
Abstract
QTLs for partial resistance to Rice yellow mottle virus (RYMV) in rice were mapped in two populations of doubled-haploid lines (DHLs) and recombinant inbred lines (RILs) derived from the same cross but evaluated for different resistance criteria (virus content and symptom severity). An integrative map was used to compare the two genetic maps and a global analysis of both populations was performed. Most of the QTLs previously identified in DHL population were confirmed with increased significance and precision. As many recent studies evidenced the role of eukaryotic translation initiation factors (eIF) of 4E and 4G families in plant susceptibility to RNA viruses, we checked if these genes co-locate with QTLs of resistance to RYMV. Their systematic in silico identification was carried out on the rice genome and their physical locations were compared to QTL positions on the integrative map. In order to confirm or not the co-locations observed, the analysis was completed by evaluation of near-isogenic lines, QTL fine mapping and sequencing of candidate genes. Three members from eIF4G family could be retained as reliable candidates whereas eIF4E genes, commonly found to govern resistances in other plant/virus interactions, were discarded. Together with the recent identification of an eIF(iso)4G as a major resistance gene, data suggests an important role of genes from eIF4G family in rice resistance to RYMV but does not exclude the contribution of factors different from the translation initiation complex.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Viruses are among the most agriculturally important and biologically intriguing groups of plant pathogens and genetic resistance can be used efficiently to protect crops from virus infection and to identify key factors in plant/virus interactions. RYMV is a Sobemovirus which causes a highly damaging rice disease in Africa (Kouassi et al. 2005). Whereas high resistance is very rare and monogenic, partial resistance is widespread in Oryza sativa upland japonica varieties and involves several quantitative trait loci (QTLs) (Ghesquière et al. 1997; Albar et al. 1998). In particular, two major QTLs mapped on chromosomes 1 and 12 were detected in different environments and for different resistance parameters. The effect of the QTL of chromosome 12 has been confirmed in the susceptible genetic background using a near-isogenic line (NIL) (Ahmadi et al. 2001).
Unlike major resistance genes, the factors that govern quantitative resistance of plants to viruses remain unknown and relatively few of them have been genetically mapped (Kang et al. 2005). Identification of the genes behind these QTLs has been described as the greatest challenge for geneticists in this century (Luo et al. 2002). In plant virus resistance, QTLs can be used alone or in combination with major genes and may improve resistance durability. In addition, their characterization will identify key genes for plant/virus interactions and, in particular, genes which would be difficult to identify by other approaches, like mutant screening. QTL positional cloning is of course possible using NILs. However, this approach is often an expensive and long-term work due to the moderate effect of QTLs on phenotypic variation (Salvi and Tuberosa 2005). For this reason, the candidate gene approach may be the most powerful way to characterise QTLs (Pflieger et al. 2001). Some obvious candidates are the genes involved in major monogenic resistance. Indeed, qualitative and quantitative resistance may be governed by different allelic forms of the same factors and the effect of a factor on resistance may depend on the genetic background. For instance, different alleles of the pvr2 resistance gene either lead to a major resistance or play a role in quantitative resistance to PVY in pepper (Ruffel and Caranta, unpublished). Moreover, the pvr2 3 allele may trigger either complete or partial resistance to PVY depending on the genetic background (Ruffel and Caranta, unpublished). Thus, as more and more is known about genes conferring qualitative resistance to virus infections, the candidate gene approach has emerged as a promising method for identifying genes associated with virus resistance QTLs.
In recent years, the outstanding role of eucaryotic translation initiation factors (eIF) of 4E or 4G families has been demonstrated in several plant/virus interactions (for review, see Robaglia and Caranta 2006). Knockout mutants in these factors, as well as naturally occurring short deletions or amino-acids substitutions, lead to recessive resistance. Whereas the exact resistance mechanism remains unknown, it probably relies on a defective interaction between these host factors and viral components, such as the VPg (genome-linked viral protein). The recent characterization of a gene encoding an eukaryotic translation initiation factor eIF(iso)4G as the only gene, Rymv1, known to confer high resistance to Rice yellow mottle virus (RYMV) and the identification of three different resistance alleles underline the key role of such factors in resistance to RYMV (Albar et al. 2006). A part of our objective was to check if these factors could also be involved in partial resistance to RYMV in rice.
We refined the resistance QTL identification and we exploited the rice genomic sequence to identify in silico all the eIF4E and eIF4G multigenic family members. Then, the combination of different genetic and candidate gene approaches enabled us to evaluate the co-locations of some of those genes with QTLs and discuss about their potential status of candidate genes.
Materials and methods
Plant materials
RYMV partial resistance was analysed in two populations derived from the cross IR64 (O. sativa indica, susceptible) × Azucena (O. sativa japonica, partially resistant). The first population comprised 174 doubled-haploid lines (DHLs) (Guiderdoni et al. 1992). A genetic map of 210 RFLP markers has been developed on this population (Huang et al. 1997) and QTLs of partial resistance to RYMV have already been mapped (Albar et al. 1998; Pressoir et al. 1998). The second population comprised 178 recombinant inbred lines (RILs) and a genetic map of 226 single sequence repeat (SSR) markers was available (Dubreuil-Tranchant et al. 2005).
