
Abstract
Diversity analyses in alfalfa have mainly evaluated genetic relationships of cultivated germplasm, with little known about variation in diploid germplasm in the M. sativa–falcata complex. A collection of 374 individual genotypes derived from 120 unimproved diploid accessions from the National Plant Germplasm System, including M. sativa subsp. caerulea, falcata, and hemicycla, were evaluated with 89 polymorphic SSR loci in order to estimate genetic diversity, infer the genetic bases of current morphology-based taxonomy, and determine population structure. Diploid alfalfa is highly variable. A model-based clustering analysis of the genomic data identified two clearly discrete subpopulations, corresponding to the morphologically defined subspecies falcata and caerulea, with evidence of the hybrid nature of the subspecies hemicycla based on genome composition. Two distinct subpopulations exist within each subsp. caerulea and subsp. falcata. The distinction of caerulea was based on geographical distribution. The two falcata groups were separated based on ecogeography. The results show that taxonomic relationships based on morphology are reflected in the genetic marker data with some exceptions, and that clear distinctions among subspecies are evident at the diploid level. This research provides a baseline from which to systematically evaluate variability in tetraploid alfalfa and serves as a starting point for exploring diploid alfalfa for genetic and breeding experiments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alfalfa is grown on over 32 million hectares worldwide (Michaud et al. 1988). Cultivated alfalfa has been improved from a complex taxonomic group known as the Medicago sativa–falcata complex, which includes both diploid (2n = 2x = 16) and tetraploid (2n = 4x = 32) taxa. Ploidy, flower color, pod shape, and pollen shape have traditionally been used to differentiate taxa in the complex. The diploid members of the complex include M. sativa subsp. falcata, with yellow flowers and sickle-shaped pods, M. sativa subsp. caerulea, with purple flowers and pods having multiple coils, and their natural hybrid, M. sativa subsp. hemicycla, with variegated flower color and partially coiled pods. The tetraploid subspecies in the complex include M. sativa subsp. sativa (the direct analogue of diploid subsp. caerulea), M. sativa subsp. falcata, and the tetraploid hybrid M. sativa subsp. varia (Quiros and Bauchan 1988). Hybridization among taxa is possible even across ploidy levels by unreduced gametes (McCoy and Bingham 1988). The genetic validity of current morphology-based classification of subspecies has not been confirmed.
The exploration of genetic diversity and population structure in alfalfa has generally focused on tetraploid breeding populations or progenitor germplasms. Nine progenitor germplasms are accepted to be the major sources of contemporary US alfalfa cultivars: falcata, Ladak, Flemish, Turkistan, Indian, African, Chilean, Peruvian, and varia (Barnes et al. 1977). M. sativa subsp. falcata and Peruvian are distinct from the others (Kidwell et al. 1994; Segovia-Lerma et al. 2003). However, modern cultivars appear to have diverged from the historical introductions based on molecular marker analyses (Maureira et al. 2004; Vandemark et al. 2006). Markers have been proven to be useful to distinguish among Italian populations and ecotypes (Pupilli et al. 1996, 2000), between Italian and Egyptian cultivars (Mengoni et al. 2000), and between diploid falcata and caerulea (Brummer et al. 1991), but less so among cultivars deriving from a single breeding program (Flajoulot et al. 2005). Sequence differences have been used to distinguish falcata and sativa or caerulea genotypes (Havananda et al. 2010).
A comprehensive study of the molecular genetic variation present in diploid germplasm would be useful for determining whether morphologically based taxonomic classifications reflect patterns of genomic differentiation. This information would be a useful baseline from which diversity among tetraploid populations could be interpreted. It would also provide information on the population structure, allelic richness, and diversity parameters of diploid germplasm to help breeders use genetic resources for cultivar development more effectively. Although alfalfa has large variability at the tetraploid level, genetic analyses of diploids are more tractable and diploid evaluations could contribute to alfalfa improvement. Therefore, we selected a broad range of unimproved diploid M. sativa accessions from throughout the Northern Hemisphere in order to elucidate the population structure of the diploid M. sativa–falcata species complex, to test the concordance between current morphology-based classification and differentiation based on SSR markers, and to infer the extent of genetic diversity that exists in diploid accessions.
