
Abstract
Sclerotinia stem rot, caused by fungus Sclerotinia sclerotiorum, is one of the most devastating diseases in rapeseed (Brassica napus L.). We report the identification of Quantitative trait loci (QTL) involved in the resistance to S. sclerotiorum in two segregating populations of DH lines: the HUA population, derived from a cross between a partially resistant Chinese winter line (Hua dbl2) and a susceptible European spring line (P1804); and the MS population, derived from a partially resistant French winter cultivar (Major) and a susceptible Canadian spring cultivar (Stellar). A petiole inoculation technique and two scoring methods, days to wilt (DW) and stem lesion length (SLL), were used for the resistance assessment. A total of eight genomic regions affecting resistance were detected in the HUA population, with four of these regions affecting both measures of resistance. Only one region, which affected both measurements, was detected in the MS population. Individual QTL explained 6–22% of the variance. At five of the QTL from both populations, alleles from the resistant parent contributed to the resistance. QTL on N2 from the HUA population had the highest LOD score and R 2 value and was detected for SLL in the first evaluation. The N12 resistance allele in Hua dbl2 was detected in a region containing a homeologous non-reciprocal transposition (HNRT) from the resistance-containing portion of N2. This result suggests that QTL in the N12.N2 HNRT enhanced the resistance of Hua dbl2 by increasing the dosage of resistance genes. The relationship of QTL from different genetic backgrounds and their associations with other agronomic traits are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sclerotinia stem rot (white mold), caused by fungal pathogen Sclerotinia sclerotiorum (Lib.) de Bary, is a major disease in many crop species worldwide. More than 400 plant species have been reported as hosts of this pathogen, including important crops such as soybean, sunflower, and alfalfa (Purdy 1979). In recent years, it has been one of the most destructive diseases of oilseed Brassica species in China, and in North Dakota and Minnesota in the United States. Yield losses can be up to 80% in severely infected fields in China (Oilcrop Research Institute, Chinese Academy of Sciences 1975), and as high as 50% in the United States (http://www.whitemoldresearch.com/HTML/canola.cfm). Breeding canola cultivars with high levels of durable resistance is a high priority for crop breeding. Although no host genotypes with complete resistance to S. sclerotiorum have been identified in any crop species, some have partial resistance (Wegulo et al. 1998; Hartman et al. 2000; Kim et al. 2000; Hoffman et al. 2002; Vuong and Hartman 2003), including some Chinese winter lines of B. napus that have consistently shown high level of resistance (Zhou et al. 1994; Zhao et al. 2004). These resistance sources could be used in genetic studies of S. sclerotiorum resistance and in breeding programs to select for cultivars with high levels of resistance.
Molecular markers and quantitative trait loci (QTL) mapping methods have been used in the identification of loci related to resistance to S. sclerotiorum in several crop species including soybean, common bean, and sunflower. In soybean [Glycine max (L.) Merr], 28 resistance QTL were identified on 14 linkage groups in five populations of recombinant inbred lines (RIL), with each QTL explaining 4–10% of the phenotypic variation (Arahana et al. 2001). In common bean (Phaseolus vulgaris L.), different resistance alleles were identified from various accessions. These QTL were located on different linkage groups and few were consistent in all the accessions surveyed (Miklas et al. 2001; Kolkman and Kelly 2003). In sunflower (Helianthus annuus L.), Mestries et al. (1998) reported that resistance to S. sclerotiorum was under polygenic control and complex, and separate QTL were identified for capitum and leaf resistance. Bert et al. (2002) reported the detection of up to 15 QTL on several linkage groups that explained 7–14% of the phenotypic variation in a population of F2:3 families in sunflower.
