
Abstract
Genetic diversity and population genetic structure of natural Oryza rufipogon populations in China were studied based on ten microsatellite loci. For a total of 237 individuals of 12 populations collected from four regions, a moderate to high level of genetic diversity was observed at population levels with the number of alleles per locus (A) ranging from 2 to 18 (average 10.6), and polymorphic loci (P) from 40.0% to 100% (average 83.3%). The observed heterozygosity (H O ) varied from 0.163 to 0.550 with the mean of 0.332, and the expected heterozygosity (H E ) from 0.164 to 0.648 with the mean of 0.413. The level of genetic diversity for Guangxi was the highest. These results are in good agreement with previous allozyme and RAPD studies. However, it was unexpected that high genetic differentiation among populations was found (R ST = 0.5199, θ = 0.491), suggesting that about one-half of the genetic variation existed between the populations. Differentiation (pairwise θ) was positively correlated with geographical distance (r = 0.464), as expected under the isolation by distance model. The habitat destruction and degradation throughout the geographic range of O. rufipogon may be the main factor attributed to high genetic differentiation among populations of O. rufipogon in China.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Common wild rice (Oryza rufipogon Griff.) is a perennial and is widely distributed in the tropics and subtropics of monsoon Asia (Vaughan 1994). As the progenitor of the Asian cultivated rice (Oryza sativa), O. rufipogon has been proven to be a valuable gene pool for rice genetic improvement and thus plays a critical role in rice breeding in the future (Chang 1984; Khush 1997). It is well established that wild rice populations harbor significantly higher genetic diversity than does the cultivated rice (Oka 1988; Provan et al. 1997; Sun et al. 2001). Based on 44 RFLP markers, for example, Sun et al. (2001) studied the genetic diversity of O. rufipogon and O. sativa from more than ten Asian countries, and found that the genetic diversity of O. rufipogon (A = 4.02, P = 97.7%, H O = 0.033) was much higher than that of cultivated rice (A = 2.34, P = 75.0%, H O = 0.014), and that a great number of genes that occurred in O. rufipogon could not be found in cultivated rice. To-date, although many studies have been conducted on cultivated rice, including assessment of genetic variation, varietal classification and identification, domestication processes and genetic mapping (See reviews in Oka 1988; Khush 1997), the genetic diversity and population genetic structure of natural populations of wild rice are less well known (Barbier 1989; Morishima and Barbier 1990; Ge et al. 1999; Xie 1999; Gao et al. 2000)
O. rufipogon was previously found in eight provinces or autonomous regions in southern China, and our recent field investigations indicated that the current geographic range of this species was significantly decreased, and the extant populations have been gradually reduced in size because of decades of habitat destruction and degradation from agriculture and urbanization (Gao et al. 2000). For a better understanding of the genetic diversity and population genetic structure, which is instructive for developing conservation management and germplasm collection, a recent decade has seen the increasing use of various genetic markers (Allozymes, RAPDs, RFLPs) in the study of O. rufipogon populations, including the natural populations in China. Ge et al. (1999) used 20 RAPD primers to investigate the genetic diversity within and between eight natural populations of the wild rice (O. rufipogon) from China and Brazil, and found that the Chinese populations showed greater polymorphism than those from Brazil at both the population and regional levels. Based on 22 allozyme loci, Gao et al. (2000) analyzed the genetic variation and population structure of 21 Chinese O. rufipogon populations sampled from seven provinces in China. The results revealed moderate allozyme variability (A = 1.33, P = 22.7%, H O = 0.033, H E = 0.068) and genetic differentiation (F ST = 0.31) among populations. Using 19 RAPD markers, Xie (1999) studied the population genetics of 29 Chinese wild populations of O. rufipogon and proposed management strategies for its conservation.