Materials and methods
Sampling of wild diploid alfalfa populations
We initially screened 256 accessions from the USDA National Plant Germplasm System using flow cytometry to identify those that were diploid. Ploidy was determined on a bulk of four genotypes per accession using previously described methods (Brummer et al. 1991) on a Cytomics FC 500 (Beckman-Coulter, Fullerton, CA) flow cytometer at the UGA Flow Cytometry Facility and has been submitted to the USDA-NPGS. If ploidy variation was observed in the bulked sample, each of the four genotypes was tested independently. As a result of the flow cytometry analysis, we selected 384 genotypes from 122 accessions found to be diploid; the tetraploid accessions and individuals were not considered further. These accessions represent the wide geographical distribution of caerulea, falcata, and hemicycla, including 57 caerulea (193 individual genotypes), 4 hemicycla (9 individuals), and 61 falcata (172 individual genotypes) according to their classification in the Germplasm Information Resource System (GRIN) when we began this study (Table 1). Accessions were represented by one to four individual genotypes, with 60 accessions having four individuals, 30 accessions having three, 16 accessions having two, and 15 accessions having one. One accession was represented by seven genotypes because we included it as both PI 641380 and W6 4794, which we later found to be the same. Seeds were germinated and plants grown in the greenhouse at Iowa State University and at the University of Georgia.
Because the main character used to distinguish among the current taxa is flower color, we recorded flower color from each of the genotypes to check agreement of flower color with the GRIN classification. Yellow flowered accessions were considered as falcata and purple flowered accessions as caerulea. Accessions with variegated flowers were listed as hemicycla. A secondary distinguishing characteristic is pod shape, with falcata having sickle-shaped (falcate) pods and caerulea having coiled pods. We harvested pods from 50 caerulea, 50 falcata, and 21 hemicycla genotypes and recorded the coil number for ten individual pods per genotype and computed a mean coiling value. Pods were scored in 1/4 coil intervals for the number of coils, ranging from completely straight (0 coils) to four coils.
DNA extraction and SSR genotyping
DNA was extracted from all 384 genotypes using young leaves from greenhouse grown plants, which were freeze-dried and ground to a powder. Genomic DNA was extracted following the CTAB method (Doyle and Doyle 1990).
We selected 89 SSR markers from those used in previous studies in alfalfa (Diwan et al. 2000; Julier et al. 2003; Robins et al. 2007) that were easy to score across the diversity in this population. The M13 tailing method described by Schuelke (2000) was used to label PCR products. Each SSR marker was amplified by PCR independently using the protocol described by Julier et al. (2003) and Sledge et al. (2005). We pooled PCR products of 4–6 reactions for genotyping on an automated ABI3730 sequencer at the UGA DNA Sequencing Facility. Allele scoring was performed using GENEMARKER software (SoftGenetics, State College, PA). Since we used diploid accessions, we scored genotypes in a biallelic genotypic format. The number of missing genotypes per marker was less than 5%. Based on marker profiles, ten of the 384 genotypes appeared to be tetraploid. We retested them with flow cytometry, confirmed their tetraploid status, and removed them from subsequent data analyses. Thus, all the results reported below are based on 374 individual diploid genotypes derived from 120 accessions.
Data analyses
In order to infer the population structure of the entire set of genotypes without regard to the preexisting subspecies classification or geographical information, we used the software program Structure (Pritchard et al. 2000). Structure provides a model-based Bayesian approach to infer population structure by using our entire SSR marker dataset to identify K clusters to which the program then assigns each individual genotype. Initially we used all 374 genotypes to deduce the optimal value of K (i.e., the number of clusters) by evaluating K = 1–10. In our model, admixture was allowed and the allele frequencies were assumed to be correlated, since it is more realistic to assume common ancestry of such closely related populations. The length of burn-in Markov Chain Monte Carlo (MCMC) replications was set to 10,000 and data were collected over 100,000 MCMC replications in each run, based on previous literature suggesting that this level is sufficient (Evanno et al. 2005). We identified the optimal value of K using both the ad hoc procedure described by Pritchard et al. (2000) and the method developed by Evanno et al. (2005).
To confirm the results obtained from Structure, we conducted two additional analyses enabling us to visualize the distribution of individual genotypes. First, we conducted a principal components analysis (PCA) using GenAlEx (Peakall and Smouse 2001) and plotted genotypic values for the first two principal component estimates. Second, we computed a genetic distance matrix (Saitou and Nei 1987) and created a neighbor-joining dendrogram, using the software program POPULATIONS version 1.2.28 (O. Langella 1999 unpublished, http://www.pge.cnrs-gif.fr/bioinfo/populations/index.php).
