
Abstract
The chloroplast and mitochondrial DNA diversity of 61 genotypes belonging to 18 Vasconcellea species, the so-called highland papayas, was studied by PCR-RFLP analysis of two non-coding cpDNA regions (trnM-rbcL and trnK1-trnK2) and one non-coding mtDNA region (nad4/1-nad4/2). This sample set was supplemented with six genotypes belonging to three other Caricaceae genera: the monotypic genus Carica, including only the cultivated papaya, and the genera Jacaratia and Cylicomorpha. Moringa ovalifolia was added as an outgroup species. The PCR-amplified cpDNA regions were digested with 18 restriction endonucleases, the mtDNA region with 11. A total of 22 point mutations and four insertion/deletions were scored in the sample. A higher level of interspecific variation was detected in the two cpDNA regions in comparison to the analysis of the mtDNA. Wagner parsimony and Neighbor-Joining analysis resulted in dendrograms with similar topologies. PCR-RFLP analysis supported the monophyly of Caricaceae, but among the 26 mutations scored, an insufficient number of markers discriminated between the different Caricaceae genera included in this study. Hence the inference of the intergeneric relationships within Caricaceae was impossible. However, some conclusions can be noted at a lower taxonomic level. The Caricaceae species were divided into two lineages. One group included only Vasconcellea spp., whereas the second included the remaining Vasconcellea spp., together with the papaya genotypes and those from the other Caricaceae genera. This may indicate a higher level of inter-fertility for the Vasconcellea species from the latter clade in interspecific crossings with papaya. The putative progenitors of the natural sterile hybrid V. × heilbornii, i.e. V. stipulata and V. cundinamarcensis, were only distantly related to V. × heilbornii. This indicates that probably none of these species was involved as the maternal progenitor in the origin of V. × heilbornii. Surprisingly, V. × heilbornii had organellar genome patterns identical with V. weberbaueri, suggesting a possible involvement of this species in the origin of V. × heilbornii. On the basis of discrepancy between morphological traits and the cpDNA profiles of some pairs of Vasconcellea species, we believe that besides V. × heilbornii, some other species have originated through interspecific hybridization. A reticulate evolution for Vasconcellea has therefore been suggested. Finally, intraspecific cpDNA variation was detected in V. microcarpa, thus providing molecular evidence for the high diversity previously indicated by morphological observations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Caricaceae are a small dicotyledonous family consisting of six genera of herbaceous, shrubby or arborescent plants. This family is, with the exception of one African genus, entirely neotropical and has its largest distribution in South America. Formerly a genus, later only a section within the genus Carica, and then again rehabilitated as a genus by Badillo (2000), Vasconcellea is now the most important genus with 21 of the 35 Caricaceae species. As a result, the genus Carica is monotypic and only includes Carica papaya L., the papaya. Ecuador, which holds at least 15 of the 21 described Vasconcellea species, must unquestionably be considered as the biodiversity center of Vasconcellea. In its southern province Loja alone, where a large part of the genotypes included in this study were collected, at least seven species are recorded (Cueva 1999; Van den Eynden et al. 1999). These species grow above 1,000 m above sea level, mostly in the wild in humid forests, but are also semi-domesticated or tolerated in local gardens. In contrast to the Vasconcellea species, which have the highlands of South America as their center of origin, C. papaya in its wild form seems to have an entirely Central American distribution and a Central American center of domestication has been advocated by several authors (Purseglove 1974; Storey 1976). The four other genera of Caricaceae are Cylicomorpha, with two African tree species, Horovitzia with one species indigenous to Mexico, Jarilla with three herbaceous species all endemic to southern Mexico and Guatemala, and finally Jacaratia, which contains seven tree species from South America (Badillo 1971, 1983, 1993, 2000).
The economic importance of the Caricaceae resides largely in the fruit production of C. papaya L., widely cultivated throughout the tropics (Heywood 1985), with Brazil being the world’s major producer (FAO 2002). Besides the commercial value of the ripe fruit and its processed products, papaya is also cultivated, although to a lesser extent, for the latex present in its plant parts (Drew et al. 1998). The different proteinases, obtained from the latex extracted from unripe fruit, have a broad spectrum of activity and are therefore widely used in food and pharmaceutical industries (Madrigal et al. 1980).
Some species of the genus Vasconcellea, the so-called highland papayas, are regarded as unexploited crops, because of their tasty, high quality fruits and as a source of proteolytic enzymes (National Research Council 1989). The babaco (V. × heilbornii ‘Babaco’) is one of these interesting Vasconcellea species. Based on morphological observations, V. × heilbornii is believed to be a natural sterile hybrid derived from crosses between V. cundinamarcensis and V. stipulata. This species probably originated in the southern and central parts of Ecuador, where the parent species grow at altitudes between 1,600 and 2,800 m (Badillo 1967; Horovitz and Jimenez 1967). These hybrids generally show some degree of parthenocarpy and are mostly propagated by clonal means (Badillo 1993). According to Badillo (2001), two varieties (V. × heilbornii var. chrysopetala and V. × heilbornii var. fructifragrans) and one cultivar (V. × heilbornii ‘Babaco’) can be distinguished. Thanks to its high yields, the babaco is of great commercial interest. At the moment, it is the only Vasconcellea species cultivated in an intensive way, albeit only in Ecuador and New Zealand (Villareal et al. 2003). Besides the interest for highland papayas as possible fruit crops, some possess interesting traits that may be useful in papaya breeding (National Research Council 1989). Intergeneric crosses between Carica papaya and Vasconcellea species are problematic due to genome incompatibility (Magdalita et al. 1997), but in recent reports embryo rescue techniques were used to overcome these incompatibility barriers (Drew et al. 1998). This and other biotechnological approaches could lead to the introduction of specific traits of Vasconcellea species into papaya such as the resistance against Papaya Ringspot Virus and cold tolerance (Manshardt and Wenslaff 1989).