In all these QTL mapping studies of S. sclerotiorum resistance, the detected QTL accounted for only a small portion of the phenotypic variation, the response was dependent on evaluation conditions, and different QTL were detected in different populations. In oilseed B. napus, Zhao and Meng (2003a) detected three QTL involved in leaf resistance at the seedling stage and three different QTL for stem resistance at plant mature stage in a segregating population from a cross between a partially resistant Chinese winter line (NingRS-1) and a winter male-sterility restorer line. However, these QTL have yet to be confirmed, since the mapping was confined to a single population of F3 families evaluated in one location.
Segregating populations of doubled haploid (DH) lines have been used extensively in QTL mapping studies because they are immortal populations, completely homozygous at every locus and can be used for replicated trial in different years and locations. Several DH populations have been developed in Brassica species and used for molecular map construction and QTL studies (Ferreira et al. 1995a, b; Toroser et al. 1995; Thormann et al. 1996; Kole et al. 2002; Quijada 2003; Udall et al. 2005). In this study, we report the identification of QTL for S. sclerotiorum resistance in two segregating populations of DH lines. The objectives were to identify QTL for S. sclerotiorum resistance in a Chinese winter line and a French winter cultivar, and to compare the resistance alleles identified in this study with those reported previously. Our results suggest that resistance of the Chinese Winter line was associated with duplication of a resistance gene through a homeologous non-reciprocal transposition (HNRT) on N12.N2(T).
Materials and methods
Parental materials and populations
Brassica napus lines, Major and RV289, were identified as partially resistant to S. sclerotiorum, and Stellar and P1804 were identified as susceptible in a previous screening study (Zhao et al. 2004). These lines had been used previously as parents to develop mapping populations (Ferreira et al. 1994; Udall 2003). RV289 is a single plant selected from Hua dbl2, provided by Dr. J. Meng from Huazhong Agricultural University, China. Major is a French winter cultivar and Stellar is a Canadian spring cultivar. P1804 is an European spring DH line having the male-sterility restorer gene of the SeedLink® transgenic system (Mariani et al. 1992). Two segregating populations of DH lines, derived from crosses of RV289 × P1804 (HUA population) and Stellar × Major (MS population) were previously developed and genotyped using molecular markers to build linkage maps (Ferreira et al. 1994; Udall 2003). Agronomic traits including seed yield, flowering time, plant height, seed quality, and blackleg resistance were segregating in these populations and had been previously analyzed (Ferreira et al. 1995a, b; Toroser et al. 1995; Thormann et al. 1996; Osborn et al. 1997, 2003; Udall et al. 2004).
Phenotypic evaluation
Randomized complete block designs were used to evaluate both populations for resistance to S. sclerotiorum. An individual plant of each line represented the experimental unit. For the HUA population, three evaluations were conducted separately in controlled greenhouse conditions at different times (planted on 27 October 2003; 12 November 2003; and 26 January 2004). Each evaluation consisted of 152 DH lines, the two parents and six other accessions with five replications. For the MS population, 104 DH lines, the two parents and two other accessions were included in one evaluation (planted on 14 May 2004) with six replications.
Seeds were planted in Scott’s Metro-mix 336p (Scott’s-Sierra Horticultural Products Co., Marysville, OH 43041, USA) into flats that held 24 8-cm2 pots. Seedlings were thinned to one plant per pot and were grown in a greenhouse under 350–450 μmol m−2 s−1 lights with 16 h photoperiod provided by sunlight and supplemental artificial lighting at a constant temperature of 21°C. Plants were watered and fertilized to achieve optimal growth.
A petiole inoculation technique was used to inoculate 4-week-old seedlings (Zhao et al. 2004). Isolate 105HT, collected from soybeans in Iowa in 1999 and provided by Glen Hartman, USDA/ARS, University of Illinois, was used as the pathogen to challenge the plants. Isolates were cultured on potato dextrose agar (PDA: 25% potato extract, 2% dextrose, 1.5% agar) and incubated at 22–25°C under 45 μmol m−2 s−1 wide spectrum lights. An 8-mm thick PDA plug that was taken from the advancing edge of the mycelium was used as inoculum to challenge plants. Responses were recorded as days to wilt (DW) each day after inoculation, and stem lesion length (SLL) in centimeters measured at day 4 post-inoculation.
