
Abstract
The low-molecular-weight (LMW) glutenin subunits are components of the highly cross-linked glutenin polymers that confer viscoelastic properties to gluten and dough. They have both quantitative and qualitative effects on dough quality that may relate to differences in their ability to form the inter-chain disulphide bonds that stabilise the polymers. In order to determine the relationship between dough quality and the amounts and properties of the LMW subunits, we have transformed the pasta wheat cultivars Svevo and Ofanto with three genes encoding proteins, which differ in their numbers or positions of cysteine residues. The transgenes were delivered under control of the high-molecular-weight (HMW) subunit 1Dx5 gene promoter and terminator regions, and the encoded proteins were C-terminally tagged by the introduction of the c-myc epitope. Stable transformants were obtained with both cultivars, and the use of a specific antibody to the c-myc epitope tag allowed the transgene products to be readily detected in the complex mixture of LMW subunits. A range of transgene expression levels was observed. The addition of the epitope tag did not compromise the correct folding of the trangenic subunits and their incorporation into the glutenin polymers. Our results demonstrate that the ability to specifically epitope-tag LMW glutenin transgenes can greatly assist in the elucidation of their individual contributions to the functionality of the complex gluten system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The manufacture of pasta, bread and some other leavened and unleavened wheat-based products depends on the capacity of wheat flour to give dough with particular characteristics of elasticity and viscosity, whose balance is described as strength.
The pasta-making quality of European durum wheat cultivars is generally limited by the low elasticity of their dough, which negatively affects some important characteristics of cooked pasta, such as firmness.
It has long been known that wheat dough elasticity is primarily determined by the glutenin fraction of the gluten proteins (Huebner and Wall 1976), which comprises high-molecular-weight (HMW) and the low-molecular-weight (LMW) glutenin subunits, linked by inter-chain disulphide bonds to form polymers with a broad molecular-weight distribution.
A series of correlative studies have shown significant associations between variation in the amount and size distribution of the glutenin polymers and the breadmaking quality of hexaploid wheat cultivars (Field et al. 1983; Gupta et al. 1995). In particular, a strong association has been shown between gluten strength and the proportion of glutenin polymers with the highest molecular masses (Gupta et al. 1993). However, differences in gluten strength also strongly correlate with allelic variation in the glutenin subunit composition (Payne et al. 1987; Pogna et al.1987, 1990). Studies of near-isogenic lines of wheat differing only in their HMW glutenin subunit composition (Popineau et al. 1994) and, more recently, of lines expressing transgenes encoding HMW glutenin subunit 1Ax1 or Dx5 (Popineau et al. 2001) have indicated that these proteins influence gluten viscoelasticity, and therefore breadmaking quality, by affecting the size distribution of the glutenin polymers and their degree of cross-linking. The effects may depend both on the total amount of HMW glutenin subunits and/or their specific aggregative properties, which appear to be highly influenced by the number and position of cysteine residues available for intermolecular disulphide bond formation.
In the case of tetraploid durum (pasta) wheat, several studies have demonstrated that LMW glutenin subunits have a greater impact on quality than HMW subunits (Payne et al. 1984; Pogna et al. 1990; Ruitz and Carillo 1995). In particular, a strong correlation exists between gluten strength and a specific pattern of LMW glutenin subunits, called LMW-2, encoded by the Glu-B3 locus. Although it is generally accepted that the high quality of cultivars with the LMW-2 subunit group results mainly from the presence of a higher amount of the LMW glutenin subunit protein than in cultivars with allelic groups of LMW subunits (Autran et al. 1987; Masci et al. 1995), it cannot be ruled out that differences in the properties of the LMW subunits present in the different allelic groups could also play a role. In fact, comparisons of the sequences of LMW glutenin subunits show that those encoded by the Glu-B3 locus have several differences when compared with subunits encoded by the orthologous loci, most notably the position of the first cysteine residue within the repetitive domain (D’Ovidio et al. 1999).