In recent years, molecular markers assessing chloroplast DNA (cpDNA) variation have proven to be a powerful tool for phylogenetic studies in a wide range of taxa, particularly at the interspecific level and higher (e.g. Tsumura et al. 1995; Cipriani et al. 1998; Bukhari et al. 1999; Parani et al. 2000). The usefulness of the chloroplast genome for molecular evolutionary studies lies in its highly conserved nature, both in terms of genome size and structure (Palmer 1987) and its predominant maternal inheritance in angiosperms, which excludes any recombination (Harris and Ingram 1991). A wide range of different methods have been applied in molecular studies of cpDNA (reviewed in Olmstead and Palmer 1994). The conserved arrangement of chloroplast genes and the availability of cpDNA sequence data from various taxa have provided the opportunity to design primers for the polymerase chain reaction (PCR) that amplify cpDNA sequences that are subject to different evolutionary rates (Ogihara et al. 1991; Taberlet et al. 1991; Demesure et al. 1995; Dumolin-Lapègue et al. 1997). Sequences with rapid evolutionary rates, yet moderate to low intraspecific polymorphism (e.g. non-coding intergenic spacer regions), are most appropriate for analyzing relationships among closely related species, whereas those with slower evolutionary rates (e.g. coding regions) may provide useful characters for studying relatively ancient divergences (Dowling et al. 1996). Compared with the chloroplast genome, plant mitochondrial DNA (mtDNA) is less studied because this genome changes rapidly in size and structure but about four times slower in primary sequence (Wolfe et al. 1987; Palmer 1992). As for the chloroplast genome, a set of universal mitochondrial primers, based on conserved coding sequences and amplifying more variable, non-coding regions, has been described (Demesure et al. 1995; Dumolin-Lapègue et al. 1997). Subsequent restriction of the obtained cpDNA and mtDNA PCR products with different restriction enzymes (PCR-RFLP) provides a fast, inexpensive and highly effective screening for DNA variation in the cytoplasmic organelles (Tsumura et al. 1995; Dumolin et al. 1995).
Despite the availability of numerous molecular marker techniques nowadays, the Caricaceae and more specifically the interesting Vasconcellea species have received little molecular genetic attention. Most molecular work focused on C. papaya using isozymes or nuclear markers for genetic diversity and mapping studies (isozymes, Morshidi 1998; RAPD, Sondur et al. 1996, Stiles et al. 1993; AFLP, Kim et al. 2002). Jobin-Décor et al. (1997) were the first to include six Vasconcellea spp. in a molecular analysis using isozymes and RAPD marker data. More recently Van Droogenbroeck et al. (2002) used the AFLP technique to perform the most complete diversity study so far comprising 95 genotypes representing at least eight Vasconcellea species, C. papaya and two Jacaratia species. In agreement with the rehabilitation of Vasconcellea as a genus based on morphological evidence (Badillo 2000) both studies illustrated the large genetic distance between C. papaya and the Vasconcellea species. Van Droogenbroeck et al. (2002) also examined a diverse set of V. × heilbornii genotypes. The specific clustering of these genotypes, together with either one of the putative parent species, provided support for the involvement of V. cundinamarcensis and V. stipulata in the origin of V. × heilbornii and suggested ongoing bi-directional introgression events between the parent species. However, so far only a single study utilized chloroplast DNA markers; Aradhya et al. (1999) applied the PCR-RFLP technique to investigate the variation in one cpDNA intergenic spacer region of papaya and 11 Vasconcellea species. The resulting cpDNA phylogeny again showed that papaya is only distantly related to the Vasconcellea species. Surprisingly, the V. × heilbornii ‘Babaco’ genotypes included in their study did not show cpDNA variation identical to either the pattern observed in V. stipulata or in V. cundinamarcensis, leaving the possible involvement of these species in the origin of V. × heilbornii unconfirmed. Finally, Olson (2002) used sequence analysis of the nuclear ITS and the chloroplast trnG locus from one to two members of four genera of Caricaceae to reconstruct intergeneric relationships. However, they overlooked the proposed rehabilitation of Vasconcellea as a genus and included V. microcarpa as the single representative of the genus Carica. In the tree resulting from the combined analysis of the ITS and trnG data sets, Cylicomorpha is the sister taxon to the rest of Caricaceae, which comprises Jarilla as the sister taxon to a Carica-Jacaratia clade (in fact a Vasconcellea-Jacaratia clade).
Despite the economical potential of the Vasconcellea species, few studies have included these wild relatives of papaya in a phylogenetic analysis. However, in the context of conservation of plant genetic resources, a better knowledge of the phylogenetic relationships within and among genera of crop species is of great importance to germplasm curators and plant breeders. Furthermore, many of the Vasconcellea species are intercompatible and can produce hybrids with various degrees of fertility. These occasionally occur spontaneously in areas where species distributions overlap (Badillo 1971). In addition, during previous ethnobotanical inventories of wild and semi-domesticated edible plants in southern Ecuador, an unrecognized variability among and within Vasconcellea spp. has been observed (Jiménez et al. 1999). Many of the collected Vasconcellea specimens observed during our field trips could not be identified at the species level using the current key of Badillo (1993), and some of them may represent interspecific hybrids (Romeijn-Peeters, personal observations). Besides, we have strong indications that inheritance of cpDNA and mtDNA is maternal in Caricaceae (Van Droogenbroeck et al., unpublished data), as in most angiosperms (Harris and Ingram 1991).
Therefore the main objectives of this study were: (1) to perform a more complete phylogenetic analysis using both cpDNA and mtDNA variation, including 18 of the 21 described Vasconcellea species and genotypes of three other genera within Caricaceae: Cylicomorpha, Carica and Jacaratia. Moringa ovalifolia (Moringaceae) was chosen as an outgroup because of its close taxonomic affinities with the family Caricaceae; (2) to investigate the involvement of V. stipulata and V. cundinamarcensis in the origin of V. × heilbornii; and (3) to identify other possible hybridization events. For these reasons we have carried out a PCR-RFLP analysis of two non-coding chloroplast regions, trnM-rbcL and trnK1-trnK2, and the non-coding mitochondrial region nad4/1-nad4/2.
Materials and methods
Plant materials and DNA extraction
Most of the plant material was collected in Loja province, southern Ecuador, from wild plants or plants tolerated in the gardens or land used by local farmers. Young leaves were collected in the field and dried with silica gel. During the field trips many specimens were found that could not be identified at the species level using the most recent key of Badillo (1993). These genotypes were excluded from the sample set. For determination at the subspecies level of the genotypes identified as V. microcarpa, shape of the leaf epidermal cells was investigated since flowers were absent and leaf morphology was too variable.
