecific hybrids. J Hered 70:185–193
Gregory WC, Krapovickas A, Gregory MP (1980) Structures, variation, evolution and classification in Arachis. In: Summerfield RJ, Bunting AH (eds) Advances in legume science. Royal Botanic Gardens, Kew, London, pp 469–481
Guerra M (2000) Patterns of heterochromatin distribution in plant chromosomes. Genet Mol Biol 23:1029–1041
Halward TM, Stalker HT, Larue EA, Kochert G (1991) Genetic variation detectable with molecular markers among unadapted germplasm resources of cultivated peanut and related wild species. Genome 34:1013–1020
He G, Prakash CS (1997) Identification of polymorphic DNA markers in cultivated peanut (Arachis hypogaea L.). Euphytica 97:143–149
Herselman L (2003) Genetic variation among Southern African cultivated peanut (Arachis hypogaea L.) genotypes as revealed by AFLP analysis. Euphytica 133:319–327
Heslop-Harrison JS, Bennett MD (1984) Chromosome order—possible implications for development. J Embryol Exp Morphol 83(Supplement):51–73
Hilu KW, Stalker HT (1995) Genetic relationships between peanut and wild species of Arachis sect. Arachis (Fabaceae): evidence from RAPDs. Plant Syst Evol 198:167–178
Husted L (1936) Cytological Studies on the Peanut, Arachis. II. Chromosome number, morphology and behavior, and their application to the problem of the cultivated forms. Cytologia 7:396–423
Isleib TG, Holbrook CC, Gorbet DW (2001) Use of Arachis sp. plant introductions in peanut cultivar development. Peanut Sci 28:96–113
Johnson DR, Wynne JC, Campbell WV (1977) Resistance of wild species of Arachis to the two spotted spider mite, Tetranychus urticae. Peanut Sci 4:9–11
Jones RN, Hegarty M (2010) Order out of chaos in the hybrid plant nucleus. Cytogenet Genome Res 126:376–389
Josefsson C, Dilkes B, Comai L (2006) Parent-dependent loss of gene silencing during interspecies hybridization. Curr Biol 16:1322–1328
Kochert G, Halward T, Branch WD, Simpson CE (1991) RFLP variability in peanut (Arachis hypogaea L.) cultivars and wild species. Theor Appl Genet 81:565–570
Kochert G, Stalker HT, Gimenes M, Galgaro L, Moore K (1996) RFLP and cytogenetic evidence for the progenitor species of allotetraploid cultivated peanut, Arachis hypogaea (Leguminosae). Am J Bot 83:1282–1291
Krapovickas A, Gregory WC (1994) Taxonomía del género Arachis (Leguminosae). Bonplandia 8:1–186
Krapovickas A, Lavia GI (2000) Advances in the taxonomy of the genus Arachis. In: Proceedings of the American Peanut Research and Education Society, vol 32. APRES, Alabama, USA, p 46
Lai Z, Gross BL, Zou Y, Andrews J, Rieseberg LH (2006) Microarray analysis reveals differential gene expression in hybrid sunflower species. Mol Ecol 15:1213–1227
Lavia GI (1996) Estudios cromosómicos en Arachis (Leguminosae). Bonplandia 9:111–120
Lavia GI (1998) Karyotypes of Arachis palustris and A. praecox (section Arachis), two species with basic chromosome number x = 9. Cytologia 63:177–181
Lavia GI (2000) Chromosome studies of wild Arachis (Leguminosae). Caryologia 53:177–181
Leitch AR, Schwarzacher T, Mosgfiller W, Bennett MD, Heslop-Harrison JS (1991) Parental genomes are separated throughout the cell cycle in a plant hybrid. Chromosoma 101:206–213
Levan A, Fredga K, Sandberg AA (1964) Nomenclature for centromeric position on chromosomes. Hereditas 52:201–220
Mallikarjuna N (2002) Gene introgression from Arachis glabrata into A. hypogaea, A. duranensis and A. diogoi. Euphytica 124:99–105
Mallikarjuna N, Pande S, Jadhav DR, Sastri DC, Rao JN (2004) Introgression of disease resistance genes from Arachis kempff-mercadoi into cultivated groundnut. Plant Breed 123:573–576