We used Analysis of Molecular Variance (AMOVA) to partition molecular genetic variance within and among accessions and among the five populations suggested by our Structure analysis. We used the 106 accessions which included two or more genotypes for the AMOVA analyses in order to be able to compute within-accession variance. We conducted the AMOVA using the software program GenAlEx 6.1 (Peakall and Smouse 2001). Several measures of diversity were computed across all accessions and also within subgroups of accessions. The diversity measures included the major allele frequency and the average number of alleles and genotypes per SSR locus, observed heterozygosity (H0), gene diversity (i.e., expected heterozygosity, He), and polymorphism information content (PIC). Computations were conducted using the software program Power Marker v3.23 (Liu 2002).
Results
Morphological analysis and correction of misclassifications
We initially defined the subspecies to which a particular accession belonged based on the classification in the GRIN system. However, accessions are occasionally misidentified in GRIN, so we clarified this assignment based on the flower color data we recorded. Falcata are typically defined as having yellow flowers, caerulea as having blue or purple flowers, and hemicycla as having variegated flowers (Lesins and Lesins 1979; Quiros and Bauchan 1988). Because one of the goals of our experiment was to test the validity of flower color to distinguish genetically discrete groups, we reclassified accessions (and genotypes) according to their flower color prior to any further analysis.
Three accessions classified as falcata in GRIN (PI464726, PI464727, PI631814) have purple flowers and should be classified as caerulea. The Afghan accession PI222198 was classified as caerulea in GRIN, but based on its yellow flower color should be classified as falcata. PI641603, which was initially classified as caerulea in GRIN, included one individual genotype with variegated flowers and on that basis, we defined the entire accession as hemicycla. These taxonomic classification corrections were used for our analysis below.
Pod shape is another morphological trait often used to differentiate alfalfa subspecies, with falcata accessions typically having sickle-shaped pods with less than one coil, caerulea pods having multiple coils, and hemicycla falling in between (Quiros and Bauchan 1988). The mean pod coiling values of our genotypes based on the taxonomic classification corrected for flower color showed that falcata had a mean of 0.2 ± 0.2 coils per pod, hemicycla 1.1 ± 0.5, and caerulea 2.0 ± 0.4, as anticipated. These values are significantly (P ≤ 0.05) different from one another based on least significant difference. However, we observed four accessions (PI464728, PI634119, PI634136, and PI634174) that dramatically deviated from flower color and pod shape expectations (see Table 1). For example, PI464728 had yellow flowers but the mean coil number per pod was 1.3, and PI641119 had purple flowers and a mean coil number per pod of 0.8.
Population structure
The second order statistics developed by Evanno et al. (2005) for Structure in order to assess the number of subpopulations identified the optimal value of K = 2 (Fig. 1a). This suggested that the set of genotypes was partitioned into two clusters, which corresponded to the genomes of the falcata and caerulea subspecies (Fig. 2a). Because individual genotypes had varying proportions of their genome from each cluster, we arbitrarily classified genotypes as follows: 0–30% caerulea genome = falcata; 31–69% caerulea genome = hemicycla, and 70–100% caerulea genome = caerulea (Fig. 2a). We also used the ad hoc procedure described by Pritchard et al. (2000), and in this analysis, a clear plateau was reached when K was five (Fig. 1b). At K = 5, two subgroups were nested within each of the falcata and caerulea subpopulations, with the hemicycla group as a separate cluster (Fig. 2b).


Diploid alfalfa population structure based on Bayesian inference among 374 individual genotypes analyzed with 89 SSR markers assuming a two clusters, K = 2 or b five clusters, K = 5, and the taxonomic association of clusters. c Identification of hemicycla genotypes whose genome composition differs from the rest of the subpopulation
Based on K = 2 and our genome proportion classification scale, 44 individuals were placed into the hemicycla group. Three accessions (PI641615, PI641619, and PI634111) initially identified in GRIN as hemicycla were indeed hybrids based on genome composition. All three individuals from PI641603 that we redefined as hemicycla based on flower color indicated a nearly even mixture of genomic backgrounds from each subspecies. Six accessions from Kazakhstan (PI634119, PI634136, PI634174, PI634176, PI641601, and 641606) and a Russian accession (PI315460) had purple flowers and were classified as caerulea in GRIN, but they had a hybrid genome pattern. The mean number of pod coils of the Kazakhstani accessions was less than or equal to one, in concordance with genomic data. The Russian accession (PI315460) had a mean pod coiling of 1.7. These results suggest that the dominant purple flower color is not sufficient to identify accessions as caerulea, and we recommend that these accessions be reclassified as hemicycla. Additionally, three of four individual genotypes belonging to the Georgian accession PI577543 showed a hybrid genome composition, suggesting that this accession should also be changed from caerulea to hemicycla.