This collection was further extended with Caricaceae genotypes obtained from various contributors (Table 1). For two species leaf material was collected in their native distribution area, dried and then sent to us: Cylicomorpha parviflora (Nairobi, Kenya) and V. chilensis (Chile). For Carica papaya, we obtained two samples collected in an area where papaya was introduced (papPHI, Philippines; papD6, Australia). All the other samples were originally collected in their native area in South America by the National Clonal Germplasm Repository (NCGR), Hilo, Hawaii, and maintained afterwards in field plantings, seed storage or tissue culture. We obtained the samples either directly from the NCGR or through the kind cooperation of other research groups, which had obtained their material from NCGR. Dr. F. Zee provided dried leaf material of V. quercifolia (querHW8), originally collected in Paraguay. Dr. R. Ming sent us DNA solution from V. horovitziana, originally collected in Ecuador. Dr. R. Drew sent us dried leaf material from V. cauliflora (cauD3), originally collected in Brazil and from V. goudotiana (goudD4), originally collected in Columbia. Finally, for some species seeds were provided by Dr. R. Drew who had obtained the material from NCGR and maintained it in vitro and in field plantings. These seeds were grown to plants and leaf material was collected for the genotypes parvDP1, goudDGa, querDQ1, querDQ2 and querDQ3. Moringa ovalifolia (Moringaceae), collected in its native distribution area (Hardap Region, Namibia), was chosen as an outgroup because of its close taxonomic affinities with the family Caricaceae (Rodman et al. 1998). In total 68 genotypes were included in the PCR-RFLP analysis, comprising genotypes of 18 different Vasconcellea species, supplemented with genotypes of three other Caricaceae genera: Carica papaya, Cylicomorpha parviflora and two Jacaratia species and finally Moringa ovalifolia as an outgroup.
Total DNA was extracted from fresh or dried leaf tissue, ground in liquid nitrogen, using the Qiagen DNAeasy Plant Mini Kit (Qiagen).
PCR-RFLP analysis
Total DNA was used as template in PCR reactions involving a set of universal primers homologous to the most conserved regions of cpDNA and mtDNA and amplifying more variable, non-coding regions.
In a first step, six pairs of chloroplast primers and one pair of mitochondrial primers were used to test amplification among six genotypes: two Vasconcellea cundinamarcensis, two V. stipulata and two Carica papaya genotypes (Table 2). All primer pairs were as described in Dumolin-Lapègue et al. (1997), except for the sixth chloroplast primer pair described in Lotfy et al. (2003), which is a new combination derived from Demesure et al. (1995), amplifying a larger region. Based on these initial results, two cpDNA regions (K1K2 and ML) and one mtDNA region (nad4/1-2) were selected (Table 2).
In a second step, the PCR products from the two cpDNA and mtDNA regions, amplified for the initial set of six genotypes, were restricted with a large number of restriction enzymes to screen for polymorphisms. Both chloroplast PCR products were restricted with 18 different restriction endonucleases. The mitochondrial region nad4/1-2 was cut with 11 restriction enzymes. Finally, eight PCR fragment/enzyme combinations were selected and used to characterize the complete set of individuals listed in Table 1.
Amplifications of these cpDNA and mtDNA regions were performed in 25 μl of a reaction mixture containing 0.2 μM of each primer, 200 μM of each of the four dNTPs, 2 mM MgCl2, 1.0 U Taq DNA polymerase (Fermentas), 1× PCR buffer containing (NH4)2SO4 (Fermentas), and 50 ng of genomic DNA. The PCR amplifications were carried out in a Mastercycler Gradient (Eppendorf) with a heated lid, using an initial cycle of 4 min at 94°C, followed by 30 cycles with 45 s at 92°C, 45 s at T an (see Table 2) and 3 min at 72°C, and finally a 10 min extension at 72°C. The success of each PCR reaction was verified by electrophoresis of 4 μl PCR product on a 1% agarose gel containing ethidium bromide in 0.5× Tris-acetate EDTA (TAE) buffer and visualization under UV light. The sizes of the PCR products were estimated by comparison with a molecular size standard (Massruler DNA ladder, Fermentas).
Seven microliters of each PCR product was restricted in a volume of 20 μl containing 10 U of restriction enzyme following the manufacturer’s instructions. For the cp regions we tested nine tetracutters (AfaI, AluI, BstOI, HinfI, TaqI, HaeIII, MboI, MseI and HhaI) and nine hexacutters (BamHI, EcoRI, EcoRV, HindIII, KpnI, PstI, SacI, StyI and ScaI). For the PCR-RFLP analysis of the mitochondrial region, we tested 11 enzymes; among them were nine tetracutters (AfaI, AluI, BstOI, DdeI, HaeIII, HhaI, HinfI, MboI and MseI) and the hexacutters BamHI and EcoRI. Restriction fragments were separated on 2% agarose gels containing ethidium bromide in 1× Tris-borate EDTA (TBE), run at 125 V for 1.5–2 h and visualized under UV light. The size of each restriction fragment was estimated by comparison with a molecular size standard (Massruler DNA ladder, Fermentas).
Data analysis
Polymorphisms occurred as point mutations and insertion/deletions. In order to determine the nature of each mutation, every PCR product was restricted with at least two different enzymes. Size variation due to insertions/deletions was assumed when similar patterns were observed with different enzymes. Polymorphic fragments were labeled by decreasing order of fragment length as visualized in the agarose gels. Haplotypes were defined according to different combinations of all detected mutations.
Detailed data analysis was performed in two ways, using PAUP* 4.0 (Swofford 2002). For phylogenetic analysis a restriction SITE data set was generated in which the presence of a restriction site is coded as 1 and the absence as 0. This data matrix was used to perform a Wagner parsimony analysis to find the shortest trees. A heuristic search was carried out one hundred times using the random-addition-sequence option in PAUP and the ACCTRAN, TBR (tree bisection-reconnection) branch swapping, MULTREES, and COLLAPSE options of the program to search for the most parsimonious tree(s). A 50% majority rule consensus tree was calculated from the most parsimonious trees using the CONTREE command in PAUP. Bootstrap values were calculated from 100 replicates using the BOOTSTRAP command in PAUP. In the present study length mutations (insertion/deletions) were not included in the phylogenetic analysis because their homologies are uncertain above species level (Soltis et al. 1991). In addition, for phenetic analysis a binary data matrix was generated, with the presence of a restriction fragment coded as 1, and its absence coded as 0. This data set is henceforth called the restriction FRAGMENT data set. From this data matrix the Nei and Li coefficient of genetic similarity (Nei and Li 1979) was calculated between each pair of genotypes after the optimality criterion was set to DISTANCE. Cluster analysis was carried out using the Neighbor-Joining (Saitou and Nei 1987) method and bootstrap values were calculated from 100 replicates using the BOOTSTRAP command in PAUP.