Efficient protocols are now available to transform wheat and have been used to express specific HMW glutenin subunit genes in both durum and bread wheats (Blechl and Anderson 1996; Barro et al.1997; He et al. 1999). This technology may also be applicable to understanding the role of the LMW subunits in determining gluten properties and quality. However, whereas the HMW subunits comprise only 3 to 5 proteins which are readily separated by SDS-PAGE, the LMW subunits comprise a complex mixture of components with similar structures and properties, including cross-reactivity with antisera. Hence, it may be difficult to identify the proteins encoded by the transgenes among the endogenous proteins.
In order to study the relationship between dough strength and the amount and properties of LMW glutenin subunits, we have transformed the durum wheat cultivars Ofanto and Svevo with different LMW glutenin alleles. Furthermore, we have tagged these proteins at their C-termini with a 14 amino-acid residue c-myc epitope, to facilitate their detection using commercially available antibodies. This has allowed the rapid and reliable assessment of transgene expression, even when the level of proteins was very low, without affecting the trafficking of the transgenic proteins and their incorporation into glutenin polymers. Whilst (c-myc) epitope-tagging is widely employed in both mammalian and yeast cell biological studies, the utility of such an approach has received only limited attention from plant scientists. Here we demonstrate the efficacy of using the c-myc epitope tag to detect the presence of a transgenic LMW glutenin subunit amongst a complex mixture of highly related glutenin proteins.
Matherials and methods
DNA manipulation
Standard DNA manipulations were performed as described in Sambrook et al. (1989). Nucleotide sequencing was performed manually using the Thermo Sequenase Radiolabelled terminator cycle-sequencing kit (Amersham, UK), following the manufacturer’s instructions. For PCR, total genomic DNA was isolated from leaf tissues using the CTAB method (Stacey and Isaac1994), while for Southern blotting the DNA extraction method reported by D’Ovidio et al. (1992) was used.
Construction of the cassette for the expression of LMW glutenin subunit genes in durum wheat
Plasmid pHMW1Dx5 and plasmid pLRPT (kindly provided by Dr. Lee Rooke, Rothamsted Research, UK), were used for the construction of the transgenic expression cassette. A 650-bp fragment of the proximal 3′ untranslated region of HMW subunit 1Dx5 was amplified by PCR from plasmid pHMWDx5, using primers containing the restriction sites XbaI and BamHI and having the following sequences: 5′-tct aga act ctc tgc agc tcg-3′; 5′- cag gat ccg agg cgt atg ctg ctg cat tt-3′. The amplification product was cloned in the transformation vector pLRPT using the same restriction sites, resulting in its insertion between the 1.3-kb fragment of the promoter region of 1Dx5 and the 0.7-kb fragment of the CaMV35S terminator. The recombinant vector was named pRDPT5.
DNA sequences of LMW glutenin subunit genes were amplified by PCR from the durum wheat cultivar Langdon using the primers and conditions described by D’Ovidio et al. (1997). The above sequences and the plasmid pLDNLMW1B- were then used as templates for the specific amplification of the coding regions of the lmw1A3-, lmw1B- and lmw1B-genes, and for the addition of the restriction sites SalI and BamHI at their 5′ and 3′ ends. Such addition allowed the cloning of the amplicons into the plasmid RMBS (kindly provided by Dr. Sean Munro, MRC Laboratory of Molecular Biology, Cambridge, UK), already containing the c-myc epitope tag inserted between the restriction sites BamHI and XbaI, and the recovery of the 3′ epitope-tagged LMW glutenin subunit genes as SalI-XbaI fragments. The tagged genes were then sub-cloned into the transformation vector pRDPT5 between the promoter and the terminator regions of the HMW subunit 1Dx5 gene. The final constructs (Fig. 1) were named pRDPT51A3*, pRDPT51B* and pRDPT51B*−.

LMW glutenin subunit-gene plasmid constructs
Plant material
Durum wheat plants cv Ofanto and Svevo were grown in 8-inch diameter pots in the glasshouse, under irradiance of ca 750 μE s−1 m2, with a 16-h light period, at 18–22°C under lighting and 14–16°C during darkness, and 50–70% relative humidity.