Although many hemicycla genotypes had a similar genome based on clustering assuming K = 5, some or all of the individual genotypes from PI315460, PI577548, PI464727, PI464728, and PI631814 showed an interesting genome composition (Fig. 2c). When K = 2, these genotypes showed mixed falcata and caerulea genomes, indicating that they are really hybrids. However, when K = 5, these genotypes did not cluster with the common “hemicycla” genome (colored pink in Fig. 2b), but rather consisted of hybrid patterns with different amounts of genome composition from the four caerulea and falcata groups. Thus, while it appears these accessions are hemicycla in the sense of having both caerulea and falcata genomes, each genotype has a distinct genome composition. One individual genotype of the Russian accession PI577548 indicated a genome composition of approximately 50% from each of the two subsp. falcata and caerulea, but the other three genotypes of the accession had a genome of predominantly caerulea. We suggest that this accession remain classified as caerulea, although its genome indicates some admixture.
The reassignment of accessions based on genome composition also resolved flower color–pod shape disagreements. All of the four accessions that showed disagreement of flower color and pod shape data (Table 1) were found to have appreciable genomic contributions from both caerulea and falcata and were therefore reclassified as hemicycla. Based on the comparison of genome composition with flower color and pod shape, we found that yellow flowers and pods with one coil or more are a strong indication that an accession is hemicycla. Falcata accessions have yellow flowers and pods with fewer than one coil. The purple flowered accessions are harder to assign to either caerulea or hemicycla based on pod coiling. In general, caerulea accessions have pods with more than 1.5 coils. If hemicycla accessions have purple flowers, they tend to have pods with fewer than 1.5 coils.
Based on the morphological analysis and genome composition data, we assigned genotypes into subspecies and subgroups (Table 1). Of the 374 genotypes, 168 were caerulea, 162 falcata, and 44 hemicycla. Within caerulea, group A had 99 genotypes and group B 69 genotypes. Falcata A had 100 genotypes and falcata B had 62 genotypes.
Principal component analyses and neighbor-joining tree
We conducted a principal components analysis to further assess the population subdivisions identified using Structure. The first principal component explained 62% and the second principal component explained 11% of the SSR variation among the 374 genotypes. Plotting the first two principal components and color coding genotypes according to the five groups identified using Structure shows the clear separation of falcata and caerulea and the intermediate position of the hybrid hemicycla (Fig. 3). Moreover, falcata accessions are clearly divided into two subgroups. Caerulea accessions form one large cluster with evidence of a weak separation of the two subpopulations visible based on the color coding (Fig. 3).

Differentiation of genotypes from five diploid alfalfa populations based on the first two principal components derived from an analysis of 89 SSR markers
Finally, we constructed a neighbor-joining tree, as a third means to visualize relationships among the diploid genotypes. The tree showed a pattern consistent with the two analyses above. Falcata and caerulea accessions are clearly separated and hemicycla genotypes show a clear hybrid pattern. The two falcata subgroups that are suggested by Structure are also indicated in the tree. The partitioning of caerulea is also evident, albeit less clearly (Fig. 4). Genotypes from the same accession are often but not exclusively in close proximity on the dendogram. In one case, genotypes of a hemicycla accession (PI577543) were placed into different subspecies. We observed a cluster that was separated from southern caerulea that includes 22 genotypes from 7 accessions collected from the eastern end of Turkey in close proximity to each other (Kars, Ağrı and Erzurum provinces) and they grouped with genotypes from two Iranian accessions without accurate provenance information. A tree with similar overall topology was also generated using the software program TASSEL (Bradbury et al. 2007; data not shown), even though this program uses a simple substitution model rather than a stepwise mutation model. In summary, with some minor differences, the same general clustering pattern was observed using a variety of independent methods, providing strong support for the subspecific relationships we propose.