Michalak P (2009) Epigenetic, transposon and small RNA determinants of hybrid dysfunctions. Heredity 102:45–50
Milla SR, Isleib TG, Stalker HT (2005) Taxonomic relationships among Arachis sect. Arachis species as revealed by AFLP markers. Genome 48:1–11
Moretzsohn MC, Hopkins MS, Mitchell SE, Kresovich S, Valls JFM, Ferreira ME (2004) Genetic diversity of peanut (Arachis hypogaea L.) and its wild relatives based on the analysis of hypervariable regions of the genome. BMC Plant Biol 4:11
Morrone JJ (2001) Biogeografía de América Latina y el Caribe. Manuales & Tesis Sociedad Entomológica Aragonesa, vol 3. Zaragoza, Spain, p 150
Moscone EA, Matzke MA, Matzke AJM (1996) The use of combined FISH/GISH in conjunction with DAPI counterstaining to identify chromosomes containing transgene inserts in amphidiploid tobacco. Chromosoma 105:231–236
Navarro G, Maldonado M (2002) Geografía ecológica de Bolivia, vegetación y ambientes acuáticos. Centro de ecología Simón Patiño, Cochabamba, Bolivia, p 719
Neves N, Castillo A, Silva M, Heslop-Harrison JS, Viegas W (1997) Genomic interactions: gene expression, DNA methylation and nuclear structure. In: Enriques-Gil N, Parker JS, Puertas MJ (eds) Chromosome Today, vol 12, pp 182–201
Paik-Ro OG, Smith RL, Knauft DA (1992) Restriction fragment length polymorphism evaluation of six peanut species within the Arachis section. Theor Appl Genet 84:201–208
Peñaloza A, Valls JFM (2005) Chromosome number and satellited chromosome morphology of eleven species of Arachis (Leguminosae). Bonplandia 14:65–72
Prado DE (1993) What is the Gran Chaco vegetation in South America? I. A review. Contribution to the study of the flora and vegetation of the Chaco. V. Candollea 48:145–172
Raina SN, Mukai Y (1999) Detection of a variable number of 18S-5.8S-26S and 5S ribosomal DNA loci by fluorescent in situ hybridization in diploid and tetraploid Arachis species. Genome 42:52–59
Raina SN, Rani V, Kojima T, Ogihara Y, Singh KP, Devarumath RM (2001) RAPD and ISSR fingerprints as useful genetic markers for analysis of genetic diversity, varietal identification, and phylogenetic relationships in peanut (Arachis hypogaea) cultivars and wild species. Genome 44:763–772
Rapp RA, Wendel JF (2005) Epigenetics and plant evolution. New Phytol 168:81–91
Robledo G, Seijo JG (2008) Characterization of Arachis D genome by FISH chromosome markers and total genome DNA hybridization. Genet Mol Biol 31:717–724
Robledo G, Lavia GI, Seijo G (2009) Species relations among wild Arachis species with the A genome as revealed by FISH mapping of rDNA loci and heterochromatin detection. Theor Appl Genet 118:1295–1307
Romero Zarco C (1986) A new method for estimating karyotype asymmetry. Taxon 35:526–530
Schwarzacher T, Ambros P, Schweizer D (1980) Application of Giemsa banding to orchid karyotype analysis. Plant Syst Evol 134:293–297
Schweizer D, Loidl J (1987) A model for heterochromatin dispersion and the evolution of C-band patterns. Chromosomes Today 9:61–74
Seijo JG, Lavia GI, Fernández A, Krapovickas A, Ducasse D, Moscone EA (2004) Physical mapping of 5S and 18S-25S rRNA genes evidences that Arachis duranensis and A. ipaënsis are the wild diploid species involved in the origin of A. hypogaea (Leguminosae). Am J Bot 91:1294–1303
Seijo JG, Lavia GI, Fernández A, Krapovickas A, Ducasse DA, Bertioli DJ, Moscone EA (2007) Genomic relationships between the cultivated peanut (Arachis hypogaea–Leguminosae) and its close relatives revealed by double GISH. Am J Bot 94:1963–1971