Durum wheat transformation
Explants prepared from immature inflorescence and scutellum explants were co-bombarded with a plasmid containing the gene of interest and plasmid pAHC25 (provided by Dr. A. Christiansen, George Maso University, Fairfax, V.), containing the uid and bar genes under the control of the maize ubiquitin promoter, using the transformation procedure described by Lamacchia et al. (2001). Plants were regenerated and selected under the herbicide phosphinotricin (3 mg/ml).
Southern analysis
Genomic DNA (10 μg) was digested with XbaI, which cuts once within the pRDPT5lmw constructs. Digested DNA was separated by electrophoresis in a 0.8% (w/v) agarose gel and transferred by capillary blotting into nylon membrane positively charged (Roche), according to the manufacturer’s instructions. Membranes were hybridized with random-primed generated probes produced using primers for the CaMV35S terminator sequence (contained in pRDPT5). The hybridised probe DNA was detected by exposure to Kodak double-emulsion films.
Protein analysis
Protein analysis was carried out on single seeds of primary transformants using the distal part with the embryo part being germinated for multiplication.
LMW glutenin subunit composition was determined by SDS-PAGE using a Tris-borate buffer system (Shewry et al. 1995), and by western and dot blotting using the anti-c-myc polyclonal antibody A-14 with an anti-rabbit alkaline phosphatase conjugated secondary antibody.
Glutenin proteins were extracted and separated by RP-HPLC, using the method described by Marchylo et al. (1989) with some modifications (Larroque et al. 2000). Samples were extracted in duplicate with single analyses of each extract being performed. Peak identity was assigned by comparison with RP-HPLC profiles of flours of the same lines, for which the identities of the peaks had been determined by SDS-PAGE and N-terminal amino-acid sequencing.
Extraction of gliadin and glutenin fractions
Monomeric subunits and small oligomers were extracted with 50 mM of sodium phosphate, pH 6.8, and 0.5% (w/v) SDS (1 mg/25 μl) for 2 h at room temperature. After centrifugation for 10 min at 10,000 g, the supernatant from each sample was recovered and divided into two aliquots, with 2-mercaptoethanol being added to one, to a final concentration of 2.5% (v/v). The protein remaining in the pellet, consisting almost exclusively of glutenins, was then extracted with the same buffer containing 5% (v/v) 2-mercaptoethanol.
Reduced and unreduced extracts were analysed by SDS-PAGE and western blotting.
Results
Stable integration of epitope-tagged LMW glutenin subunit genes in durum wheat
Three sequences encoding LMW subunits were used for durum wheat transformation. Gene lmw1B was obtained by PCR amplification from the durum wheat cultivar Langdon (D’Ovidio et al. 1997) and encodes a 330 amino-acid subunit from the Glu-B3 locus. A mutant form of this gene, lmw1B-, in which a cysteine residue at position 45 of the mature protein was mutated to arginine, was also used. This cysteine residue is one of two, that are likely to be involved in the formation of inter-chain disulphide bonds (Keck et al. 1995), and hence the mutation should affect the ability of the protein to be incorporated into glutenin polymers. Gene lmwA3, encoding a protein of 280 amino acids, was also amplified from cv Langdon and corresponds to a gene from the Glu-A3 locus.
The promoter and terminator from the HMW subunit 1Dx5 gene of bread wheat were used to ensure strong expression of the transgenes only in the starchy endosperm cells (Lamacchia et al. 2001). The CaMV35S promoter, already present on the plasmid pLRPT used for the construction of pRDPT5, was not removed, since it was the only sequence on the transformation construct that did not have any similarity with the genomic DNA of durum wheat, but was long enough to allow good hybridisation in Southern blotting. To allow specific detection of the transgenic subunits while minimising the possibility of compromising their correct folding and trafficking, a sequence encoding a single c-myc epitope-tag was fused at the C-termini of the genes via insertional cloning.