Statistical analysis
Normality of frequency distributions was checked using the UNIVARIATE procedure of SAS (SAS Institute 2001). Error variances of the individual evaluations for the HUA population were heterogeneous as determined by the Bartlett’s test. Thus, the three evaluations were analyzed separately.
Least square means for DW and SLL were calculated for each DH line in each population using the MIXED procedure of SAS (SAS Institute 2001). Pearson’s correlation coefficients between DW and SLL were calculated using the CORR procedure of SAS for the means of each line. The narrow-sense heritability of DW and SLL in the HUA population and the broad sense heritability of DW and SLL in the MS population were estimated and exact 95% confidence intervals were calculated according to Knapp et al. (1985). QTL associated with DW and SLL in each population and each evaluation were identified using the CIM procedure within the QTL Cartographer suite of computer programs (QTLcart V1.16; Basten et al. 2002). The likelihood of a QTL and its corresponding effect at every 2 cM was estimated. Four to nine cofactors for each trait were selected by SRmapqtl using a forward–backward regression algorithm (cofactor in for P<0.01; cofactor out for P>0.05). Cofactors within 10 cM on either side of the QTL test site were not included in the Zmapqtl QTL model (Model 6) to avoid the inclusion of putative tail effects of the tested chromosome position. A 1,000-permutation test of shuffling the phenotypes means with the genotypes was performed to estimate a significance threshold of the test statistic for a QTL based upon a 5% experiment-wise error rate (Doerge and Churchill 1996). In the HUA population, the LOD thresholds for significance were 2.84, 2.86, and 2.88 for SLL in the three evaluations, respectively, and 3.0 for DW in all three evaluations. In the MS population, the LOD thresholds for significance were 3.2 for SLL and 2.8 for DW in one evaluation.
Results
Phenotypic data and segregation in the populations
Both RV289 and Major showed higher level of resistance than P1804 and Stellar measured by DW and SLL except in evaluations 2 and 3 of the HUA population where RV289 and P1804 were not significantly different for SLL and DW2. The narrow-sense heritabilities of DW and SLL were 65 and 79%, respectively, in the HUA population and the broad-sense heritabilities of DW and SLL were 73 and 77% in the MS population, indicating that genetic variance accounted for a large portion of the phenotypic variance of S. sclerotiorum resistance. There was transgressive segregation in both populations with some lines showing higher levels of resistance or susceptibility than the two parents (Table 1). The most susceptible lines started wilting by day 3, and all plants had wilted by day 7 after inoculation. This result indicates that the resistance to S. sclerotiorum exhibited in both parents was partial, and the resistance mechanism could involve a delay in the disease progress.
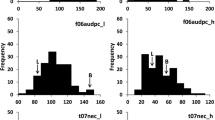
The DH lines of both populations showed continuous distribution for DW and SLL, suggesting multi-genic control of resistance, and the two populations were normally distributed in all the evaluations, except for DW in evaluation 1 of the HUA population which was skewed toward the resistant parent RV289 (Fig. 1). The DW and SLL scores within each evaluation were significantly correlated in the HUA population (r DW1,SLL1 = −0.64; r DW2,SLL2 = −0.75; r DW3,SLL3 = −0.63) and in the MS population (r DW,SLL = −0.65). SLL among three evaluations in HUA population were moderately correlated (r SLL1,SLL2 = 0.58; r SLL1,SLL3 = 0.52; r SLL2,SLL3 = 0.60), consistent with a significant treatment by evaluation interaction (P<0.01). This interaction was mainly due to the response of intermediate resistant lines to the pathogen challenge in each evaluation, while highly resistant or susceptible lines were consistent among evaluations.