NILs were developed using two different strategies in order to analyse specific QTLs in more detail. First, NILs were derived from heterogeneous inbred families (HIFs), i.e. families still segregating for a target locus, selected from the F7–F9 generations of RILs screened with SSR markers, as described by Tuinstra et al. (1997). The second strategy consisted in a classical back-crossing process in order to introgress a target region from the donor parent into the recurrent parent. This process was previously used to develop a pair of NILs with IR64 versus Azucena alleles for the RYMV resistance QTL of chromosome 12 (IR64 versus NIL-QTL12) in the IR64 background (BC4 level) by Ahmadi et al. (2001). A population of 649 F2 was derived from the cross NIL-QTL12 × IR64 in order to fine-map this QTL. A second introgressed line was developed in an Azucena resistant background to analyse a region of chromosome 1 which was thought to contain a resistance QTL.
Phenotypic evaluations
The DHL population has already been evaluated for partial resistance to RYMV (Albar et al. 1998). The virus content in plants measured 7 days after mechanical inoculation in a growth chamber by ELISA (VC2) was retained as the resistance criterion in the present study.
The other resistance evaluations performed in this study were based on symptom intensity. Plants cultivated in greenhouse were mechanically inoculated 10–15 days after sowing, as described in Ndjiondjop et al. (1999). Symptoms were observed between 7 and 35 days after inoculation, using a symptom severity scale. The Area Under Symptoms Progression Curve (AUSPC) was calculated as AUSPC = ∑((S i + S (i+1))(T (i+1)–T i))/2, where S i corresponds to the symptom score at T i date.
For the RIL population, the resistance evaluation assay consisted in two randomized complete trials. Each trial contained the 178 RILs, with six inoculated plants per line. As no trial effect was detected for controls, the AUSPC mean of the two tests was used for the analysis.
For HIF analysis, a single trial comprising six randomly laid out repetitions of four plants for each progeny was performed to compare the different NIL pairs.
For fine mapping of QTL12, only the progenies of individuals recombined in the target region were evaluated to determine their allelic status on the QTL. Evaluations were divided into five independent trials, each one containing two repetitions of ten plants of progenies to be tested and four repetitions of each control (IR64, NIL-QTL12 and the progeny of a non-recombinant heterozygous F2). For each test, the most discriminating date between controls (depending on the test, between 7 and 16 days after inoculation) was retained for analysis. Given the expected heterogeneity of heterozygous plant progenies, the non-parametric Kolmogorov–Smirnov test was used to test the homogeneity of the score distribution obtained for different samples: first, the homogeneity between lines of the same genotype was confirmed (P = 0.01) and then each genotype to be tested was compared to the controls (P = 0.001). Statistical results were compared with the individual genotype to determine allelic status at QTL12.
For introgressed line segregating for the QTL1 region, progenies were compared in a single trial comprising four randomly laid out repetitions of six plants for each progeny.
DNA extractions and molecular markers
The DNA of DHLs has already been extracted by Albar et al. (1998). DNA of additional material was extracted from fresh leaves using the protocol described in Edwards et al. (1991). SSR markers were amplified and detected on LI-COR Genotyper as described by Albar et al. (2006).
Thirty-one SSR markers were used to complete available genetic maps (on QTLs or candidate gene regions for both DHL and RIL populations and on four marker-poor regions of the genetic map for the DHL population) and in the course of NIL development (Table S1). These markers are either published markers that have been experimentally tested (Albar et al. 2006; Temnykh et al. 2000, 2001; Panaud et al. 1996) or in silico designed markers (McCouch et al. 2002), or markers that have been newly designed on the Nipponbare sequence, as described by McCouch et al. (2002). In the latter case, primers were designed on SSR flanking regions by Primer3 (Rozen and Skaletsky 2000). Results obtained on ten markers designed by McCouch et al. (2002) and 50 markers newly designed in this study were very consistent, except for the QTL12 region: in both cases 80% gave an amplification product and respectively, 50 and 64% of them revealed polymorphism between Azucena and IR64. In the specific QTL12 region, only seven polymorphic markers were obtained from 26 markers which gave an amplification product.
Genetic map, QTLs and statistical analysis
An integrative map was used to compare and analyse both DHL and RIL populations. The map was based on the RIL genetic map and on different versions of the DHL genetic map (http://www.gramene.org/db/cmap/viewer): Cornell SSR 2001 (Temnyckh et al. 2000), IRRI IR64/Azu DH QTL 2003 (Ramalingam et al. 2003), Cornell IR64/Azu DH QTL 2001 (Temnykh et al. 2000) and CIRAD IR64/Azu DH QTL 2003 (Sallaud et al. 2003). SSR markers of the RIL genetic map were present on the rice Gramene and TIGR assembly of pseudo-molecules and served as framework markers to integrate other markers. As sequence information was available for most of the markers, they were mapped in silico and interpolated in their respective intervals spanned by the framework SSR markers. The markers that were not physically mapped were genetically interpolated. Mapping errors and slight order discrepancy could then be solved. More than 95% of the markers gave consistent projections on the integrative map and accurate positions between framework markers. The resulting genetic integrative map was therefore enriched by a considerable number of markers (1,729) in a physically correct order and with exact genetic correspondence (Dubreuil-Tranchant et al. 2005). Candidate genes and markers added in the current study were placed on this integrative map according to both their position on rice pseudo-molecules and to their genetic mapping using Mapmaker/Exp v3.0. In the DHL population, data from 17 markers, including ten markers of chromosome 6, did not fit with the integrative map and these markers had to be removed from the analysis. The resulting maps included 197 markers for the DHL population and 231 for the RIL population.