Results
PCR amplification and restriction analysis
In a preliminary survey including papaya and two Vasconcellea species, only two out of six cpDNA regions were strongly amplified for all six genotypes (K1K2 and ML). The use of the mitochondrial primer pair nad4/1-2 also resulted in a strong amplification of this region for papaya and the two Vasconcellea species tested (Table 2). These three sets of primers gave amplified products in all genotypes.
In order to find enzymes that revealed polymorphisms among the three selected amplified regions of the different taxa, the PCR products of the six genotypes included in the amplification test were cut with several restriction enzymes. Both chloroplast regions (K1K2 and ML) were restricted with nine tetracutters and nine hexacutters. For K1K2, seven of these did not have any restriction sites (HhaI, HindIII, KpnI, PstI, SacI, StyI, ScaI) and only three (AfaI, EcoRV and ScaI) revealed polymorphism among the six genotypes included in this restriction test. For the ML region, 15 of the 18 restriction enzymes tested were able to recognize at least one restriction site; the hexacutters EcoRV, HindIII and KpnI had no recognition site within ML. A single tetracutter (MseI) and one hexacutter (PstI) detected variation among the six genotypes selected for the restriction test.
For the PCR-RFLP analysis of the mitochondrial region, we tested nine tetracutters and two hexacutters. All had at least one recognition site within nad4/1-2, but only BstOI, DdeI and HinfI showed informative restriction profiles for the six genotypes included in the restriction test. Summarizing, eight fragment/enzyme combinations were informative and were selected to fingerprint the complete data set: K1K2/EcoRV, K1K2/ScaI, K1K2/AfaI, ML/PstI, ML/MseI and nad4/1-2/HinfI, nad4/1-2/BstOI, nad4/1-2/DdeI.
PCR-RFLP haplotypes
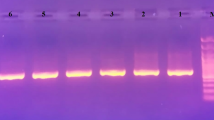
When these eight selected fragment/enzyme combinations were applied to analyze the complete sample set given in Table 1, many polymorphic fragments were detected (Table 3). Eighteen point mutations and two indels were identified using the cpDNA fragment/enzyme combinations. Of these, ten point mutations were found with the combination K1K2/AfaI, and two indels could be confirmed with at least two restriction enzymes. The high level of polymorphism detected with the combination K1K2/AfaI is illustrated in Fig. 1A. Lanes 11 and 12 of Fig. 1A also clearly illustrate the indel specific for the V. goudotiana genotypes. The fragment of about 270 bp observed in the genotypes shown in lanes 1–4 and lane 6 (band X in Fig. 1A) is replaced in the V. goudotiana genotypes (lanes 11 and 12) by a fragment with an approximate length of 200 bp (band X′ in Fig. 1A). This deletion of approximately 70 bp was also observed in the restriction patterns of the region K1K2 restricted with the enzymes EcoRV and ScaI. The other indel found in the region K1K2 was detected with both ScaI and AfaI, and represented only a slight difference in size (illustrated for ScaI in Fig. 1B). Due to this very small size variation, this indel could not be verified in the combination K1K2/EcoRV. Although the amplified ML fragment is about 500 bp larger than K1K2, the enzymes PstI and MseI revealed only five point mutations (Table 3).

A Restriction fragment patterns of cpDNA from 14 Vasconcellea taxa and Carica papaya detected by the fragment/enzyme combination K1K2/AfaI. M Massruler DNA ladder (size in base pairs). Polymorphic bands used for haplotype definition (found in Tables 3, 4) are labeled on the photo. The alternative fragment of band X is indicated as band X′ (without insertion or with a deletion of about 70 bp). Lanes 1–15 represent the following taxa: 1 V. cundinamarcensis, 2 V. palandensis, 3 V. monoica, 4 V. longiflora, 5 V. stipulata, 6 V. cauliflora, 7 V. × heilbornii ‘Babaco’, 8 V. weberbaueri, 9 V. quercifolia, 10 V. parviflora, 11 V. goudotiana (goudD4), 12 V. goudotiana (goud24), 13 C. papaya, 14 V. candicans. B Detection of one indel with fragment/enzyme combination K1K2/ScaI (band III). M Massruler DNA ladder (size in base pairs). Lanes 1–4 represent the following accessions: 1 V. candicans (can113, 510 bp), 2 V. candicans (can171, 510 bp), 3 V. parviflora (parvDP1, 540 bp), 4 V. chilensis (chil, 560 bp)
Combination of the different variants detected in the cpDNA regions K1K2 and ML from all investigated genotypes allowed the identification of 15 different chloroplast haplotypes (Table 4). Of the 11 haplotypes detected among the Vasconcellea species, six were specific for a single described species. The two Jacaratia species, Carica papaya and Moringa ovalifolia also have a private chloroplast haplotype. The remaining five haplotypes were found in two or more species. Only V. microcarpa displayed intraspecific variation at the cpDNA level. The subspecies microcarpa had the same haplotype as V. monoica, V. palandensis and V. omnilingua (haplotype A) while subspecies baccata and heterophylla shared haplotype B with three other species.
For the mitochondrial nad4/1-2 region, four point mutations and one indel were detected. This indel was specific for Moringa ovalifolia, illustrating its distant relationship to the Caricaceae. The combination of the data obtained from the mutations detected in the mitochondrial region nad4/1-2 resulted in the identification of five different mitochondrial haplotypes (mitotypes; Table 5). Species sharing the same chloroplast haplotype always had the same mitotype. Two major groups containing 18 of the 21 species investigated were found, differing from each other by two point mutations. The first group (mitotype A) contained only Vasconcellea genotypes (eight species) while the second (mitotype C) included the genotypes of the three other Caricaceae genera (Carica papaya, Cylicomorpha parviflora and the two Jacaratia species) together with seven Vasconcellea spp. Two remaining minor haplotypes were assigned to V. cundinamarcensis having a private mitotype (mitotype B) with one extra point mutation in comparison to mitotype A, while V. quercifolia and V. chilensis (mitotype D) were separated from the species sharing mitotype C by one point mutation. Finally Moringa ovalifolia could be distinguished from the species with mitotype C by its specific indel (mitotype E). None of the investigated species displayed intraspecific variation for the mtDNA-region investigated.