Neighbor-joining dendrogram of 374 individual genotypes from 120 wild diploid accessions of M. sativa. Falcata A (lowland falcata) = brown; Falcata B (upland falcata) = green; Caerulea A (southern caerulea) = red; Caerulea B (northern caerulea) = light blue; and Hemicycla = dark blue
Classification of genotypes
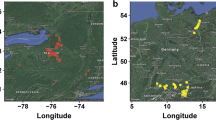
All three approaches clearly separated falcata from caerulea along with confirmation of the hybrid nature of hemicycla. Differentiation within each of subspecies was also evident; falcata and caerulea were further subdivided into two groups. In order to interpret the separation within subspecies, we noted the habitat and geographical location from which each accession was collected. Not all accessions had accurate provenance information in GRIN, but the available information revealed patterns of ecological differentiation that corresponded to the genetic groupings. The two caerulea groups largely correspond to Northern and Southern regions of Eurasia (Fig. 5). Caerulea A corresponded of Southern caerulea whereas caerulea B corresponded to Northern caerulea. All Russian, Georgian, Armenian, and Former Soviet Union accessions for which we do not have exact collection location information fell into the Northern group (Fig. 5). Accessions that were received from Canada but with an unknown original location of collection (PI571551 and PI571552) were also in the northern group. The Southern group included Turkish, Iranian, and Uzbekistani accessions, representing the southern end of the natural distribution of caerulea. Accessions from Kazakhstan were clustered into both groups depending on the location of collection. The accessions that were collected from northern Kazakhstan (Aqtobe) grouped with the Northern cluster, whereas the accessions collected from southern Kazakhstan (Dzumbul) were placed in the southern group. Three Kazakh accessions without exact collection location information (PI631922, PI631925, and PI577541) clustered with Northern caerulea. Turkish accessions generally clustered with the Southern group, but Turkish accession PI464726 grouped with the Northern cluster (Fig. 5).

Map indicating the collection locations of the caerulea and falcata genotypes evaluated in this experiment
The separation of 56 falcata accessions into two groups based on Structure analysis was more evident compared with the separation of caerulea, but this clear differentiation was not related to any pattern of regional separation that we could determine, unlike the case for caerulea accessions. GRIN provided precise location of collection information for 13 of 19 accessions in the first cluster (falcata A). Of these, six were collected from locations around rivers or wetlands, two accessions were gathered from coastal areas, two were from areas surrounded by dense forest, and three were collected from low-elevation areas currently inhabited by humans. We have precise collection location information for 18 of 33 accessions in the second cluster (falcata B). Fourteen of these were collected from rocky mountain ranges or dry slopes. The two Mongolian accessions (PI641543 and PI641544) were collected from high elevations (732 and 762 m, respectively) around wheat fields. The Ukrainian accession PI634106 was collected from a small peninsula with elevation of around 100 m; however, from a habitat described as a north slope, moderately steep, and on a cliff. The Russian accession PI631666 was collected from a moist stream terrace.
Based on this information, we can infer that the first group of falcata (falcata A) was collected from lowlands and locations close to river basins or the sea coast and we named this ecotype as the lowland ecotype. The second group (falcata B) was collected from dry slopes of mountain ranges and/or high elevations; therefore, we denoted them as the highland ecotype. Sinskaya (1961) classified former USSR falcata accessions into ecotypes based on a combination of geography and ecology. She identified a floodland ecotype, steppe ecotype, submontane ecotype, mountain ecotype, and forest-steppe ecotype, and these may relate in general terms to our upland and lowland ecotypes.
All individuals from five accessions (PI577558, PI538987, PI631568, PI577555, and PI631707) showed a hybrid genome pattern between the two falcata groups. Accession PI631568 was collected from a valley between a lowland plain and the Italian Alps. The two Russian accessions PI577558 and PI538987 were collected from the border of a river basin and dry plains. The Ukrainian accession PI577555 was collected from a cultivated plain at 100 m elevation, very close to the edge of a wetland. The Chinese accession PI631707 was collected from a mountain base at an elevation of 1,700 m but close to the Yining River basin in Xinjiang. All of these locations represent potential hybrid zones.
Analysis of molecular variance
In our AMOVA analysis, 19% of the total genetic variance was explained by the five groups, 16% was among accessions and the remaining 65% resided within accessions (Table 2). Although most of the genetic variance was among individuals within an accession, the differentiation of the accessions and groups was highly significant (P = 0.001). The pairwise ΦPT values (analogous to F ST) ranged from 0.059 (between the hemicycla and caerulea B subgroups) to 0.294 (between caerulea A and falcata B), and each of the pairwise ΦPT values was significantly different from zero according to tests based on 9,999 random permutations (P < 0.0001) (Table 3).