Frequency distributions of doubled haploid lines from the HUA (a, b) and the MS (c, d) populations in response to one evaluation of S. sclerotiorum resistance. Days to wilt were measured each day post-inoculation and stem lesion length was measured in centimeters on the 4th day post-inoculation. Days to wilt and stem lesion length of the four parents RV289, P1804, Major, and Stellar are shown
Identification of QTL for S. sclerotiorum resistance
Seven distinct QTL for SLL were identified in the three separate evaluations of the HUA population. In each evaluation, the number of QTL identified varied from 2 to 6, with each QTL explaining 6–22% of the phenotypic variation (Table 2, Fig. 2). For three QTL (Sll14b, Sll16, and Sll19), P1804 (the susceptible parent) contributed the resistance allele. Sll16 was detected in all three evaluations at approximately the same position of linkage group N16 and accounted for 6–15% of the phenotypic variation. Sll14b and Sll19 were detected only in evaluation 1 and each explained 8% of the phenotypic variation. For the remaining QTL (Sll2, Sll5, Sll12, and Sll14a), RV289 (the resistant parent) contributed the resistance allele. Sll12, located on the top of N12 in an interval of 12 cM, was detected in the first evaluation explaining 6% of the phenotypic variation, and the same genomic region had elevated LOD scores of 2.3 and 2.1, respectively, in evaluations 2 and 3, which were just below threshold values. Sll14a was detected approximately at the same positions in two evaluations and it explained approximately 6% of the phenotypic variation. Thus, N14 may contain two segregating QTL regions in the HUA population (Sll14a and Sll14b) that affect resistance, but their allelic effects were in opposite directions. Sll5 had a significant effect in evaluation 2, explaining 10% of the phenotypic variation, and an effect just below the LOD threshold (LOD score of 2.6) in evaluation 1. The QTL located on the top of N2 explained the largest proportion of the variance and had the highest LOD score in this study; however, it was only significant in one evaluation. QTL alleles on N2 and N12 were located in a HNRT region in which a segment of N2 had been transposed to N12 via a previous homeologous exchange in the Hua dbl2 parent (Udall et al. 2005). Thus, the resistance alleles on N2 and N12 might be identical by descent.

Quantitative trait loci for S. sclerotiorum resistance measured by days to wilt and stem lesion length detected in the HUA population. Only the linkage groups with significant QTL are illustrated. The numbers of linkage groups (N) are shown on the top of each linkage group and distance in cM are shown on the left. Peak effects of QTL are shown by a small triangle. Bars on either side indicate 1-LOD score confidence intervals. Rectangle bars indicate QTL for SLL and rounded-corner bars represent QTL for DW. Solid bars indicate resistance alleles are from RV289 and open bars indicate resistance alleles are from P1804. Homeologous marker loci on N2 and N12 are shown by straight lines. The shaded region on N12 indicates the approximate chromosome portion originally from N2 (B. rapa origin) (HNRT, see text for explanation). Numbers on the top of each bar indicate the evaluations in which QTL were detected
Four significant QTL for DW were identified in the three evaluations of the HUA population. In each evaluation, the number of QTL identified varied from 1 to 3, with each QTL explaining 6–12% of the phenotypic variation. QTL Dw12 was detected in the evaluations 1 and 3 and had the resistance allele from RV289. It was located on the top of N12 and had a 1-LOD confidence interval that overlapped with Sll12. The other three QTL (Dw3, Dw16, and Dw19) were only detected in single evaluations, and the P1804 allele contributed to the resistance. Dw16 and Dw19 had overlapping 1-LOD confidence intervals with QTL Sll16 and Sll19 (Table 2, Fig. 2). There was a fifth QTL of DW with a LOD score just below the threshold (2.1) on linkage group N2, which had the same allelic effect and a 1-LOD confidence interval that overlapped with Sll2 (data not shown).