Phylogenetic and phenetic analysis
The phylogenetic analysis was started by comparing the results obtained from a PAUP analysis using only the 18 cpDNA point mutations with that of the combined data set, including both the 18 cpDNA and 4 mtDNA point mutations. Since only minor differences were found, together with the fact that we have strong indications that both genomes are inherited maternally in Caricaceae (Van Droogenbroeck et al., unpublished data), only the results derived from the combined data set will be further considered.
The Wagner parsimony analysis of this data set containing 22 point mutations, of which 17 were parsimony-informative, resulted in 66 equally parsimonious trees with a length of 31 steps, a consistency index of 0.71 (0.65 excluding uninformative characters), a homoplasy index of 0.29 and a retention index of 0.97. Figure 2 shows the 50% majority-rule consensus tree generated from the most parsimonious trees.

A 50% majority-rule consensus tree obtained from the 66 most parsimonious trees based on PCR-RFLP restriction SITE data derived from two chloroplast regions and one mitochondrial region. Accessions are labeled according to the codes listed in Table 1. Only bootstrap values above 50% are shown
The in-group taxa were divided into three clades and a group of two species forming a polytomy. The first clade (moderate bootstrap support of 53%) is the largest, comprising only Vasconcellea species and is further subdivided into four subgroups. The first subgroup, clade 1A, is formed by the genotypes sharing haplotype B (see Table 6). Within this group we find V. longiflora, V. pulchra and remarkably also two subspecies of V. microcarpa, i.e. subsp. baccata and heterophylla. Subgroup 1B holds only V. cundinamarcensis (haplotype D), while the species having haplotypes A and C were united in the unresolved subgroup 1C. In this cluster, the third subspecies of V. microcarpa included in this study, i.e. subsp. microcarpa, is placed together with V. monoica, V. palandensis, V. omnilingua and V. goudotiana. Subgroup 1D, holding V. cauliflora, is the basal lineage of this subcluster. The second largest clade includes five Vasconcellea species, comprising the two subspecies of V. × heilbornii, together with the genotypes from the other genera of the family. The V. × heilbornii genotypes share all 22 point mutations with V. weberbaueri and form clade 2A. Sister to this clade is the clade 2B, containing V. candicans as well as C. papaya in separate, species-specific subclusters. In clade 2C, V. quercifolia and V. chilensis are grouped together. The remaining two clades hold Cylicomorpha parviflora (2D) and the two Jacaratia species (2E). Clade 3 is specific for V. stipulata and group 4 finally consists of an unresolved group of V. parviflora and V. crassipetala genotypes.
In addition to the phylogenetic analysis using restriction site presence or absence, a restriction FRAGMENT data set was generated in which the presence or absence of each polymorphic restriction fragment was scored. In total 67 fragments were scored of which 45 were polymorphic (Table 7). This data set was used to perform a phenetic analysis using the Neighbor-Joining clustering method. Overall, the topology of the NJ tree (Fig. 3) is very similar to the 50% majority-rule consensus tree of the 66 most parsimonious trees except for some small differences. These differences mostly stem from the fact that the restriction FRAGMENT data set included polymorphisms generated by insertion/deletion events. Vasconcellea chilensis, the group of V. stipulata genotypes, as well as the V. parviflora-V. crassipetala complex now form distinct subclusters within clade 2, leaving only two major groups in the NJ analysis. Another difference between the two dendrograms is the separate grouping of the V. goudotiana genotypes (1E) in the NJ tree.

Neighbor-joining tree based on PCR-RFLP restriction FRAGMENT data derived from two chloroplast regions and one mitochondrial region. Accessions are labeled according to the codes listed in Table 1. Groups also identified in Fig. 2 carry the same label as in Fig. 2, bold labels indicate that the specific group was not identified as such in the parsimony analysis. Cluster 2B was split into two clusters, 2B a and b because the Cylicomorpha and Jacaratia genotypes took another position in comparison to Fig. 2. Only bootstrap values above 50% are shown
Discussion
Restriction fragment or restriction site data set?
Restriction fragment data sets based on fragment presence/absence are seldomly used for phylogenetic purposes since the same event could be counted two or more times. If one fragment is cut into two smaller ones due to a point mutation resulting in the gain of a specific restriction site, and the corresponding polymorphic fragments are scored, the same information is counted three times. By doing so, the assumption of independence among characters is violated. Moreover, in the absence of a restriction map reconstruction, two fragments of equal length coming from different parts of the sequence cannot be distinguished and the number of informative characters could therefore be underestimated. However, there is evidence that the latter risk is negligible with short sequences and when working with closely related species (Bremer 1991). On the other hand, when working with a restriction site data set, insertion/deletions events should be excluded because their homologies are uncertain above species level (Soltis et al. 1991). In addition, when large cpDNA or mtDNA regions are restricted with tetracutters, restriction profiles are often complex banding patterns with many fragments. This, in combination with a data set containing a large number of different species, often makes it difficult to construct a restriction map. In that case also a restriction site-based data set may contain linked or biased information. So, neither of the two data sets may in fact represent the correct relationships among the samples investigated. By using both the phenetic and phylogenetic analyses and comparing the results, a better estimation of the true genetic relationships can be inferred in comparison to the result obtained with either one of the approaches (Grant 2003).
In our case, the clustering obtained with the two methods was very similar, but the NJ tree showed better resolution. This is probably due to the extra information derived from the insertion/deletion events, which were not included in the phylogenetic analysis. Twelve percent of the mutations detected in this study were insertion/deletion events. Similar levels of insertion/deletions were found in comparable PCR-RFLP studies [e.g. Cipriani et al. 1998 (13%); Nwakanma et al. 2003 (13%)].