Diversity measurements
We computed diversity statistics across all 374 individual genotypes, within each of the three subspecies, and within the two subgroups within subspecies. The overall number of alleles per SSR locus across all genotypes ranged from 6 to 53 with a mean of 19.3; the mean number of genotypes per SSR locus was 52.8 (Table 4). Although caerulea had more genotypes than falcata (168 vs. 162), both the average number of alleles and average number of genotypes per SSR locus were higher in falcata. No obvious differences were evident among the subpopulations with either subspecies when taking the different number of individuals into account. Over all genotypes, observed heterozygosity was 0.46, ranging from 0.12 to 0.84, and was slightly higher in falcata than caerulea, with hemicycla intermediate. Gene diversity, a measurement of expected heterozygosity, was higher in all cases compared with observed heterozygosity. The PIC mean polymorphism level of loci over all genotypes was 0.71, again with falcata higher than caerulea, indicating more allelic diversity.
Discussion
The classification of taxa in the M. sativa–falcata complex as species or subspecies has been controversial (Sinskaya 1961; Lesins and Lesins 1979; Ivanov 1988; and Quiros and Bauchan 1988). Sinskaya (1961) denoted caerulea, hemicycla, falcata, and sativa as species and considered them along with a number of other taxa as a “circle of species.” Sinskaya (1961) first divided taxa based on ploidy, assuming that taxa within the same ploidy level were more closely related. Lesins and Lesins (1979) also classified falcata and sativa as different species of Medicago; however, they relegated hemicycla and caerulea to the subspecific level. They also noted that there exist no hybridization obstacles between falcata and sativa despite their obvious morphological distinctness. More recently, all these taxa have been given subspecific status within the M. sativa–falcata complex (Quiros and Bauchan 1988), and this nomenclature has been adopted by the NPGS and used in GRIN. Based on several different analyses of our molecular marker data, we found that falcata and caerulea were clearly distinguished at the genetic marker level, and that hemicycla, the putative hybrid between caerulea and falcata, had a hybrid genome pattern. The separation between diploid falcata and caerulea was previously observed in a narrow germplasm examination using RFLPs (Brummer et al. 1991), as well as in a more recent examination using nuclear and organellar DNA sequences (Havananda et al. 2010). However, another study evaluating two nuclear genes did not show clear differentiation between the subspecies (Muller et al. 2005). Neither of these experiments explicitly addressed the status of hemicycla genotypes. In addition to the clear separation of the three subspecies, we found that falcata and caerulea are each further clustered into two distinct groups. The two caerulea groups corresponded to northern and southern locales, reflecting geographic distribution. Based on available provenance information, we suggest that ecogeography underlines the differentiation of the two falcata clusters, which we designated as lowland (falcata A) and upland (falcata B) ecotypes.
We initially used the NPGS-GRIN nomenclature for accessions, but noted a number of misclassifications based on flower color and ploidy of each individual genotype. Although flower color is an informative character for assigning individual genotypes to either caerulea or falcata, it is difficult to separate either from hemicycla. Because morphological traits are governed by a small number of genes, they may fail to give a clear view of hybridization events. Therefore, we conclude that in order to accurately define a genotype’s subspecific status, genomic data are needed. Since the degree of genome admixture is continuous, we defined an arbitrary genome composition cut-off point to classify an individual as hybrid or not. If a genotype did not have more than 70% of its genome derived from either falcata or caerulea, then we defined it as hemicycla, regardless of flower color.
The individual genotypes classified as sativa or falcata based on phenotype, but which we denoted as hemicycla based on genome composition, derived from locations of sympatry between caerulea and falcata. Therefore, extensive gene flow between the subspecies in these regions is expected. Two regions of sympatry we identified are the Kars province of northeastern Turkey and Aqtobe, Kazakhstan, where almost the entire USDA hemicycla collection was obtained. Little differentiation between falcata and caerulea or sativa was noted based on mitochondrial DNA variation (Muller et al. 2003). Although many of the accessions Muller et al. (2003) evaluated were tetraploid, their results suggest that gene flow between the subspecies occurs and our results are congruent with that explanation. Most of the accessions denoted as hemicycla in GRIN that we evaluated are in fact tetraploid and hence belong to M. sativa subsp. varia.