One significant region was detected on N3 in the MS population which affected both DW and SLL. This QTL explained 22 and 11% of the phenotypic variation for DW and SLL, respectively, and alleles from Major (the resistant parent) contributed to S. sclerotiorum resistance for both measurements.
Discussion
Correlations between DW and SLL
A petiole inoculation technique was used for disease evaluation in this study, and two scoring methods, DW and SLL, were used to measure the resistance. Correlations between DW and SLL were higher within each evaluation than were the correlations of single measurements between evaluations. Significant treatment by environment interactions was detected, and the interaction was more significant for DW than for SLL. The three evaluations were conducted under greenhouse conditions, and each was planted at different times of year using artificial lights as a supplement to sunlight. Thus, different environmental conditions could have had different effects on plant growth and the progress of disease development.
Quantitative trait loci for S. sclerotiorum resistance identified in this study
A total of nine distinct QTL regions were detected on seven linkage groups in the B. napus genetic map, eight QTL in the HUA population, and one in the MS population. At four of the QTL in the HUA population, the combined effect of alleles from the resistant donor parent RV289 increased resistance by reducing the SLL an average of 1.8 cm for the three evaluations. The resistance alleles at the remaining 4 QTL detected in the HUA population were from P1804, suggesting it contains cryptic alleles for resistance. In the MS population, only one significant QTL was detected and it was located in a different genomic region than the QTL detected in the HUA population. Additional QTL might have been detected in the MS population if it had been evaluated multiple times. The QTL identified in these two populations need to be verified by deriving independent segregating lines and testing these lines for resistance.
The four QTL regions on N2, N12, N16, and N19 in the HUA population and the one QTL region in the MS population were associated with both DW and SLL. For each region, the DW and SLL QTL had overlapping 1-LOD confidence intervals and the resistance alleles came from the same parent, suggesting that these regions contained loci which controlled both traits. The remaining QTL on N3, N5, and N14 in the HUA population were detected only for either DW or SLL. This could be due to specific environmental conditions that limited the detection of corresponding QTL, or that these regions contain specific QTL for either DW or SLL. DW scores recorded how fast the plant initiated the response to S. sclerotiorum infection, while SLL measured the response after disease progress. Therefore, these measurements might reflect two different mechanisms which are controlled in part by different genes.
Chromosomal rearrangements and resistance
Brassica napus is hypothesized to have originated by hybridization of B. rapa and B. oleracea (Song and Osborn 1992; U 1935). N2 is of B. rapa origin and N12 is the homeologous chromosome from B. oleracea (Parkin et al. 1995). De novo and pre-existing HNRT have been observed for these chromosomes that presumably arose from a homeologous exchange between N2 and N12, and the resistant parent RV289 has a pre-exiting HNRT on N12 [N12.N2(T)] (Udall et al. 2005). Since we detected alleles for resistance from RV289 on both N2 and N12 in the region of this HNRT, the resistance on N12.N2(T) may be due to an identical B. rapa allele from N2. Thus, this rearrangement might have increased resistance in RV289 by increasing the dosage of resistance alleles. Among the segregating lines of the HUA population, three classes (representing marker loci pX115 to pW251 from top of N2 to the middle of N12) were present with respect to allelic dosage on N2 and N12: class 1 had N2 and N12 alleles from RV289 (4 doses of the resistant B. rapa allele), class 2 had N2 alleles from RV289 and N12 alleles from P1804 or N2 alleles from P1804 and N12 alleles from RV289 (2 doses of the resistant B. rapa allele), and class 3 had N2 and N12 alleles from P1804 (0 doses of the resistant B. rapa allele). Based on the result from evaluation 1, class 1 genotypes showed higher resistance (SLL=3.5 cm) than class 3 genotypes (SLL=5.2 cm), and class 2 genotypes were intermediate (SLL=4.6 cm). Other Chinese winter accessions with high levels of resistance, such as CK821, seem to have a similar HNRT as detected by PCR-based markers developed from RFLP markers in this region (data not shown). For these genotypes, the association between genome rearrangements in this region and resistance needs further investigation.