Equimolar amounts of the pRDPT5lmw plasmids and the plasmid pAHC25 were co-bombarded into immature scutellum and inflorescence explants, and with transformants selected on medium containing the herbicide phosphinotricine. PCR analysis of putative transformants led to the identification of 11 transgenic plants containing both the bar selectable marker gene and the gene of interest. Transformation efficiencies varied between 0.6 and 3.1% while the overall co-transformation efficiency was of 64%. Two lines transgenic for pRDPT51B*, three for pRDPT51A3* and one for pRDPT51B−* were obtained in the cultivar Ofanto while two lines transgenic for pRDPT51B*, two lines transgenic for pRDPT51A3* and one for pRDPT51B−* were obtained in the cultivar Svevo (Table 1).
Southern blotting using the restriction endonuclease XbaI, which linearizes the plasmid containing the genes of interest, and hybridization with a probe corresponding to the CaMV35S terminator sequence, showed that all the primary transformants contained multiple inserts (Fig. 2A,B). The banding patterns, however, were different, confirming that the plants were derived from independent transformation events and could therefore be considered as independent lines.

Southern analysis of XbaI-cut genomic DNA from transgenic Ofanto (A) and Svevo (B) lines containing the pRDT5lmw transgenes, and from untransformed control plants, hybridized with a probe prepared by random priming of the CaMV35S terminator sequence. A Lane 1 = Control DNA Ofanto; lane 2 = 1061; lane 3=1093; lane 4 = 124la; lane 5 = 1241b; lane 6 = 1241c; lane 7 = 1284. B Lane 1 = 1044; lane 2 = Control DNA Svevo; lane 3 = 1115; lane 4 = 1235; lane 5 = 1275; lane 6 = 1279
Hybridising fragments larger in size than the plasmid used for transformation (6.4 kb), which were presumed to correspond to full-length versions of the genes, were detected in DNA from all of the transgenic lines. However, hybridising bands of the same size or smaller than the transformation constructs, indicating the presence of truncated and/or rearranged forms of the genes, were also detected in the same lines.
Detection of the transgenic LMW glutenin subunit in seeds of the transformed plants
The ability to use a polyclonal antibody against the c-myc epitope-tag for dot immunoblotting greatly simplified the analysis of the transformants for transgene expression. Analysis of 25–50 seeds of each primary transformant showed expression of the transgenes with no signal being obtained with seeds from the control untransformed plants. However, dot immunoblotting is not quantitative so it was not possible to discriminate between homozygous seeds expressing three copies of the transgenes in the triploid endosperm or heterozygotes expressing one or two copies.
The separation of the total protein extracts of the transgenic lines by SDS-PAGE allowed the identification of the transgenic subunits by staining with Coomassie Blue in lines 1061, 1093 and 1275 expressing pRDPT51B*, lines 1241a, 1044 and 1297 expressing pRDPT51A3*, and line 1115 expressing pRDPT51B−* (see examples in Fig. 3A, B, D). However, the transgenic proteins in lines 1241b, 1241c, 1235 and 1284 were not identifiable by Comassie staining (see example in Fig. 3C), probably because they were expressed at lower levels than the endogenous subunits, and could only be detected by western blotting with the anti-myc antibody.

SDS-PAGE (left) and western blot (right) of T1 seeds from the transgenic lines 1093 (A) and 1061 (B), expressing pRDPT51B*; 1284 (C), expressing pRDPT51B*-; 1241a (D), expressing pRDPT51A3*. A sample of four seeds representative for each line are shown. Arrows indicate the position of the transgenic LMW-GS, as confirmed by the corresponding western blotting using anti-c-myc antibody
The relative mobilities of the bands corresponding to the transgenic proteins were consistent with none of the inserted genes having undergone rearrangements. This suggests that the rearranged copies of the transgenes identified by Southern blotting were either not translated, or the proteins were not sufficiently stable or abundant to be detected by the c-myc antibody.
Quantification of the transgenic subunit encoded by lmw1B in two independent homozygous transgenic lines
More detailed analyses were carried out on the two independent lines of cultivar Ofanto expressing lmw1B*, which is the longest of the two LMW glutenin genes used in the transformation experiments and contains two cysteine residues that are thought to be available for intermolecular disulphide-bond formation.