A particularly interesting result of our experiment is that of the two types of hemicycla genomes we identified in our Structure analysis. In addition to a clear hemicycla group with a common genomic constitution (colored pink in Fig. 2), we also identified a second group consisting of a number of individuals that appear to be recent hybrids and whose ancestries could be identified (Fig. 2c). In some species, hybrids are differentiated from their parents by morphological and/or ecological characteristics that enable the hybrids to form their own distinct population and ultimately species (Gross et al. 2003; Ma et al. 2006; Arnold et al. 1990). In our experiment, the core hemicycla group appears to have diverged over the time and formed a unique population, suggesting that it is a true subspecies of the M. sativa–falcata complex. The other hemicycla individuals have genomes that are simply mixtures of the two subspecies. These hybrids could have occurred in the field within sympatric populations of falcata and caerulea, or they could have been produced in seed increases at NPGS following collection. In any case, further investigation on the development and persistence of hybrids, and whether the newly formed hybrids would evolve toward the hemicycla genome or revert to one parental subspecies would be an interesting research avenue to pursue.
Most outcrossing plant species show large amounts of intra-population genetic variation, and cultivated alfalfa is no exception (e.g., Flajoulot et al. 2005). Our experiment is the first genome-wide marker evaluation of diploid alfalfa germplasm, and we showed abundant variation within the wild gene pool, even within accessions, despite only assaying 2–4 individual genotypes in most of them. Previous genetic diversity assessment studies in alfalfa have focused mainly on tetraploid cultivated germplasm. Although comparisons between experiments for characteristics such as the average number of alleles per SSR marker is fraught with many difficulties, we noted more alleles in our 374 genotypes than in similarly sized populations of more narrow cultivated material. For example, Flajoulot et al. (2005) evaluated 209 tetraploid individuals (essentially the same number of chromosomes as we evaluated in our experiment) from seven cultivars and one breeding pool using eight SSR markers and found that the number of alleles per SSR locus ranged from 3 to 24 with a mean of 14.9, as compared with our 19.3 average (Table 4).
Although the cultivated pool has fewer alleles, it is perhaps more surprising that the number remains so high, particularly since these materials derived from only one breeding program representing years of selective breeding. In any case, abundant allelic variation exists in the wild diploid germplasm, and presumably some of it will be useful for breeding programs, adding desirable alleles for key traits. We are currently pursuing experiments to link this extensive genetic diversity with agronomically important traits to effectively incorporate diploid alleles into cultivar development programs. Finally, identifying desirable alleles in diploid germplasm will be considerably easier than in tetraploids, based on more favorable genetic segregation ratios and more robust genetic mapping capabilities in diploids. Therefore, the utility of diploid germplasm for mining useful alleles is probably larger than has previously been assumed based on classical breeding techniques.
References
Arnold ML, Hamrick JL, Bennett BD (1990) Allozyme variation in Louisiana irises: a test for introgression and hybrid speciation. Heredity 65:297–306
Barnes DK, Bingham ET, Murphy RP, Hunt OJ, Beard DF, Skrdla WH, Teuber LR (1977) Alfalfa germplasm in the United States: genetic vulnerability, use, improvement, and maintenance. USDA Tech Bull 1571
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brummer EC, Kochert G, Bouton JH (1991) RFLP variation in diploid and tetraploid alfalfa. Theor Appl Gen 83:89–96
Diwan N, Bouton JH, Kochert G, Cregan PB (2000) Mapping of simple sequence repeat (SSR) DNA markers in diploid and tetraploid alfalfa. Theor Appl Genet 101:165–172
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Flajoulot S, Ronfort J, Baudouin P, Barre P, Huguet T, Huyghe C, Julier B (2005) Genetic diversity among alfalfa (Medicago sativa) cultivars coming from a breeding program, using SSR markers. Theor Appl Genet 111:1420–1429
Gross BL, Schwarzbach AE, Rieseberg LH (2003) Origin(s) of the diploid hybrid species Helianthus deserticola (Asteraceae). Am J Bot 90:1708–1719