Another chromosomal rearrangement also might have influenced the segregation of resistance. The susceptible parent P1804 has a pre-exiting homeologous reciprocal transposition (HRT) between N7 (B. rapa origin) and N16 (B. oleracea origin) (Udall et al. 2005), and the transgenes for herbicide resistance and male-fertility restoration are located in this region on N7. In the development of these lines, application of herbicide was used to select for DH lines having the male-fertility restorer gene; and thus, the entire population was fixed for P1804 alleles in this region of N7 (Udall et al. 2005). Due to the HRT in P1804, the alleles on N7 are of B. oleracea origin. DH lines that inherit the normal N16 chromosome from RV289 (B. oleracea origin) have greater intergenome homozygosity and lower seed yield (Osborn et al. 2003; Udall 2003). Thus, the increase in susceptibility associated with RV289 allele on N16 may be due to a loss of general vigor resulting from greater intergenome homozygosity.
Quantitative trait loci resistance and other agronomic traits
Resistance QTL on N2 and N12 were also in the same genomic regions as QTL for flowering time (Udall 2003), although the correlation between flowering time and S. sclerotiorum resistance was not very high (r=0.52). We do not yet know whether the S. sclerotiorum resistance and flowering time QTL correspond to the same locus with pleiotropic effects or whether they represent the effects of closely linked genes. From the results of our previous screening experiments, we found that RV289 and other Chinese winter accessions showed high levels of resistance to S. sclerotiorum both at the seedling and flowering stages when internodes are more elongated (Zhao et al. 2004). Thus, the resistance expressed in RV289 does not appear to be due entirely to difference in internode length associated with flowering time. Furthermore, we found QTL located on N5, N14, and N16 that were not near flowering time QTL (Udall 2003), and the QTL for resistance we found in the MS population was not located near a flowering time QTL (Osborn et al. 1997). The introgression of the N2 and N12 resistance alleles into another background and breakage of the potential linkages between genes for resistance and flowering time may help dissect these associations.
Breeding cultivars with canola quality (low erucic acid content and low glucosinolate content in the seed) have become an essential part of oilseed B. napus breeding. Zhao and Meng (2003b) reported that two QTL for S. sclerotiorum resistance had overlapping confidence interval with QTL for seed glucosinolate content with resistance linked to high glucosinolate. Whether this relationship is due to linkage or pleiotropy remains to be seen. Although both the HUA and MS populations segregated for glucosinolate content (Toroser et al. 1995; Udall 2003), we did not find correspondence between the locations of QTL for seed glucosinolate content and S. sclerotiorum resistance in either population.
Quantitative trait loci for S. sclerotiorum resistance from different sources
Six QTL for S. sclerotiorum resistance in B. napus were reported previously in a population derived from NingRS-1 × H5200; three conferred leaf resistance at the seedling stage and three others were involved in stem resistance at plant mature stage (Zhao and Meng 2003a). None of these QTL appears to correspond to the nine QTL reported here for the HUA and MS populations. Zhao and Meng (2003a) detected an effect on N17 where we also found a weak effect in the HUA population (one evaluation, LOD score of 2.5). These may represent the same resistance locus, and there may be additional loci in common among the parents that were undetected because of unmapped genomic regions, inability to detect small effects, or genotype × environment interactions which could be identified by further study. The fact that the remaining QTL were specific for each population suggests that S. sclerotiorum resistance in Hua dbl2, Major, and NingRS-1 is due, at least in part, to alleles at different QTL. Combining the different resistant alleles characterized in these studies may ultimately provide high levels of resistance that could be useful for breeding resistant canola cultivars. Additional studies are needed to determine the individual and combined effects of these QTL alleles in different genetic backgrounds.