QTL analyses of single populations were performed using composite interval mapping with the Windows QTL Cartographer v2.5 program (http://statgen.ncsu.edu/∼shchwang/).
An additional QTL analysis was performed in a multicross design on the data sets of both DHL and RIL populations and is henceforth referred to as the multicross analysis. The phenotypic data, AUSPC and VC2 in RIL and DHL populations respectively, were independently centred and normalised. Data were then analysed with the MCQTL software package (Jourjon et al. 2005), using the iterative QTL mapping method. The two populations were considered as derived from different parents.
The LOD threshold was set by calculating 1,000 permutations and an overall Type I error of 5%. Confidence intervals were delimited by the difference between 2-LOD and the maximum LODscores.
In silico analysis of genes from eIF4E and eIF4G families
Rice (O. sativa, Nipponbare) and Arabidopsis (Arabidopsis thaliana) genomes were screened in silico for genes from eIF4E and eIF4G families. An initial search was carried out on TIGR (http://www.tigr.org/tdb/e2k1/osa1/domain_search.shtml) and riceGAAS websites (http://ricegaas.dna.affrc.go.jp/rgadb/) for rice, and on TAIR website (http://www.arabidopsis.org/) for Arabidopsis, using keywords (IF4E and MIF4G) or PFAM accessions (PF01652 and PF02854) corresponding to eIF4E and eIF4G central domains, respectively. In rice, TIGR annotations (release 4) prevailed over riceGAAS ones when two different annotations were found for the same locus. All the annotations we found were then used as queries for the TBLASTN search on TIGR and TAIR gene annotations. The annotations obtained were challenged using the SMART program (Letunic et al. 2006) and only those corresponding to predicted protein sequences with eIF4E and MIF4G domains were retained for further analysis. This data set was completed with protein sequences from other plants found by BLASTP searches on the NCBI website (http://www.ncbi.nlm.nih.gov/BLAST/) using the predicted protein sequences obtained in rice and Arabidopsis as queries.
Alignments of amino acid sequences were performed using the CLUSTALX program (Thompson et al. 1997). Phylogenetic trees were constructed using the neighbor-joining method (Saitou and Nei 1987) in CLUSTALX. The robustness of the nodes were assessed by bootstrap proportion analysis (Felsenstein 1985) computed after 1,000 replicates. The phylogenetic trees were edited using the TreeView v1.6.6 program (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html) and manually optimized for better viewing clarity.
Sequence analyses of the eIF4E and eIF4G-like (chr. 12) candidate genes
The cDNA of the eIF4E (chr. 1) and eIF4G-like (chr. 12) candidate genes were sequenced in IR64 and Azucena varieties. Total RNA was isolated from leaf tissue with TRIzol reagent (Invitrogen, Paisley, UK). Retrotranscription was performed with an oligo(dT) primer, using ImproII retrotranscriptase (Promega) and the protocol recommended by the manufacturer. Sequencing reactions were carried out with Applied Biosystems BigDye terminator and analysed on an ABI 3100 sequencer (Applied Biosystems, Foster City, CA, USA). The sequences are registered in the EMBL data library under accession numbers AM411440 (IR64) and AM411441 (Azucena) for eIF4E and AM778049 (IR64) and AM778048 (Azucena) for eIF4G-like.
Results
QTL analysis of partial resistance to RYMV
The genetic bases of partial resistance to RYMV were studied in the IR64 × Azucena DHL and RIL populations (Fig. 1, Table S2). Compared to the study of Albar et al. (1998), no additional QTL was detected in the DHL population despite the improved genetic map. QTLs for symptoms from the RIL population were located in the regions of QTLs for virus content from the DHL population, suggesting that the same factors underlie these two traits thus justifying the multicross analysis. However, new minor QTLs (R 2 < 6%) were identified on chromosomes 6 and 7 in the RIL population and were confirmed in the multicross analysis. In addition, multicross analysis enabled the detection of a minor QTL (R 2 = 2.7%) on chromosome 4. For all QTLs, the favourable allele for resistance came from the partially resistant parent Azucena.