Recently, microsatellites or simple sequence repeats (SSRs) have become the markers of choice for a wide spectrum of genetic, population and evolutionary studies in many plants, including rice (Zhao and Kochert 1992; Wu and Tanksley 1993; Panaud et al. 1996). In addition to the technical simplicity, relatively low cost and high power of genetic resolution, microsatellites have some advantages over other markers: abundant, codominant, interspersed throughout the genome and highly polymorphic in plant species (reviewed in McCouch et al. 1997). Microsatellite markers have been developed and utilized in the study of cultivated rice, including variety identification and germplasm conservation (Olufowote et al. 1997; Garland et al. 1999), genetic diversity (Yang et al. 1994; Davierwala et al. 2000), and gene and quantitative trait locus analysis (Xiao et al. 1996). They have also been applied to study the wild relatives of rice (Xiao et al. 1996; Ishii et al. 2001). In the present study, we used microsatellite markers to investigate the genetic diversity and population genetic structure of natural populations of O. rufipogon in China. Such information will be of great significance in developing conservation strategies and providing the basis for germplasm collection of this important species. In addition, we were interested in comparing the results from studies with different marker systems and to evaluate the utility of microsatellite markers in terms of the population genetics study.
Materials and methods
Population sampling and DNA extraction
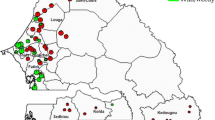
The plant materials used in this study consist of 12 populations sampled throughout the natural distribution of the species in China (Fig. 1). These populations are distributed in different habitats from the north to south in China (Fig. 1, Table 1). The Dongxiang populations (P01, P03) are the most northern populations in the entire distribution region of this species. Nineteen to thirty five individuals were randomly collected at an interval of at least 5 m to prevent collecting ramets from a single genet, except for two Hainan populations (P47 and P49) where samples were collected at 3-m intervals. Leaves were harvested, dried with silica gel and stored in paper bags. Total DNA was extracted using the CTAB method. The detailed protocol for leaf preservation and DNA extraction were described by Xie (1999).

Distribution of 12 O. rufipogon populations in China. Numbers correspond to the populations in Table 1
Primer screening and amplification of microsatellites
One hundred and thirty four microsatellite primer pairs which were developed in O. sativa were used for primer screening for their utility in O. rufigopon populations, including 98 RM and 36 OSR primers. The RM primers were designed using rice sequences from genomic libraries and GenBank (Wu and Tanksley 1993; Panaud et al. 1996; Chen et al. 1997; Temnykh et al. 2000), while the OSR primers were derived from the DDBJ database (Akiga et al. 1996). Some of OSR loci were found in RM loci and have integrated with RM loci (Temnykh et al. 2000).
PCR amplification was performed in PTC-200 (MJ Research Inc.). Twenty five microliter reactions contained 10 mM of Tris-HCl (pH 8.3), 50 mM of KCl, 2.0 mM of MgCl2, 0.2 mM of dNTP, 0.2 μM of primers, approximately 50 ng of genomic DNA and 0.75 units of Taq polymerase (TAKARA, Japan). The profile was 5 min at 94 °C; two cycles of 40 s at 94 °C, 40 s at 61 °C, 80 s at 72 °C; two cycles of 40 s at 94 °C, 40 s at 59 °C, 80 s at 72 °C; 34 cycles of 40 s at 94 °C, 40 s at 57 °C, 80 s at 72 °C and 10 min at 72 °C, for final extension. PCR products were separated on 6% polyacrylamide denaturing gels and the banding patterns were visualized using the silver-staining protocol according to the manufacturer's instructions (Promega, USA). Multiple loading (multiplexing), which is the sequential loading of independent PCR-amplification products on a single PAGE gel at an interval of about 30 min, was used for reducing cost and sparing time for single loci.
Statistical analysis
After silver staining of polyacrylamide gels, a cluster of two to five discrete bands was apparent for most of markers that could be amplified successfully. The most intensely amplified band for microsatellite markers was selected as the representative band at that locus (Cho et al. 2000). Data were entered in the form of single-individual genotypes. Genetic polymorphism for each population was assessed by calculating the mean number of alleles per locus (A), the percentage of polymorphic loci (P), the mean observed heterozygosity (H O ) and the mean expected heterozygosity under Hardy-Weinberg equilibrium (H E ). Deviation from the Hardy-Weinberg equilibrium was assessed at each locus for every population and per locus across all populations. To investigate linkage disequilibrium, Ohta's two-locus analyses of population subdivision (D-stastistics) for multiple populations were performed (P ≤ 0.05) (Ohta 1982). The above analyses were calculated using genetic analysis package POPGENE version 1.3 (Yeh et al. 1999).