References
Arahana VS, Graef GL, Specht JE, Steadman JR, Eskridge KM (2001) Identification of QTLs for resistance to Sclerotinia sclerotiorum in soybean. Crop Sci 41:180–188
Basten CJ, Weir BS, Zeng ZB (2002) QTL Cartographer, Version 1.16. Department of Statistics, North Carolina State University, Raleigh
Bert PF, Jouan I, Tourvieille de Labrouhe D, Serre F, Nicolas P, Vear F (2002) Comparative genetic analysis of quantitative traits in sunflower (Helianthus annuus L.) 1. QTL involved in resistance to Sclerotinia sclerotiorum and Diaporthe helianthi. Theor Appl Genet 105:985–993
Doerge RW, Churchill GA (1996) Permutation tests for multiple loci affecting a quantitative character. Genetics 142:285–294
Ferreira ME, Williams PH, Osborn TC (1994) RFLP mapping of Brassica napus using doubled haploid lines. Theor Appl Genet 89:615–621
Ferreira ME, Satagopan J, Yandell BS, Williams PH, Osborn TC (1995a) Mapping loci controlling vernalization requirement and flowering time in Brassica napus. Theor Appl Genet 90:727–732
Ferreira ME, Rimmer SR, Williams PH, Osborn TC (1995b) Mapping loci controlling Brassica napus resistance to Leptosphaeria maculans under different screening conditions. Phytopathology 85:213–217
Hartman GL, Gardner ME, Hymowitz T, Naidoo GC (2000) Evaluation of perennial Glycine species for resistance to soybean fungal pathogens that cause Sclerotinia stem rot and sudden death syndrome. Crop Sci 40:545–549
Hoffman DD, Diers BW, Hartman GL, Nickell CD, Nelson RL, Pedersen WL, Cober ER, Graef GL, Steadman JR, Grau CR, Nelson BD, del Rio LE, Helms T, Anderson T, Poysa V, Rajcan I, Stienstra WC (2002) Selected soybean plant introductions with partial resistance to Sclerotinia sclerotiorum. Plant Dis 86:971–980
Kim HS, Hartman GL, Manandhar JB, Graef GL, Steadman JR, Diers BW (2000) Reaction of soybean cultivars to Sclerotinia stem rot in field, greenhouse, and laboratory evaluations. Crop Sci 40:665–669
Knapp SJ, Stroup WW, Ross WW (1985) Exact confidence intervals for heritability on a progeny mean basis. Crop Sci 25:192–194
Kole C, Thormann CE, Karlsson BH, Palta JP, Gaffney P, Yandell B, Osborn TC (2002) Comparative mapping of loci controlling winter survival and related traits in oilseed Brassica rapa and B. napus. Mol Breed 9:201–210
Kolkman JM, Kelly JD (2003) QTL conferring resistance and avoidance to white mold in common bean. Crop Sci 43:539–548
Mariani C, Gossele V, de Beuckeleer M, de Block M, Goldberg RB, de Greef W, Leemans J (1992) A chimeric ribonuclease-inhibitor gene restores fertility to male-sterile plants. Nature 357:384–387
Mestries E, Gentzbittel L, de Labrouhe DT, Nicolas P, Vear F (1998) Analyses of quantitative trait loci associated with resistance to Sclerotinia sclerotiorum in sunflowers (Helianthus annuus L.) using molecular markers. Mol Breed 4:215–226
Miklas PN, Johnson WC, Delorme R, Gepts P (2001) QTL conditioning physiological resistance and avoidance to white mold in dry bean. Crop Sci 41:309–315
Oilcrop Research Institute, Chinese Academy of Sciences (1975) Sclerotinia disease of oilseed crops. Agriculture Press, Beijing
Osborn TC, Kole C, Parkin IA, Sharpe AG, Kuiper M, Lydiate DJ, Trick M (1997) Comparison of flowering time genes in Brassica rapa, B. napus and Arabidopsis thaliana. Genetics 146:1123–1129
Osborn TC, Butruille DV, Sharpe AG, Pickering KJ, Parkin IAP, John PS, Lydiate DJ (2003) Detection and effects of a homoeologous reciprocal transposition in Brassica napus. Genetics 165:1569–1577
Parkin IAP, Sharpe AG, Keith DJ, Lydiate DJ (1995) Identification of A and C genomes of amphidiploid Brassica napus (oilseed rape). Genome 38:1122–1131
Purdy LH (1979) Sclerotinia sclerotiorum: history, diseases and symptomatology, host range, geographic distribution and impact. Phytopathology 69:887–890
Quijada PA (2003) Introgression of germplasm from winter into spring Brassica napus: detection and confirmation of quantitative trait loci. PhD Thesis. University of Wisconsin.