Genetic locations of QTL positions (left) and gene annotations from eIF4E and eIF4G families (right) on the integrative map. QTLs of DHL, RIL and multicross analyses are represented by filled ovals covering a 2-LOD confidence interval in height and proportional to phenotypic variation explained in width. The double-arrows symbolise suspected duplications
The QTLs of chromosomes 1 (QTL1), 2 and 12 (QTL12) were confirmed as the most important ones and the multicross analysis enabled us to identify their locations more precisely (Fig. 1, Table S2). Whereas the addition of new markers only slightly reduced the QTL confidence interval in the DHL population, the multicross analysis appeared to reduce it greatly. For instance, the confidence interval of QTL1 was reduced from 20 or 23 cM in the DHL and RIL populations, respectively, to 11 cM in the multicross analysis. A similar result was observed for QTL12, with a confidence interval reduced from 17 or 20 to 12 cM. As previously observed (Albar et al. 1998), the role of chromosome 2 in resistance to RYMV appears to be complex. Three different intervals contain QTLs. In the first one, QTLs were found in DHL and RIL populations and by the multicross analysis. Whereas we cannot exclude that the QTLs identified in the RIL and in the DHL population are distinct, the partial overlapping of confidence intervals suggests a single QTL may be involved in both populations. According to this hypothesis, and as observed for QTL1 and QTL12, multicross analysis would reduce the confidence interval of this QTL. The last two QTLs were detected in the DHL population but not in the RIL population and were still significant in the multicross analysis. In these cases, the confidence intervals increased in the multicross analysis.
Identification of genes from eIF4E and eIF4G families in rice genome
Rice genomic sequence (O. sativa ssp. japonica cv. Nipponbare) was analysed to identify genes from eIF4E and eIF4G families and to compare them to their homologs in Arabidopsis and in other plants.
Three putative rice genes were identified in the eIF4E family. All were supported by cDNA or EST. Their predicted protein sequences were compared to those of five putative Arabidopsis genes and of 12 genes from other plants, including five eIF4E genes reported to be involved in virus resistance (Fig. 2a). Three groups corresponding to eIF4E, eIF(iso)4E and new cap binding protein genes were identified. One rice gene was identified in each group: rice eIF4E maps on chromosome 1, eIF(iso)4E on chromosome 10, and the novel cap-binding protein gene on chromosome 3.

Relationships between genes from eIF4E (a) and eIF4G (b) families in plants. The neighbor-joining unrooted tree and bootstrap proportions using 1,000 replicates were obtained from a total protein alignment. Bootstrap values higher than 950 are represented as black dots. Accession numbers are given and virus resistance genes or mutant names are in bold. Rice and Arabidopsis gene annotations are respectively in bold and underlined. The rice chromosome that carries the gene annotation is also indicated. Concerning the eIF4G family (b), predicted protein sequences and domains are represented. The grey zone on the phylogenetic tree indicates architecture with a trichotomy and a low boostrap value (476)
Eleven putative rice genes were identified in the eIF4G family. All were supported by cDNA or EST. Their predicted protein sequences were compared to those of nine putative Arabidopsis genes and wheat eIF(iso)4G (Fig. 2b). Clustering with previously described genes enabled the identification of one rice eIF4G gene (Os07g36940) on chromosome 7, and two rice eIF(iso)4G genes (Os02g39840 and Os04g42140) on chromosomes 2 and 4, the latter corresponding to the major resistance gene Rymv1 (Albar et al. 2006). The eight remaining predicted rice proteins of the eIF4G family were distant from eIF4G and eIF(iso)4G groups. They are henceforth referred to as eIF4G-like. Although they appeared to be distributed in five groups, the trichotomy feature and a weak bootstrap value (476) impaired reliable clustering. Arabidopsis ortholog genes were found in all groups except one. eIF4G-like predicted proteins generally shared homology with proteins involved in RNA machinery or possessed domains characteristic of such proteins, as for instance MA3 domain (PF02847) from eIF4G family (Pontig 2000), or UPF2 domain (PF04050) involved in regulation of nonsense transcripts (Mendell et al. 2000) (Fig. 2b).
Genomic distribution of homologous eIF(iso)4G and eIF4G-like genes suggested ancestral duplications depending on the organisation of the rice genome (Fig. 1). First, a tandem duplication of an eIF4G-like gene might have occurred on chromosome 6. Second, another duplication was suspected between two homologous eIF4G-like genes on chromosomes 11 and 12. Third, regions of about 1.2 Mb containing both eIF(iso)4G and eIF4G-like genes were identified on chromosome 2 and on chromosome 4. They are the result of the extensive duplication between these chromosomes (Guyot and Keller 2004), which might have been preceded by an initial tandem duplication.
Positional relationships between RYMV resistance QTLs and genes from eIF4E and eIF4G families
The locations of the QTLs were compared to those of genes from eIF4E and eIF4G families. Three interesting co-locations were detected and concerned the QTLs of chromosomes 1, 2 and 12 (Fig. 1). First, QTL1 mapped near the translation initiation factor gene eIF4E, largely involved in plant/virus interactions, and near the semi-dwarfing gene sd-1, previously reported as a possible candidate (Albar et al. 1998) (Fig. 4a). Second, two candidate genes, eIF(iso)4G and eIF4G-like, mapped in the confidence interval of a minor QTL of chromosome 2. Finally, another eIF4G-like gene was located inside the confidence interval of QTL12.