Although the number of mutations detected may be considered low for a phylogenetic reconstruction, a low level of homoplasy was expected, as reported for other taxonomic reconstructions carried out at low hierarchical levels (Jansen et al. 1992). However, homoplastic characters, which are characters with multiple changes resulting from parallel or convergent evolution and from character-state reversals, occurred often. This increased the number of equally parsimonious trees produced by the phylogenetic analysis. The conflicts between trees resulted in few clades supported in the 50% majority rule consensus tree and low to moderate bootstrap support for the clusters identified. Both the parsimony and phenetic analysis therefore gave insufficient information about the intergeneric relationships and the evolution of Caricaceae; however, some important results at a lower taxonomic level can be noted.
Caricaceae phylogeny
Only one phylogenetic analysis based on chloroplast DNA variation among Caricaceae, including Carica papaya, Jacaratia mexicana and 11 Vasconcellea species, has been published so far (Aradhya et al. 1999). Our study is the first to include data from mtDNA variation of Caricaceae.
The present results suggest the existence of two lineages within Caricaceae. The first one comprises only Vasconcellea species, while the second group holds the remaining Vasconcellea species together with genotypes from Carica, Cylicomorpha and Jacaratia. This subdivision into two lineages is in contrast with the results of Aradhya et al. (1999) and the proposed rehabilitation of Vasconcellea as a separate genus by Badillo (2000). Detailed comparison with the results of Aradhya et al. (1999) is difficult for two reasons: (1) the smaller set of species used in their analysis might influence the individual position of genotypes (e.g. Sanderson and Donoghue 1989) and (2) and probably more importantly, they investigated a larger number of characters derived from a single cpDNA region, whereas we investigated fewer characters from multiple regions. However, the mutations we detected were not sufficiently numerous for clarifying the intergeneric relationships within Caricaceae.
Nevertheless, the observed clustering may have some implications for plant breeding. Van Raamsdonk et al. (2003) proposed the hypothesis that a shorter distance between two species in a cpDNA tree, compared to their distance in a tree based on nuclear data, would predict inter-fertility at a certain level. This hypothesis is based on the situation that chloroplast capture, i.e. a short distance in cpDNA tree, is the result of effective inter-fertility to some extent. The validity of this hypothesis can be verified by comparison of the clustering found here with results on intergeneric and interspecific Caricaceae crosses; Drew et al. (1998) found that it was more difficult to cross papaya with V. cauliflora or V. goudotiana (both belonging to clade 1, Fig. 3) than with V. quercifolia or V. parviflora (belonging to the same clade as papaya, Fig. 3). Breeding programs aimed at producing intergeneric hybrids between papaya and Vasoncellea spp. should therefore focus on the wild Vasconcellea species belonging to the same clade as papaya.
Vasconcellea phylogeny
In the present study, the species V. longiflora, V. pulchra, V. horovitziana and two subspecies of V. microcarpa, i.e. subsp. baccata and heterophylla, grouped together, all growing in or at the edges of cloud forests. The first two species are considered as close relatives based on morphology (Badillo 1993), but for the two subspecies of V. microcarpa, the clustering within this group was unexpected since they clustered separately from the third V. microcarpa subspecies, i.e. subsp. microcarpa. A second subgroup within Vasconcellea holding multiple species is the morphologically diverse complex V. monoica, V. palandensis, V. omnilingua and V. microcarpa subsp. microcarpa. In a previous AFLP analysis (Van Droogenbroeck et al. 2002), V. monoica, and V. palandensis also clustered together, confirming their close genetic relationship. The clustering of V. microcarpa subsp. microcarpa is in congruence with the results obtained by Aradhya et al. (1999) where the complex V. monoica-V. microcarpa subsp. microcarpa was the only well-supported subgroup that could be identified among the Vasconcellea species analyzed.
In addition several pairs of species, which are placed far away from each other on morphological grounds (Badillo 1993), appear to be closely related in terms of the cpDNA analysis. For instance, V. parviflora and V. crassipetala are morphologically clearly distinct (flower color, fruits; Badillo 1993), but they do not show any differences in chloroplast DNA restriction site or fragment data. A second case is the pair V. weberbaueri and V. × heilbornii. The degree of similarity based on morphological traits relies mainly on the expression of nuclear genes and thus traces a biparental lineage, whereas that traced by cpDNA data is maternal in Caricaceae (Van Droogenbroeck et al., unpublished data). The most likely explanation for the incongruence between morphological and cpDNA classification is that one species within each pair or species complex is a natural hybrid having another species from the group as the maternal parent. If, besides V. × heilbornii, other taxa are indeed natural interspecific hybrids, a reticulate evolution must be postulated for Vasconcellea. The occurrence of natural hybridization within Vasconcellea has already been suggested by Badillo (1971) and is further supported by the fact that most Vasconcellea species easily cross in interspecific controlled crosses (Mekako and Nakasone 1975; Drew et al. 1998).
Intraspecific variation within V. microcarpa
In agreement with Aradhya et al. (1999) who revealed intraspecific cpDNA variation in V. microcarpa, we also detected intraspecific cpDNA differences among the V. microcarpa genotypes included in this work. These results provide molecular evidence for the highly diverse nature of V. microcarpa observed using morphological characteristics. All three V. microcarpa subsp. microcarpa genotypes group together, whereas the subsp. heterophylla and baccata are placed in another cluster suggesting that subsp. microcarpa could be considered as a distinct species. The intraspecific variation observed in cpDNA in V. microcarpa may have resulted from introgression (‘chloroplast capture’) involving other Vasconcellea species, possibly V. longiflora or V. pulchra, as suggested by Aradhya et al. (1999) and is supported by the sympatric distribution observed for these three species during our collection trips. However, additional studies and extensive sampling of all described subspecies of V. microcarpa are needed to resolve the exact phylogenetic relationships within this diverse group of genotypes.