Simpson CE (2001) Use of wild Arachis species/introgression of genes into A. hypogaea L. Peanut Sci 28:114–116
Simpson CE, Starr JL (2001) Registration of ‘COAN’ peanut. Crop Sci 41:918
Singh AK (1986) Utilization of wild relatives in the genetic improvement of Arachis hypogaea L. Part 8. Synthetic amphidiploids and their importance in interspecific breeding. Theor Appl Genet 72:433–439
Singh AK, Moss JP (1982) Utilization of wild relatives in genetic improvement of Arachis hypogaea L. Part 2. Chromosome complements of species of section Arachis. Theor Appl Genet 61:305–314
Singh AK, Moss JP (1984) Utilization of wild relatives in genetic improvement of Arachis hypogaea L. Part 5. Genome analysis in section Arachis and its implications in gene transfer. Theor Appl Genet 68:355–364
Singh AK, Smartt J (1998) The genome donors of the groundnut/peanut (Arachis hypogaea L.) revisited. Genet Resour Crop Evol 45:113–118
Smartt J, Gregory WC, Gregory MP (1978) The genomes of Arachis hypogaea. 1. Cytogenetic studies of putative genome donors. Euphytica 27:665–675
Spichiger R, Calenge C, Bise B (2004) Geographical zonation in the Neotropics of tree species characteristic of the Paraguay–Paraná Basin. J Biogeogr 31:1489–1501
Stalker HT (1991) A new species section Arachis of peanuts with D genome. Am J Bot 78:630–637
Stalker HT, Dalmacio RD (1981) Chromosomes of Arachis species, section Arachis (Leguminosae). J Hered 72:403–408
Stalker HT, Wynne JC (1979) Cytology of interspecific hybrids in section Arachis of peanuts. Peanut Sci 6:110–114
Stalker HT, Dhesi JS, Parry DC, Hahn JH (1991) Cytological and interfertility relationship of Arachis section Arachis. Am J Bot 78:238–246
Stebbins GL (1971) Chromosomal evolution in higher plants. Addison-wesley, Reading, MA
Subramanian V, Gurtu S, Nageswara Rao RC, Nigam SN (2000) Identification of DNA polymorphism in cultivated groundnut using random amplified polymorphic DNA (RAPD) assay. Genome 43:656–660
Tallury SP, Hilu KW, Milla SR, Friend SA, Alsaghir M, Stalker HT, Quandt D (2005) Genomic affinities in Arachis section Arachis (Fabaceae): molecular and cytogenetic evidence. Theor Appl Genet 111:1229–1237
Valls JFM, Simpson CE (2005) New species of Arachis from Brazil, Paraguay, and Bolivia. Bonplandia 14:35–64
Acknowledgments
This work was supported by Consultative Group on International Agricultural Research, Challenge Program, subprogram “Trait capture for crop improvement”, period 2005-2008; and Agencia Nacional de Promoción Científica y Técnica, PICT 2005 Nº34458 and PICT 2007 Nº1356, Argentina. We would like to thank Dirección General de Biodiversidad, Ministerio de Desarrollo Sostenible y Planificación, Bolivia, for authorizing botanical collections; INTA Manfredi Station, Córdoba, Argentina; the Texas Agriculture Experimental Station, Stephenville, Texas, USA; and Embrapa Recursos Genéticos e Biotecnologia (CENARGEN), Brasília-DF, Brazil, for providing seeds of some accessions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Heslop-Harrison.
Rights and permissions
About this article
Cite this article
Robledo, G., Seijo, G. Species relationships among the wild B genome of Arachis species (section Arachis) based on FISH mapping of rDNA loci and heterochromatin detection: a new proposal for genome arrangement. Theor Appl Genet 121, 1033–1046 (2010). https://doi.org/10.1007/s00122-010-1369-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1369-7