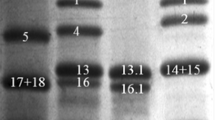
T2 homozygous transgenic plants of lines 1061 and 1093 were grown together in the glasshouse with T2 null plants, derived from T1 hemizygous plants of the same lines, and untransformed control plants of cv Ofanto. Mature T3 seeds were analysed by dot-immunoblotting to verify the stability of transgene expression and homozygosity. Seeds from the lines were then bulked, weighed and used for all successive analyses as single homogeneous samples. Glutenin extracts from the two transgenic and control lines were analysed by RP-HPLC, a reproducible, high-resolution procedure that is extensively used to separate and quantify cereal proteins (Marchylo et al. 1988; Masci et al. 1995).
The RP-HPLC chromatograms of the Ofanto control and null samples, and the Ofanto control and transgenic samples, are compared in Fig. 4, while the peak identities and elution times determined from these chromatograms are shown in Table 2. The same table also reports the quantification of the peak areas, which are expressed as percentages of the total peak area (mean values) to compensate for differences in the amounts of storage proteins between the five samples. Since each of the peaks contains specific subunits, the percentage values represent the contributions of specific subunits to the total glutenin fractions. Each peak was compared separately determining the standard error of difference between the two means (SED), the least significant difference (LSD) and the F-probability to test for significant differences. In every case, the F-probability values indicated that significant differences were present between the transgenic and control lines. Comparison of the data for the Ofanto control line, and the 1061 and 1093 null lines, showed no significant differences for peaks 1, 3, 4, 5 and 8 (see Table 2). However, the mean for peak 2 of the Ofanto control was significantly lower than those for either of the null lines (p<0.01), while the converse was observed for peaks 6 (p<0.01) and 7 (p=0.05). No significant differences were observed between the null lines.

Reversed-phase HPCL chromatograms of control and transgenic lines of Ofanto. Peak numbers correspond to those shown in Table 2
Comparison of the 1061 null and 1061 transgenic lines showed no significant differences between peaks 3, 5, 6 and 7, all corresponding to LMW subunits (Table 2). However, peaks 1 (p=0.035) and 2 (p=0.001), corresponding to HMW subunits, were significantly lower in the transgenic samples, while peak 8, corresponding to LMW subunits, was significantly higher (p<0.001).
Comparison of the 1093 null and the 1093 transgenic lines showed a more complex pattern of differences. Significant differences were observed for all eight peaks, with the transgenic line having higher values for peaks 1 (p=0.031), 2 (p<0.001) and 8 (p=0.001), and lower values for peaks 3 (p=0.018), 4 (p=0.002), 5 (p=0.001), 6 (p<0.001) and 7 (p<0.001). The fact that peak 8 gave consistently higher values in both transgenic lines, when compared to the Ofanto control and the null lines, strongly suggests that this peak contains the transgenic subunit.
The decrease in peak 7 of the 1093 transformant was particularly marked, as this corresponds to the major LMW subunit band observed by SDS-PAGE (Fig. 5, arrow1).

SDS-PAGE of reduced total protein extracts of seeds from cv Ofanto (lanes 1,2), null (lanes 3,4) and transgenic (lanes 5,6) lines of Ofanto 1061 and null (lanes 7,8) and transgenic (lanes 9,10) lines of Ofanto 1093. Black arrows indicate the transgenic subunit; grey arrows indicate the positions of the down-regulated subunits in line 1093
Figure 5 also shows that at least one other major subunit (arrow 2), possibly corresponding to peak 6, is also down-regulated.
The transgenic LMW glutenin subunit contributed about 7–8% of the total glutenins in line 1061 (calculated as the difference between the means of the peak 8 area values of the transgenic and the Ofanto control/ null lines), while in the transgenic line 1093 it represented just over 15% of the total glutenin fraction and was the most abundant of the glutenin subunits. However, the higher proportion of the transgenic protein in line 1093 probably relates to the reduced expression of the endogenous LMW glutenin subunits rather than to a higher level of expression of the transgene, the absolute areas of peak 8 (data not shown) indicating similar levels of expression in the two lines.