The origin of V. × heilbornii
Certainly the most surprising result of this PCR-RFLP study is the close relationship between V. × heilbornii and V. weberbaueri, moderately supported by bootstrap confidence in the phenogram. Preliminary results of the sequence analysis of the matK region are in perfect agreement with this PCR-RFLP study and illustrate the same close relationship between these two species (Kyndt et al., in preparation). However, in the AFLP analysis this close relationship was not as evident since V. weberbaueri, together with V. parviflora, was placed in a subcluster between V. stipulata and V. cundinamarcensis (Van Droogenbroeck et al. 2002). As mentioned above, the shorter distance between V. × heilbornii and V. weberbaueri in our cpDNA phylogeny in comparison to their distance in the AFLP-based phenogram may predict inter-fertility at a certain level (Van Raamsdonk et al. 2003). Unfortunately Aradhya et al. (1999) did not include V. weberbaueri, but they observed that the two babaco genotypes in their study neither showed the cp haplotype of V. stipulata nor that of V. cundinamarcensis. This was also observed in our study; for three out of the five cpDNA-region/enzyme combinations analyzed, V. × heilbornii had a PCR-RFLP pattern different from both of that of its two putative parent species V. cundinamarcensis and V. stipulata, but identical to that of V. weberbaueri. For the remaining two cpDNA-region/enzyme combinations, V. × heilbornii shared its PCR-RFLP pattern with V. stipulata, V. weberbaueri and some other species. These results also illustrate the importance of including different regions randomly spread over the chloroplast genome, in combination with several restriction enzymes, when using PCR-RFLP to investigate cpDNA diversity and infer phylogenies. In both the cladogram and the phenogram, both putative parent species suggested by Badillo (1967) were only distantly related to V. × heilbornii and these results suggest that probably none of these two species was involved as the maternal progenitor in the origin of V. × heilbornii. Based on the geographical distribution of V. weberbaueri and the results obtained here, a common maternal ancestor for V. × heilbornii and V. weberbaueri is very likely.
References
Aradhya MK, Manshardt RM, Zee F, Morden CW (1999) A phylogenetic analysis of the genus Carica L. (Caricaceae) based on restriction fragment length variation in a cpDNA intergenic spacer region. Genet Res Crop Evol 46:579–586
Badillo VM (1967) Acerca de la naturalezea hibrida de Carica pentagona, C. chrysopetala y C. fructifragrans, frutales del Ecuador y Colombia. Rev Fac Agron Univ Centr Venez 4:3–14
Badillo VM (1971) Monografia de la familia Caricaceae. Publicada por la Associacion de Profesores, Univ Centr Venez, Venezuela
Badillo VM (1983) Caricaceae. In: Harling G, Sparre B (eds) Flora of Ecuador. Balogh Scientific Books, Illinois, pp 27–47
Badillo VM (1993) Caricaceae. Segundo esquema. Rev Fac Agron Univ Centr Venez 43:1–111
Badillo VM (2000) Carica L. vs. Vasconcella St. Hil. (Caricaceae): con la rehabilitación de este último. Ernstia 10:74–79
Badillo VM (2001) Nota correctiva Vasconcellea St. Hil y no Vasconcella (Caricaceae). Ernstia 11:75–76
Bremer B (1991) Restriction data from chloroplast DNA for phylogenetic reconstruction: is there only one accurate way of scoring? Plant Syst Evol 175:39–54
Bukhari YM, Koivu K, Tigerstedt PMA (1999) Phylogenetic analysis of Acacia (Mimosaceae) as revealed from chloroplast RFLP data. Theor Appl Genet 98:291–298
Cipriani G, Testolin R, Gardner R (1998) Restriction-site variation of PCR-amplified chloroplast DNA regions and its implications for the evolution and taxonomy of Actinidia. Theor Appl Genet 96:389–396
Cueva E (1999) Recolección, clasificación y estudio etnobotánico de los recursos fitogenéticos arbóreos y arbustivos nativos, productores de frutos comestibles, de la provincia de Loja. Dissertation, Universidad Nacional de Loja, Ecuador
Demesure B, Sodzi N, Petit RJ (1995) A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol Ecol 4:129–131
Dowling TE, Moritz C, Palmer JD, Rieseberg LH (1996). Nucleic acids III: analysis of fragments and restriction sites. In: Hillis et al. (eds) Molecular systematics. Sinauer, Sunderland, Mass., pp 249–320
Drew RA, O’Brien CM, Magdalita PM (1998) Development of interspecific Carica hybrids. Acta Hortic 461:285–292
Dumolin S, Demesure B, Petit RJ (1995) Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet 91:1253–1256
Dumolin-Lapègue S, Pemonge MH, Petit RJ (1997) An enlarged set of consensus primers for the study of organelle DNA in plants. Mol Ecol 6:393–397
Food and Agriculture Organization (2002) Statistical databases of the Food and Agriculture Organization of the United Nations. http://apps.fao.org
Grant V (2003) Incongruence between cladistic and taxonomic systems. Am J Bot 90:1263–1270
Harris SA, Ingram R (1991) Chloroplast DNA and biosystematics: the effects of intraspecific diversity and plastid transmission. Taxon 40:393–412
Heywood VH (1985) Flowering plants of the world. Croom Helm, London
Horovitz S, Jimenez H (1967) Cruzamientos interespecificos e intergenericos en Caricaceas y sus implicaciones fitotecnicas. Agron Trop 17:323–344
Jansen RK, Michaels HJ, Wallace RS, Kim K-J, Keeley SC, Watson LE, Palmer JD (1992) Chloroplast DNA variation in the Asteraceae: phylogenetic and evolutionary implications. In: Soltis PE, Soltis DE, Doyle JJ (eds) Molecular systematics of plants. Chapman and Hall, New York, pp 252–279
Jiménez Y, Romero J, Scheldeman X (1999) Colección, caracterización y descripción de Carica × heilbornii nm. pentagona B., Carica pubescens (A.D.C.) Solms-Laub y Carica stipulata B., en la provincia de Loja. Rev Difus Técn Cient Fac Cienc Agric Univ Nac Loja 29:43–54
Jobin-Décor MP, Graham GC, Henry RJ, Drew RA (1997) RAPD and isozyme analysis of genetic relationships between Carica papaya and wild relatives. Genet Res Crop Evol 44:471–477