NIL-based verification of QTL positions and co-locations with candidate genes
In order to confirm the co-locations observed between QTLs and candidate genes or to obtain more precise QTL positions, we developed NILs differentiated by QTL flanking regions or, more specifically, by candidate gene alleles using HIFs or a classical backcross process.
Concerning the co-location of chromosome 2, one HIF segregating for eIF4G-like but not for eIF(iso)4G was obtained but evaluation of this HIF for RYMV symptom development did not reveal significant difference between parental alleles (Student’s t test P > 0.05).
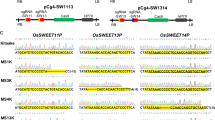
Concerning the co-location of chromosome 12, two HIFs segregating for eIF4G-like were obtained. Significant differences (Student’s t test P < 0.05) for RYMV symptoms evaluation were observed in these families (Fig. 3a), confirming the co-location of eIF4G-like and QTL12. A NIL of the IR64 variety introgressed with the Azucena allele in the QTL12 region (NIL-QTL12) has already been developed and confirmed the effect of QTL12 in the IR64 background (Ahmadi et al. 2001). This line was used to fine-map QTL12. Marker data indicated that Azucena introgression was contained in an 8.2 Mb interval, between markers RZ816 and AB25 (Fig. 3b). Then 649 F2 derived from the cross IR64 × NIL-QTL12 were screened with five SSR markers defined in the introgression. Forty individuals recombined between markers AB1 and AB28 located at the extremities of the target interval. Their progenies were evaluated for resistance to RYMV. Phenotype was clearly established for 23 progenies which indicated that QTL12 was located in a 2.23 Mb interval between markers MV4 and AB28 (Fig. 3b). The maximum LODscore observed in the multicross analysis and the eIF4G-like candidate gene still mapped within this candidate region. The mapping strategy thus confirmed this eIF4G-like gene as a candidate for QTL12. The coding region of eIF4G-like gene was sequenced on the cDNA of IR64 and Azucena. It corresponded to 2,364 nucleotides in Azucena and to 2,304 nucleotides in IR64. Five InDels and four substitutions, located in the first exon before the MIF4G domain, distinguished Azucena from IR64 predicted amino-acids sequences (data not shown).

QTL12 characterization a RYMV symptom evaluations of HIFs developed for eIF4G-like and introgressed line for the QTL12 region (NIL-QTL12). Black and white bars show Azucena and IR64 alleles at the target locus, respectively. Confidence intervals are given. Asterisks indicate a significant difference (Student’s t test, P < 0.05) between lines of Azucena versus IR64 alleles at the candidate gene or locus for QTL12. b Fine genetic and physical mapping of the QTL12 region. The upper part of the figure is a physical representation of the QTL12 region in NIL-QTL12, where the most informative markers are indicated and the Azucena introgressed fragment is coloured black. The positions of the eIF4G-like candidate and of the confidence interval for QTL12 obtained in the multicross analysis are shown. The number of recombinants observed in the NIL-QTL12 × IR64 population are indicated under the corresponding intervals. The lower part of the figure represents the physical fragments accepted (in white) or rejected (hatched), as QTL12 containing fragments, based on the comparison of phenotype and genotype for one to nine recombinants per interval, as indicated on the right. All data are in agreement with the location of QTL12 between MV4 and AB28 markers
Concerning the co-locations of chromosome 1, two HIFs were obtained for sd-1 and two for eIF4E (Fig. 4a). Evaluation of RYMV symptoms revealed no significant differences between parental alleles for each of these HIFs (Student’s t test P > 0.05) (Fig. 4b), which did not support the involvement of eIF4E and sd-1 genes in the effect of QTL1. In addition, sequencing of the coding region of the rice eIF4E gene did not reveal any nucleotidic polymorphism between Azucena and IR64, and no variation in transcript accumulation was detected by semi-quantitative RT-PCR for this gene (data not shown). These results do not support eIF4E gene as a factor governing QTL1. As the above results led us to think that QTL1 was located between sd-1 and eIF4E genes, we performed two successive back-crosses from a RIL with Azucena recurrent parent to introgress this candidate region from IR64 in an Azucena background. A preliminary experiment managed directly on BC2F2 individuals suggested an effect of QTL1 on resistance to RYMV only in interaction with QTL12 (data not shown). Next we selected a plant (BC2F1-hQTL1) heterozygous both for QTL1 and QTL12 regions (Fig. 4a) to evaluate the respective effect of both QTLs on resistance. Two to four BC2F3 progenies were tested for each category of the BC2F2 genotype (Fig. 4b). On the one hand, as expected, the effect of QTL12 was significant whatever the allelic status on QTL1 region (Fig. 4b) (Student’s t test P = 0.007 for IR64 allele on QTL1 and P = 0.013 for Azucena allele). On the other hand, the effect of QTL1 was significant in progenies homozygous for the IR64 allele on QTL12 (P = 0.007) but not for progenies homozygous for the Azucena allele on this QTL (P = 0.427) (Fig. 4b), confirming the previous observation. These results are in agreement with the duplicate epistasis observed by Pressoir et al. (1998) between QTL1 and QTL12 regions for the impact of RYMV inoculation on plant height and heading date. Thus, our results suggest that QTL1 is controlled by a genetic factor located between sd-1 and eIF4E genes, which could be evidenced only when the susceptible allele was present on QTL12.