Incorporation of the transgenic subunit in the glutenin polymers
The subunit encoded by lmw1B contains two cysteines that are thought to form interchain disulphide bonds allowing incorporation into high molecular mass polymers. In order to confirm that this was the case, sequential extraction of monomeric and polymeric proteins from the transgenic lines 1061 and 1093 was carried out as described in the Materials and methods section. The fraction extracted with SDS contains both monomers and oligomers, and comparison of the blotting results obtained with unreduced and reduced samples indicated that the LMW subunit was predominantly present in oligomers (Fig. 6B). Similarly, it was also present in the fractions extracted with SDS+2-mercaptoethanol, which correspond to the subunits of polymers (Fig. 6B). Only traces were detected as monomers in the unreduced fractions (grey arrow in Fig. 6B), which migrated slightly faster than the reduced polymers, due probably to the presence of intra-chain disulphide bonds. The distribution of the transgenic subunit between oligomers and polymers also differed in the two transgenic lines, being present in about equal amounts in the two fractions of line 1061 but more abundant in the oligomers (about 2.5-times as estimated by densitometric scanning) in line 1093. Figure 6A also shows that line 1093 contains a lower proportion of reduced polymeric protein which presumably relates to the down-regulation of endogenous LMW subunits shown in Figs. 4 and 5, and discussed above.

SDS-PAGE (A) and western blotting (B) with anti-c-myc antibody of reduced and unreduced fraction from flour of Ofanto control (1) 1061 transgenic (2) and 1093 transgenic (3) lines. The black arrows (A) indicate the bands having different intensity of staining in the three lines. The grey arrow (B) indicates the unreduced monomeric subunit, while the black arrows (B) indicates the reduced form. The unreduced form of the subunit migrates faster probably due to its more compact confirmation determined by the presence of intramolecular disulphide bonds
Discussion
The aim of this work was to test the validity of using epitope-tagging in order to detect the expression of gluten protein transgenes in the wheat endosperm without compromising the stability of their encoded proteins and their normal folding, polymerisation and trafficking. We have clearly demonstrated that the addition of the epitope does not compromise the expression of the transgenes, as all of the lines that tested PCR positive for the gene of interest were also positive for transgene expression, unequivocally determined by the use of the anti-c-myc antibody. Furthermore, because of the high sensitivity of the immunological assay, it has been possible to more accurately assess the efficiency of the HMW subunit 1Dx5 gene-promoter in driving the expression of the LMW glutenin subunit coding-regions.
The HMW subunit 1Dx5 gene-promoter had been previously used in transgenic durum wheat for the expression of its cognate gene (He et al. 1999) and to drive the expression of the UidA (Gus) reporter gene (Lamacchia et al. 2001), with high levels of expression of the transgenes being reported in both cases.
In this work a range of expression levels were observed compared with endogenous LMW glutenin subunits. In four of the 11 transgenic lines, the expression was, in fact, so low that it was not possible to identify the transgenic protein by Coomassie-Blue staining of SDS-PAGE. The use of the anti-c-myc antibody has meant that the laborious RT-PCR analysis of developing seeds, in order to identify low levels of transgene expression, could be avoided and the selection of the transgenic and null seeds, for the multiplication of the lines was greatly simplified. The expression levels achieved in the best lines were comparable with those of the endogenous HMW subunit 1Dx5 and HMW subunit transgenes expressed under the control of the same gene-promoter (Barro et al. 1997). Similar levels have also been achieved by using a wheat LMW subunit promoter to drive the expression of pea legumin in transgenic wheat (Stoger et al. 2001) and this promoter could also be used for future studies.