Havananda T, Brummer EC, Maureira-Butler IJ, Doyle JJ (2010) Relationships among diploid members of the Medicago sativa (Fabaceae) species complex based on chloroplast and mitochondrial DNA sequences. Syst Bot 35:140–150
Ivanov AI (1988) Alfalfa. Amerind Publishing, New Delhi
Julier B, Flajoulot S, Barre P, Cardinet G, Santoni S, Huguet T, Huyghe C (2003) Construction of two genetic linkage maps in cultivated tetraploid alfalfa (Medicago sativa) using microsatellite and AFLP markers. BMC Plant Biol 3:9
Kidwell KK, Austin DF, Osborn TC (1994) RFLP evaluation of nine Medicago accessions representing the original germplasm sources for North American alfalfa cultivars. Crop Sci 34:230–236
Lesins K, Lesins I (1979) Genus Medicago (Leguminasae): A taxogenetic study. Kluwer, Dordrecht
Liu J (2002) POWERMARKER—a powerful software for marker data analysis. Raleigh, NC: North Carolina State University, Bioinformatics Research Center. http://www.powermarker.net
Ma X-F, Szmidt AE, Wang X-R (2006) Genetic structure and evolutionary history of a diploid hybrid pine Pinus densata inferred from the nucleotide variation at seven gene loci. Mol Biol Evol 23:807–816
Maureira IJ, Ortega F, Campos H, Osborn TC (2004) Population structure and combining ability of diverse Medicago sativa germplasms. Theor Appl Genet 109:775–782
McCoy TJ, Bingham ET (1988) Cytology and cytogenetics of alfalfa. In: Hanson AA, Barnes DK, Hill RR (eds) Alfalfa and alfalfa improvement. ASA-CSSA–SSSA, Madison
Mengoni A, Gori A, Bazzicalupo M (2000) Use of RAPD and microsatellite (SSR) variation to assess genetic relationships among populations of tetraploid alfalfa, Medicago sativa. Plant Breed 119:311–317
Michaud R, Lehman WF, Rumbaugh MD (1988) World distribution and historical development. In: Hanson AA, Barnes DK, Hill RR (eds) Alfalfa and alfalfa improvement. ASA-CSSA-SSSA, Madison
Muller MH, Prosperi JM, Santoni S, Ronfort J (2003) Inferences from mitochondrial DNA patterns on the domestication history of alfalfa (Medicago sativa). Mol Ecol 12:2187–2199
Muller MH, Poncet C, Prosperi JM, Santoni S, Ronfort J (2005) Domestication history in the Medicago sativa species complex: inferences from nuclear sequence polymorphism. Mol Ecol 15:1589–1602
Peakall R, Smouse PE (2001) GenAlEx V6: genetic analysis in excel population genetic software for teaching and research. Australian National University, Canberra. http://wwwanueduau/BoZo/GenAlE
Pritchard JK, Stevens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pupilli F, Businelli S, Paolocci F, Scotti C, Damiani F, Arcioni S (1996) Extent of RFLP variability in tetraploid populations of alfalfa, Medicago sativa. Plant Breed 115:106–112
Pupilli F, Labombarda P, Scotti C, Arcioni S (2000) RFLP analysis allows for the identification of alfalfa ecotypes. Plant Breed 119:271–276
Quiros CF, Bauchan GR (1988) The genus Medicago and the origin of the Medicago sativa complex. In: Hanson AA, Barnes DK, Hill RR (eds) Alfalfa and alfalfa improvement. ASA-CSSA-SSSA, Madison
Robins JG, Luth D, Campbell TA, Bauchan GR, He C, Viands DR, Hansen JL, Brummer EC (2007) Genetic mapping of biomass production in tetraploid alfalfa (Medicago sativa L). Crop Sci 47:1–10
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Segovia-Lerma A, Cantrell RG, Conway JM, Ray IM (2003) AFLP-based assessment of genetic diversity among nine alfalfa germplasms using bulk DNA templates. Genome 46:51–58
Sinskaya EN (1961) Flora of cultivated plants of the U.S.S.R. XIII perennial leguminous. National Science Foundation, Washington DC
Sledge MK, Ray IM, Jiang G (2005) An expressed sequence tag SSR map of tetraploid alfalfa (Medicago sativa L). Theor Appl Genet 111:980–992
Vandemark GJ, Ariss JJ, Bauchan GA, Larsen RC, Hughes TJ (2006) Estimating genetic relationships among historical sources of alfalfa germplasm and selected cultivars with sequence related amplified polymorphisms. Euphytica 152:9–16
Acknowledgments
We would like to thank Donald Wood, Jonathan Markham, and Frank Newsome for the technical assistance; the Turkish Government for funding MS; the USDA-DOE Plant Feedstock Genomics for Bioenergy award 2006-35300-17224 (to ECB and JJD) and National Science Foundation award DEB-0516673 (to JJD) for funding this research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. -B. Veyrieras.
Rights and permissions
About this article
Cite this article
Şakiroğlu, M., Doyle, J.J. & Charles Brummer, E. Inferring population structure and genetic diversity of broad range of wild diploid alfalfa (Medicago sativa L.) accessions using SSR markers. Theor Appl Genet 121, 403–415 (2010). https://doi.org/10.1007/s00122-010-1319-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1319-4