Characterization of QTL1. a Representation of the genotypes of lines developed for characterization of QTL1. The LODscore curve of multicross analysis and the positions and the names of candidate genes and genetic markers are indicated. b Evaluation of RYMV symptoms in HIFs and in lines developed from BC2F1-hQTL1. Black and white bars show Azucena and IR64 alleles at the target locus, respectively. Confidence intervals are given. Asterisks indicate a significant difference (Student’s t test, P < 0.05). For BC2F1-hQTL1 evaluation, both genotypes at QTL1 and QTL12 were considered and two to four progenies were tested for each category of genotype, as indicated inside the bars
Discussion
This study refined the QTL analysis of partial resistance to RYMV in rice and investigated if genes from eIF4E and eIF4G families participate in the control of this resistance. For this purpose, we combined QTL analysis with a candidate gene approach. QTL analysis can only reveal loci and the underlying genes for which polymorphism between the parents of the population studied is linked to differential expression of the trait. For instance, eIF(iso)4G is known to be linked to resistance to RYMV in rice (Albar et al. 2006) but this gene does not co-locate with a QTL in IR64 × Azucena population. Similarly, other rare polymorphisms may be involved in partial resistance but may not exist between IR64 and Azucena parents. However, partial resistance to RYMV is characteristic of O. sativa japonica subspecies and considering the genetic structuration of japonica and indica subspecies into O. sativa (Glaszmann 1987), a single indica × japonica cross, such as IR64 × Azucena, is assumed to maximise polymorphism and to sample most of the genes underlying partial resistance to RYMV.
QTL mapping is often limited by the number of individuals analysed; it may impair the detection of minor QTLs and the precise location of major ones. For these reasons, we improved QTL mapping in DHL and RIL populations by analysing them together. This multicross analysis required a combination of both genetic and phenotypic data. Concerning the mapping strategy, problems were generally encountered when comparing QTLs obtained on different genetic maps due to the difficulties in aligning the maps. Recently several mapping tools were developed to compare genetic maps, such as Biomercator (Arcade et al. 2004), cMap (Fang et al. 2003) and CMTV (Sawkins et al. 2004). A similar approach was performed manually here to align the different rice genetic maps with pseudo-molecules and to construct an integrative map. This map was a quite powerful tool to compare the QTLs in our populations and to directly map candidate genes to identify co-locations with QTLs. Concerning the phenotypic data, resistance to RYMV was evaluated based on the virus content in the plant in the DHL population and on symptom development in the RIL population. The combination of these two criteria in the multicross analysis supposes that the same genetic factors control both of them. However, this is not always the case. For instance, the Arabidopsis SBB1 locus is involved in symptom development after Spring beauty latent virus (SBLV) infection but has no effect on virus accumulation (Fujisaki et al. 2004). Few data are available on the comparison of QTLs of virus content and symptom development. Our previous data on a collection of rice varieties (unpublished results) and on the DHL population (Albar et al. 1998) strongly suggested these two traits are correlated. Moreover, in this study, QTL mapping in DHL and RIL populations on the integrative map pinpointed exactly the same loci on chromosomes 1 and 12. We thus considered that these resistance criteria were partially controlled by the same major QTLs. The multicross analysis enabled considerable more precision in the confidence intervals for major QTLs on chromosomes 1 and 12. Yet, results obtained in single populations suggest a complex role of chromosome 2 in resistance to RYMV and results of the multicross analysis on this chromosome should be interpreted with caution, as different QTLs might control the two resistance criteria. Precise dissection of the QTLs of this region would require the production of several set of NILs, so that the segregating regions successively overlap and cover the whole of chromosome 2. The detection in multicross analysis of minor QTLs that were not detected in the DHL population could also be explained by increased power of detection associated with the analysis of a larger number of individuals. Multicross analysis may be thus an appropriate tool to detect QTLs with minor effects and to improve QTL confidence intervals.
It is generally believed that QTL mapping does not accurately locate genes that explain polygenic traits on the genome. However, based on several publications, Price (2006) recently reported that QTL position is accurate to within 2 cM or less for major QTLs and to within 3 cM or less for minor QTLs. So, we can be relatively confident about the positions of the QTLs, especially those obtained by multicross analysis, and on co-locations between QTLs and underlying candidate genes. In order to further verify (or not) the co-locations observed, the step following QTL mapping was the study of NILs which segregate for the target region. In this study, the target regions were either candidate QTL regions, candidate genes, or both. It should be noted that this is the first time that the development of HIFs is directly based on candidate genes. Introgressed lines and HIF strategies both proved to be reliable for QTL12 in this study. However, the absence of a significant difference in a family does not necessarily imply that the target region does not control a variation in the character. Indeed, as already highlighted by Tuinstra et al. (1997) and as suggested here by evaluation of BC2F2-hQTL1, genetic background may influence the detection of a QTL effect. Thus, it would be better to analyse a candidate gene or region for a QTL in a relatively large set of HIFs, if available.