The epitope was added as a single copy at the C-termini of the LMW glutenin subunit proteins, which contrary to the N-terminus does not undergo processing. Also, the c-myc tag is only 14 amino acids long and we therefore considered that it was unlikely to affect the conformation of the transgenic subunit and interfere with its polymerisation. The results of the analysis, carried out on the two independent transgenic lines expressing pRDPT51B*, have shown that the transgenic protein is indeed incorporated in the glutenin polymers. However, in one of the transgenic lines the transgenic subunit was incorporated less efficiently and was concentrated in the oligomeric protein fraction. This same transgenic line also showed clear evidence of the down-regulation of the endogenous LMW glutenin subunits that participate in the formation of large polymers. This is presumed to result from sense or transgene-mediated suppression (Meyers and Sadler 1996; Fagard and Vaucheret 2000), a phenomenon often seen in transgenic plants when transgenes related to endogenous genes are introduced into the genome. We therefore consider that the poor incorporation of the transgenic subunit in that line results from the associated down-regulation of endogenous subunits and not from mis-folding or incorrect trafficking of the proteins. Consequently, although lines 1061 and 1093 express the same transgene at similar levels, they differ in the ability to form glutenin polymers. This would be predicted to affect the viscoelasticity and processing quality of dough made from the lines.
References
Autran JC, Laignelet B, Morel MH (1987) Characterisation and quantification of low-molecular-weight glutenins in durum wheats. Biochimie 69:699–711
Barro F, Rooke L, Bekes F, Gras P, Tatham AS, Fido R, Lazzeri PA, Shewry PR, Barcelo P (1997) Transformation of wheat with high-molecular-weight subunit genes results in improved functional properties. Nature Biotech 15:1295–1299
Blechl AE, Anderson OD (1996) Expression of a novel high-molecular-weight glutenin subunit gene in transgenic wheat. Nature Biotech 14:875–879
D’Ovidio R, Tanzarella OA, Porceddu E (1992) Nucleotide sequence of a low-molecular-weight glutenin from Triticum durum. Plant Mol Biol 18:781–4
D’Ovidio R, Simeone M, Masci S, Porceddu E (1997) Molecular characterization of a LMW-GS gene located on chromosome 1B and the development of primers specific for the Glu-B3 complex locus in durum wheat. Theor Appl Genet 95:1119–1126
D’Ovidio R, Marchitelli C, Cardelli LE, Porceddu E (1999) Sequence similarity between allelic Glu-B3 genes related to quality properties of durum wheat. Theor Appl Genet 98:455–461
Fagard M, Vaucheret H (2000) (Trans) gene-silencing in plants: how many mechanisms? Ann Rev Plant Physiol Plant Mol Biol 51:167–194
Field JM, Shewry PR, Miflin BJ (1983) Solubilisation and characterisation of wheat gluten proteins: correlation between the amount of aggregated proteins and baking quality. J Sci Food Agric 34:370–377
Gupta RB, Khan K, MacRichie F (1993) Biochemical basis of flour properties in bread wheats. I. Effects of variation in the quantity and size distribution of polymeric protein. J Cereal Sci 18:23–41
Gupta RB, Popineau Y, Lefebvre J, Cornec M, Lawrence GJ, MacRichie F (1995) Biochemical basis of flour properties in bread wheats. II. Changes in polymeric protein formation and dough/gluten properties associated with the loss of low Mr or high Mr glutenin subunits. J Cereal Sci 21:103–116
He GY, Rooke L, Steele S, Bekes F, Gras P, Tatham AS, Fido R, Barcelo P, Shewry PR, Lazzeri P (1999) Transformation of pasta wheat (Triticum turgidum L. var. durum) with high-molecular-weight glutenin subunit genes and modification of dough functionality. Mol Breed 5:377–386
Huebner FR, Wall JS (1976) Fractionation and quantitative differences of glutenin from wheat varieties varying in baking quality. Cereal Chem 53:258–269
Keck B, Kohler P, Wieser H (1995) Disulphide bonds in wheat gluten: cystine peptides derived from gluten proteins following peptic and thermolitic digestion. Z Lebensm Unters Forsch 200:432–439