Song KM, Osborn TC (1992) Polyphyletic origins of Brassica napus – new evidence based on organelle and nuclear RFLP analyses. Genome 35:992–1001
Thormann CE, Romero J, Mantet J, Osborn TC (1996) Mapping loci controlling the concentrations of erucic and linolenic acids in seed oil of Brassica napus L. Theor Appl Genet 93:282–286
Toroser D, Thormann CE, Osborn TC, Mithen R (1995) RFLP mapping of quantitative trait loci controlling seed aliphatic-glucosinolate content in oilseed rape (Brassica napus L.). Theor Appl Genet 91:802–808
U N (1935) Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn J Bot 7:384–452
Udall JA (2003) A genetic study of oilseed Brassica napus L.: mapping chromosome rearrangements and quantitative trait loci. PhD thesis. University of Wisconsin-Madison
Udall JA, Quijada PA, Polewicz H, Vogelzang R, Osborn TC (2004) Phenotypic effects of introgressing Chinese winter and resynthesized Brassica napus L. germplasm into hybrid spring Canola. Crop Sci 44:1990–1996
Udall JA, Quijada PA, Osborn TC (2005) Detection of chromosomal rearrangements derived from homoeologous recombination in four mapping populations of Brassica napus L. Genetics 169:967–979
Vuong TD, Hartman GL (2003) Evaluation of soybean resistance to Sclerotinia stem rot using reciprocal grafting. Plant Dis 87:154–158
Wegulo SN, Yang XB, Martinson CA (1998) Soybean cultivar responses to Sclerotinia sclerotiorum in field and controlled environment studies. Plant Dis 82:1264–1270
Zhao J, Meng J (2003a) Genetic analysis of loci associated with partial resistance to Sclerotinia sclerotiorum in rapeseed (Brassica napus L.). Theor Appl Genet 106:759–764
Zhao J, Meng J (2003b) Detection of loci controlling seed glucosinolate content and their association with Sclerotinia resistance in Brassica napus. Plant Breed 122:19–23
Zhao J, Peltier AJ, Meng J, Osborn TC, Grau CR (2004) Evaluation of sclerotinia stem rot resistance in oilseed Brassica napus using a petiole inoculation technique under greenhouse conditions. Plant Dis 88:1033–1039
Zhou LC, Yu Q, Liu SY, Zhou BW (1994) Evaluation of Sclerotinia resistance in rapeseed. Chin Oilseed Crop (supplement) 4:69–72
Acknowledgements
We thank Dr. Glen Hartman, USDA/ARS, University of Illinois for providing the S. sclerotiorum isolate. This work was funded by the USDA Sclerotinia Initiative.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. Möllers
Rights and permissions
About this article
Cite this article
Zhao, J., Udall, J.A., Quijada, P.A. et al. Quantitative trait loci for resistance to Sclerotinia sclerotiorum and its association with a homeologous non-reciprocal transposition in Brassica napus L. . Theor Appl Genet 112, 509–516 (2006). https://doi.org/10.1007/s00122-005-0154-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-005-0154-5