In this study, it is the relevancy of different approaches, namely, QTL detection in different mapping populations, NILs development and sequence data, which can converge to declare or not a peculiar gene as a reliable candidate gene. For QTL1, the global information seems very consistent to discard eIF4E as a candidate gene. Combined with the absence of QTL detection on chromosomes that carry eIF(iso)4E and the new cap-binding protein gene, the results do not support a major involvement of genes from eIF4E family in QTLs of partial resistance to RYMV. It is probably more difficult to conclude at this stage for the chromosome 2 and the two candidate genes from eIF4G family, since the reproducibility of QTL detection between the two mapping populations was only partial. Nevertheless, because of the orthology (one of them at least) with Rymv1 gene, we can provisionally retain these genes as candidates, which will require further confirmation by classical NILs development and evaluation. Last, an eIF4G-like gene was confirmed as a candidate for QTL12 by HIF evaluation and by mapping to a candidate region of 2.23 Mb. This gene may have no function in translation initiation but the polymorphisms between the amino-acids sequences of the two parents are encouraging to consider this gene may be involved in RYMV partial resistance by its MIF4G or MA3 domain in a similar way as Rymv1 gene. The aim of our next study will be to further validate this co-location by continuing fine genetic mapping and by studying the eIF4G-like candidate gene.
Finally, in addition to the role of eIF(iso)4G (chr.4) in high resistance (Albar et al. 2006), we have identified three other members of the eIF4G family which can be reasonably considered as good candidates to focus the genetic studies later on, while members of the eIF4E family do not seem directly involved in QTLs of partial resistance to RYMV. Moreover, this analysis gives large room for other factors explaining partial resistance, in particular for QTL1, and highlights the diversity of plant/virus interactions. It would be of great interest to identify these factors to see if, like other eIF or the poly-A binding protein, they play a role in the translation initiation complex or if they are involved in other functions. In all cases, characterization of the QTLs involved in partial resistance to RYMV would provide new insights in plant/virus interactions.
References
Ahmadi N, Albar L, Pressoir G, Pinel A, Fargette D, Ghesquière A (2001) Genetic basis and mapping of the resistance to Rice Yellow Mottle Virus. III. Analysis of QTL efficiency in introgressed progenies confirmed the hypothesis of complementary epistasis between two resistance QTLs. Theor Appl Genet 103:1084–1092
Albar L, Lorieux M, Ahmadi N, Rimbault I, Pinel A, Sy A, Fargette D, Ghesquiere A (1998) Genetic basis and mapping of the resistance to Rice yellow mottle virus. I. QTLs identification and relationship between resistance and plant morphology. Theor Appl Genet 97:1145–1154
Albar L, Bangratz-Reyser M, Hebrard E, Ndjiondjop M-N, Jones M, Ghesquiere A (2006) Mutations in the eIF(iso)4G translation initiation factor confer high resistance of rice to Rice yellow mottle virus. Plant J 47:417–426
Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, Joets J (2004) BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics 20:2324–2326
Dubreuil-Tranchant C, Ahmadi N, Courtois B, Lorieux M, McCouch SR, Glaszman JC, Ghesquière A (2005) New resources and integrated map for IR64 × Azucena, a reference population in rice. Poster presented to generation challenge programme 2005 annual research meeting Sept 29th–Oct 1st, Roma, Italy
Edwards K, Johnstone C, Thompson C (1991) A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucl Acids Res 19:1349
Fang Z, Polacco M, Chen S, Schroeder S, Hancock D, Sanchez H, Coe E (2003) cMap: the comparative genetic map viewer. Bioinformatics 19:416–417
Felsenstein J (1985) Confidence limits on phylogenies: an approach using bootstrap. Evolution 39:783–791
Fujisaki K, Hagihara F, Azukawa Y, Kaido M, Okuno T, Mise K (2004) Identification and characterization of the SSB1 locus involved in symptom development by Spring beauty latent virus infection in Arabidopsis thaliana. Mol Plant Microbe Interact 17:967–975
Ghesquiere A, Albar L, Lorieux M, Ahmadi N, Fargette D, Huang N, McCouch SR, Notteghem JL (1997) A major quantitative trait locus for Rice yellow mottle virus resistance maps to a cluster of blast resistance genes on chromosome 12. Phytopathology 87:1243–1249
Glaszmann JC (1987) Isozyme and classification of Asian rice varieties. Theor Appl Genet 74:21–30
Guiderdoni E, Galinato E, Luistro J, Vergara G (1992) Anther culture of tropical japonica × indica hybrids of rice (Oryza sativa L.). Euphytica 62:219–224
Guyot R, Keller B (2004) Ancestral genome duplication in rice. Genome 47:610–614
Huang N, Parco A, Mew T, Magpantay G, McCouch SR, Guiderdoni E, Xu J, Subudhi P, Angeles ER, Khush GS (1997) RFLP mapping of isozymes, RAPD and QTLs for grain shape, brown planthopper resistance in a doubled haploid rice population. Mol Breed 3:105–113