Lamacchia C, Shewry PR, Di Fonzo N, Forsyth JL, Harris N, Lazzeri P, Napier JA, Halford NG, Barcelo P (2001) Endosperm specific activity of a storage protein gene promoter in transgenic wheat seed. J Exp Bot 52:243–250
Larroque OR, Bekes F, Wrigley CW, Rathmell WG (2000) Analysis of gluten proteins in grain and flour blends by RP-HPLC. In: Shewry PR, Tatham AS (eds) Wheat gluten. RSC, Cambridge, UK, pp 136–139
Marchylo BA, Kruger JE (1989) Quantitative reversed-phase high-performance liquid chromatographic analysis of wheat storage proteins as a potential quality prediction tool. J Cereal Sci 9:113–130
Marchylo BA, Hatcher DW, Kruger JE (1988) Identification of wheat cultivars by reversed-phase high-performance liquid chromatography of storage proteins. Cereal Chem 65:28–40
Masci S, Lew EJ-L, Lafiandra D, Porceddu E, Kasarda DD (1995) Characterisation of low-molecular-weight glutenin subunits in durum wheat by reversed-phase high performance liquid chromatography and N-terminal sequencing. Cereal Chem 72:100–104
Meyer P, Saedler H (1996) Homology-dependent gene silencing in plants. Ann Rev Plant Physiol Plant Mol Biol 47:23–48
Payne PI, Jackson EA, Holt LM (1984) The association between γ-gliadin 45 and gluten strength in durum wheat varieties: a direct casual effect or the result of genetic linkage? J Cereal Sci 2:73–81
Payne PI, Nightingale MA, Krattiger AF, Holt LM (1987) The relationship between HMW glutenin subunit composition and the breadmaking quality of British-grown wheat varieties. J Sci Food Agric 40:51–65
Pogna N, Mellini F, Dal Belin Peruffo A (1987) Glutenin subunits of Italian common wheats of good breadmaking quality and comparative effects of high-molecular-weight glutenin subunits 2 and 5, 10 and 12 on flour quality. In: Borghi B (ed) Hard wheat: agronomic, technological, biochemical and genetic aspects. Commission of the European Communities, pp 53–69
Pogna N, Autran JC, Mellini F, Lafiandra D, Feillet P (1990) Chromosome 1B-encoded gliadins and glutenin subunits in durum wheat: genetics and relationship to gluten strength. J Cereal Sci 11:15–34
Popineau Y, Cornec M, Lefebre J, Marchylo B (1994) Influence of high Mr glutenin subunits on glutenin polymers, and rheological properties of glutens and gluten subfractions on near-isogenic lines of wheat Sicco. J Cereal Sci 19:231–241
Popineau Y, Deshayes G, Lefebre J, Fido R, Tatham AS, Shewry PR (2001) Prolamin aggregation, gluten viscoelasticity, and mixing properties of transgenic wheat gluten lines expressing 1Ax and 1Dx high-molecular-weight glutenin subunit transgenes. J Agric Food Chem 49:395–401
Ruitz M, Carillo JM (1995) Separate effects on gluten strength of Gli-1 and Glu-3 prolamin genes on chromosome 1A and 1B in durum wheat. J Cereal Sci 21:137–144
Sambrook J, Fritsch FE, Maniatis T (1989) Molecular cloning : a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, New York
Shewry PR, Tatham AS, Fido RJ (1995) Separation of plant proteins by electrophoresis. In: Jones H (ed) Methods in molecular biology, Vol 49. Humana Press, New Jersey, pp 399–422
Singh NK, Sheperd KW (1987) Solubility behaviour, synthesis, degradation and subcellular location of a new class of disulphide-linked proteins in wheat endosperm. Aust J Plant Physiol 14:245–252
Stacey J, Isaac PG (1994) Isolation of DNA from plants. Methods Mol Biol 28:9–15
Stoger E, Parker M, Christou P, Casey R (2001) Pea legumin overexpressed in wheat endosperm assembles into an ordered paracrystalline matrix. Plant Physiol 125:1732–1742
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by J. W. Snape
Rights and permissions
About this article
Cite this article
Tosi, P., D’Ovidio, R., Napier, J.A. et al. Expression of epitope-tagged LMW glutenin subunits in the starchy endosperm of transgenic wheat and their incorporation into glutenin polymers. Theor Appl Genet 108, 468–476 (2004). https://doi.org/10.1007/s00122-003-1459-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-003-1459-x