Kim MS, Moore PH, Zee F, Fitch MMM, Steiger DL, Manshardt RM, Paull RE, Drew RA, Sekioka T, Ming R (2002) Genetic diversity of Carica papaya as revealed by AFLP markers. Genome 45:503–512
Lotfy S, Luro F, Carreel F, Froelicher Y, Rist D, Ollitrault P (2003) Application of cleaved amplified polymorphic sequence method for analysis of the cytoplasmic genome among Aurantioideae intergeneric somatic hybrids. J Am Soc Hortic Sci 128:225–230
Madrigal L, Ortiz AN, Cooke RD, Fernandez RH (1980) The dependence of crude papain yields on different collection (‘Tapping’) procedures for papaya latex. J Sci Food Agric 31:279–285
Magdalita PM, Drew RA, Adkins SW, Godwin ID (1997) Morphological, molecular and cytological analysis of Carica papaya×C. cauliflora interspecific hybrids. Theor Appl Genet 95:224–229
Manshardt RM, Wenslaff TF (1989) Interspecific hybridization of papaya with other species. J Am Soc Hortic Sci 114:689–694
Mekako HU, Nakasone HY (1975) Interspecific hybridization among six Carica species. J Am Soc Hortic Sci 100:237–242
Morshidi M (1998) Genetic control of isozymes in Carica papaya L. Theor Appl Genet 103:89–94
National Research Council (1989) Highland papayas. In: Ruskin FR (ed) Lost crops of the Incas: little-known plants of the Andes with promise for worldwide cultivation. National Academy Press, Washington D.C., pp 252–261
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5267–5273
Nwakanma DC, Pillay M, Okoli BE, Tenkouano A (2003) Sectional relationships in the genus Musa L. inferred from the PCR-RFLP of organelle DNA sequences. Theor Appl Genet, DOI 10.1007/s00122-003-1340-y
Ogihara Y, Terachi T, Sasakuma T (1991) Molecular analysis of the hot spot region related to length mutation in wheat chloroplast DNAs. I. Nucleotide divergence of genes and intergenic spacer regions located in the hot spot region. Genetics 129:321–332
Olmstead RG, Palmer JD (1994) Chloroplast DNA systematics: a review of methods and data analysis. Am J Bot 81:1205–1224
Olson ME (2002) Intergeneric relationships whitin the Caricaceae-Moringaceae clade (Brassicales) and potential morphological synapomorphies of the clade and its families. Int J Plant Sci 163:51–65
Palmer JD (1987) Chloroplast DNA evolution and biosystematic uses of chloroplast DNA variation. Am Nat 130:S6–S29
Palmer JD (1992) Comparison of chloroplast and mitochondrial genome evolution in plants. In: Herrmann R (ed) Cell organelles. Springer, Berlin Heidelberg New York, pp 99–133
Parani M, Lakshmi M, Ziegenhagen B, Fladung M, Senthilkumar P, Parida A (2000) Molecular phylogeny of mangroves VII. PCR-RFLP of trnS-psbC and rbcL gene regions in 24 mangrove and mangrove-associated species. Theor Appl Genet 100:454–460
Purseglove JW (1974) Tropical crops: dicotyledons. Longman, London, pp 86–104
Rodman JE, Soltis PS, Soltis DE, Sytsma KJ, Karol KG (1998) Parallel evolution of glucosinolate biosynthesis inferred from congruent nuclear and plastid gene phylogenies. Am J Bot 85:997–1006
Saitou N, Nei M (1987) The neighbour-joining method: a new method reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sanderson MJ, Donoghue MJ (1989) Patterns of variation in levels of homoplasy. Evolution 43:1781–1795
Soltis DE, Soltis PS, Collier TG, Edgerton ML (1991) Chloroplast DNA variation within and among genera of the Heuchera group (Saxifragaceae): evidence for chloroplast transfer and paraphyly. Am J Bot 78:1091–1112
Sondur SN, Manshardt RM, Stiles JI (1996) A genetic linkage map of papaya based on randomly amplified polymorphic DNA markers. Theor Appl Genet 93:547–553
Stiles JI, Lemme C, Sondur S, Morshidi MB, Manshardt RM (1993) Using randomly amplified polymorphic DNA for evaluating genetic relationships among papaya cultivars. Theor Appl Genet 85:697–701
Storey WB (1976) Papaya, Carica papaya. In: Simmonds NW (ed) Evolution of crop plants. Longman, London, pp 21–24
Swofford DL (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods), Version 4.0. Sinauer, Boston
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Tsumura Y, Yoshimura K, Tomaru N, Ohba K (1995) Molecular phylogeny of conifers using RFLP analysis of PCR-amplified specific chloroplast genes. Theor Appl Genet 91:1222–1236
Van den Eynden V, Cueva E, Cabrera O (1999) Plantas silvestres comestibles del sur del Ecuador. Ediciones Abya-Yala, Quito, Ecuador
Van Droogenbroeck B, Breyne P, Goetghebeur P, Romeijn-Peeters E, Kyndt T, Gheysen G (2002) AFLP analysis of genetic relationships among papaya and its wild relatives (Caricaceae) from Ecuador. Theor Appl Genet 105:289–297
Van Raamsdonk LWD, Ensink W, Van Heusden AW, Vrielink-Van Ginkel M, Kik C (2003). Biodiversity assessment based on cpDNA and crossability results in selected species of Allium subgenus Rhizirideum. Theor Appl Genet 107:1048–1058
Villarreal L, Dhuique-Mayer C, Dornier M, Ruales J, Reynes M (2003) Evaluation de l’intérêt du babaco (Carica pentagona Heilb.). Fruits 58:39–52
Wolfe KH, Li WH, Sharp PM (1987) Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast and nuclear DNAs. Proc Natl Acad Sci USA 84:9054–9058
Acknowledgements
The authors thank Dr. Drew, Dr. Fichet, Dr. Thuan, Dr. Muasya, Dr. Zee and Dr. Ming for providing leaf material or DNA. This research project was funded by the Flemish Fund for Scientic Research (FWO-Vlaanderen Project no. 3G005100). Appreciation for assistance during sample collection and cooperation in Ecuador is extended to Naturaleza & Cultura Internacional. The experiments comply with current Belgian laws and regulations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. Nybom
Rights and permissions
About this article
Cite this article
Van Droogenbroeck, B., Kyndt, T., Maertens, I. et al. Phylogenetic analysis of the highland papayas (Vasconcellea) and allied genera (Caricaceae) using PCR-RFLP. Theor Appl Genet 108, 1473–1486 (2004). https://doi.org/10.1007/s00122-003-1575-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-003-1575-7