Population differentiation was analyzed for polymorphic loci by F-statistics (Wright 1978). Methods and theory for quantifying genetic differentiation varied for different authors. Slaktin's R ST is an estimator of gene differentiation accounting for variance in allele size and defined for genetic markers undergoing a Stepwise Mutation Model (SMM) (Slaktin 1995). Under this model, alleles can only mutate by the gain and loss of one repeat, and alleles of similar size are assumed to be more closely related to each other than those of very different sizes. As indicated by Olsen and Schaal (2001), SMM-based measures of population differentiation are expected to be most accurate for populations that diverged long enough ago that current genetic differentiation reflects mutations accumulated since divergence. Under the Infinite Allele Model (IAM), however, it was assumed that most new mutations gave rise to new distinguishable alleles. θ statistics considers the IAM model and is another commonly used estimator of gene differentiation (Collevatti et al. 2001). θ statistics may be interpreted as the correlation between allele frequencies of different individuals in the same population in the IAM model (Weir and Cockerham 1984), and is thought to be more appropriate for recently diverged populations (Olsen and Schaal 2001).
For purposes of comparison, genetic differentiation was quantified using both θ statistics and R ST , that were estimated with the program FSTAT ver. 2.9.3 (Goudet 2001) without assuming random mating within samples. A significance test of population differentiation (Pairwise θ) and genotypic disequilibrium was performed by randomizing genotypes among samples to obtain the log-likelihood G-statistics (Goudet et al. 1996). Significance tests were performed by bootstrapping over loci with a 95% nominal confidence interval. The sequential Bonferroni correction (Rice 1989) was implemented for the multiple tests. An unbiased estimate of R ST followed Goodman (1997) in which the overall R ST estimates were based on averaging the variance component across loci, rather than averaging across individual locus estimates of R ST .
Population relationships were inferred using the UPGMA clustering method on the basis of Nei's (1978) unbiased genetic distance with POPGENE. The tree was subsequently visualized with TREEVIEW ver. 1.52 (Page 1996). To test the hypothesis of 'isolation by distance', a matrix of Weir's pairwise linearized θ was obtained by the Partial Mantel Test using FSTAT software and correlated to a geographical distance matrix.
Results
Microsatellite polymorphisms and genetic diversity
After screening 134 primer pairs against five individuals, ten primer pairs that produced clear bands were selected for further survey, i.e. RM38, RM212, RM215, RM228, RM253, OSR2, OSR16, OSR20, OSR22 and OSR32. The ten loci were dispersed in cultivated rice chromosomes 1, 6, 7, 8, 9, 10 and 12; the location of OSR16 was as yet unclear. The number of alleles varied widely among the ten loci and are summarized in Table 3. All of the ten loci displayed polymorphism among 12 populations with a total of 106 alleles identified. The most variable locus (RM228) had 18 alleles, and OSR16 produced only two alleles across 12 populations. The remaining loci had more than six alleles. Six to ten alleles were resolved for three loci (RM215, RM212 and OSR2), and 11–16 alleles were identified for four loci (RM38, OSR20, RM253 and OSR2). Genotypic linkage disequilibrium tests showed no significant values (P < 0.05), suggesting an absence of linkage disequilibrium between loci (data not shown). The table of allelic frequencies of each population is available from the corresponding author on request.
Genetic parameters in all the ten microsatellite loci for 12 populations are given in Table 2. Mean observed and expected heterozygosity ranged from 0.163 (P03) to 0.550 (P25) and from 0.164 (P45) to 0.648 (P25), respectively. Population P45 exhibited the lowest level of polymorphism (P = 40%, H E = 0.164), whereas three populations (P19, P52, P62) exhibited the greatest level of polymorphism (P = 100%) and high values of expected heterozygosity (H E = 0.370–0.582). The A values ranged from 1.4 in P45 to 4.9 in P19 with the mean value of 3.35 at the population level. Among regions, the percentage of polymorphic loci is the highest in Guangxi (93.3%), and the lowest in Jiangxi (70.0%). The A value ranged from 4.0 (Guangdong) to 1.95 (Jiangxi). The highest observed heterozygosity was observed in Guangdong (H O = 0.385), and the lowest, 0.176, in Jiangxi populations. At the species level, high genetic diversity was observed with all the loci being polymorphic and 10.6 alleles per locus. The fixation index (F) ranged from 1 to –1, indicating a deficiency or excess of heterozygotes. Although two populations (P05 and P45) possessed significantly negative fixation indices (see discussion below), most populations have the F values significantly positive or close to zero (Table 2).