Jourjon M-F, Jasson S, Marcel J, Ngom B, Mangin B (2005) MCQTL: multi-allelic QTL mapping in multi-cross design. Bioinformatics 21:128–130
Kang B-C, Yeam I, Jahn MM (2005) Genetics of plant virus resistance. Annu Rev Phytopathol 43:581–621
Kouassi NK, N’Guessan P, Albar L, Fauquet CM, Brugidou C (2005) Distribution and characterization of Rice yellow mottle virus: a threat to African farmers. Plant Dis 89:124–133
Letunic I, Copley RR, Pils B, Pinkert S, Schultz J, Bork P (2006) SMART 5: domains in the context of genomes and networks. Nucl Acids Res 34: 257–260
Luo ZW, Wu C-I, Kearsey MJ (2002) Precision and high-resolution mapping of quantitative trait loci by use of recurrent selection, backcross or intercross schemes. Genetics 161:915–929
McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, DeClerck G, Schneider D, Cartinhour S, Ware D, Stein L (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
Mendell JT, Medghalchi SM, Lake RG, Noensie EN, Dietz HC (2000) Novel Upf2p orthologues suggest a functional link between translation initiation and nonsense surveillance complexes. Mol Cell Biol 20:8944–8957
Ndjiondjop MN, Albar L, Fargette D, Fauquet C, Ghesquière A (1999) The genetic basis of high resistance to Rice yellow mottle virus (RYMV) in cultivars of two cultivated rice species. Plant Dis 83:931–935
Panaud O, Chen X, McCouch SR (1996) Development of microsatellite markers and characterization of simple sequence length polymorphism (SSLP) in rice (Oryza sativa L.). Mol Gen Genet 252:597–607
Pflieger S, Lefebvre V, Causse M (2001) The candidate gene approach in plant genetics: a review. Mol Breed 7:275–291
Ponting CP (2000) Novel eIF4G domain homologues linking mRNA translation with nonsense-mediated mRNA decay. Trends Biochem Sci 25:423–426
Pressoir G, Albar L, Ahmadi N, Rimbault I, Lorieux M, Fargette D, Ghesquiere A (1998) Genetic basis and mapping of the resistance to Rice yellow mottle virus. II. Evidence of a complementary epistasis between two QTLs. Theor Appl Genet 97:1155–1161
Price AH (2006) Believe it or not, QTLs are accurate! Trends Plant Sci 11:213–216
Ramalingam J, Vera Cruz CM, Kukreja K, Chittoor JM, Wu JL, Lee SW, Baraoidan M, George ML, Cohen MB, Hulbert SH, Leach JE, Leung H (2003) Candidate defense genes from rice, barley, and maize and their association with qualitative and quantitative resistance in rice. Mol Plant Microbe Interact 16:14–24
Robaglia C, Caranta C (2006) Translation initiation factors: a weak link in plant RNA virus infection. Trends Plant Sci 11:40–45
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sallaud C, Lorieux M, Roumen E, Tharreau D, Berruyer R, Svestasrani P, Garsmeur O, Ghesquiere A, Notteghem JL (2003) Identification of five new blast resistance genes in the highly blast-resistant rice variety IR64 using a QTL mapping strategy. Theor Appl Genet 106:794–803
Salvi S, Tuberosa R (2005) To clone or not to clone plant QTLs: present and future challenges. Trends Plant Sci 10:297–304
Sawkins MC, Farmer AD, Hoisington D, Sullivan J, Tolopko A, Jiang Z, Ribaut JM (2004) Comparative map and trait viewer (CMTV): an integrated bioinformatic tool to construct consensus maps and compare QTL and functional genomics data across genomes and experiments. Plant Mol Biol 56:465–480
Temnykh S, Park WD, Ayres N, Cartinhour S, Hauck N, Lipovich L, Cho YG, Ishii T, McCouch SR (2000) Mapping and genome organization of microsatellite sequences in rice (Oryza sativa L.). Theor Appl Genet 100:697–712
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:1441–1452
Thompson J, Gibson T, Plewniak F, Jeanmougin F, Higgins D (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res 25:4876–4882
Tuinstra MR, Ejeta G, Goldsbrough PB (1997) Heterogeneous inbred family (HIF) analysis: a method for developing near-isogenic lines that differ at quantitative trait loci. Theor Appl Genet 95:1005–1011
Acknowledgments
This work was supported in part by the GENOPLANTE program TRANSVIR. Arnaud Boisnard was supported by a fellowship from the French Ministry of Research and Deless Thiémélé by a fellowship from the French-speaking University network (AUF). We gratefully acknowledge Thierry Mathieu and Sophie Perez for plant care and Nour Ahmadi for having provided us genotypes data of RIL population. Stéphane Jouannic’s help with protein sequence alignment and phylogenetic tree was greatly appreciated. We thank Romain Guyot and Mathias Lorieux for critical reviews of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Y. Xue.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Boisnard, A., Albar, L., Thiéméle, D. et al. Evaluation of genes from eIF4E and eIF4G multigenic families as potential candidates for partial resistance QTLs to Rice yellow mottle virus in rice. Theor Appl Genet 116, 53–62 (2007). https://doi.org/10.1007/s00122-007-0646-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-007-0646-6