Population genetic structure and differentiation
Genetic differentiation of populations was measured with different statistics, and is given in Table 3. The mean weighted R ST and θ were 0.5199 and 0.491, respectively, indicating that about half of the total genetic variation resided among populations under both SMM and IAM models. Although R ST showed a slightly higher genetic differentiation than θ, both estimates indicated a significant differentiation among populations both in allele size and frequency.
The pairwise populations and the test of pairwise population differentiation are shown in Table 4. Across all loci, all of the comparisons between populations showed significant differentiation (P < 0.01). The results indicated that 12 populations were completely different even after the Bonferroni corrections based on the adjusted P-value.
Cluster analysis
A phenogram was generated by UPGMA to show the genetic relationships of the populations studied and was presented in Fig. 2. The phenogram indicated that the populations from the same region clustered together and were clearly separated into two major clusters. One cluster consisted of populations from Hainan island (P45, P47, P49, P52), and the other included populations from Jiangxi, Guangdong and Guangxi with the populations from each region clustered together. Pairwise θ values were positively correlated with geographical distance (r = 0.464, P = 0.05), which corroborates the findings of the cluster analysis.

A UPGMA dendrogram based on Nei's distance showing genetic relationships among the 12 populations of O. rufipogon in China
Discussion
Genetic diversity
Our microsatellite survey of 12 natural populations of O. rufipogon from China indicates a high level of genetic variation both at species (A = 10.6; P = 100%; H E = 0.787) and at population levels (A = 3.35; P = 83.3%; H E = 0.413). In our previous allozyme and RAPD studies on more than 20 wild populations of O. rufipogon that were collected from similar geographic areas in China, a moderate genetic diversity was found for both allozymes (A = 1.33, P = 22.7%, H E = 0.068) (Gao et al. 2000) and RAPDs (P = 32.9%) (Xie 1999). It seems that the three marker systems produced different genetic profiles, in that apparently higher genetic diversity was generated by microsatellites. This discrepancy could be attributed to the different marker systems. As pointed out by many recent studies, microsatellites possessed hypervariability and higher resolving power among various genetic markers (Powell et al. 1996; Davierwala et al. 2000; Guadagnuolo et al. 2001). In their studies on 238 accessions of landraces and cultivars of cultivated rice, Yang et al. (1994) detected 93 alleles with ten SSR loci that was much larger than those detected using other types of markers. Davierwala et al. (2000) used three different marker systems to estimate the genetic diversity of 42 Indian elite rice varieties, and found that RAPD, ISSR and sequence-tagged SSR resulted in mean heterozygosity values of 0.429, 0.675 and 0.882, respectively. Similarly, higher genetic diversity was observed for microsatellite loci (A = 4.07; H E = 0.50) than for allozyme loci (A = 1.53; H E = 0.20) in a perennial species (Freville et al. 2001). This tendency was also evident in many other plant species (Powell et al. 1996; Streiff et al. 1998; Sun et al. 1999; Guadagnuolo et al. 2001). Therefore, the higher level of polymorphism associated with SSR is to be expected partly because of the unique mechanism responsible for generating SSR allelic diversity by replication slippage (Powell et al. 1996), and partly because of the limited number of polymorphic loci and representation of only variation in the coding regions for the allozyme loci (Guadagnuolo et al. 2001). These results also indicate that microsatellites are sufficiently informative and powerful to assess the genetic variability of both natural populations of O. rufipogon and derived rice cultivars, and thus are particularly useful for germplasm evaluation and evolutionary studies of crop plants.
Using 44 RFLP markers, Sun et al. (2001) evaluated the genetic diversity of 122 accessions of O. rufipogon and 75 entries of O. sativa from more than ten Asian countries, and concluded that O. rufipogon populations from China possess the highest genetic diversity, followed by those from South Asia and Southeast Asia. In the present study, microsatellite data indicate that Guangxi and Guangdong maintain higher genetic diversity than the other regions, suggesting that South China may be the center of genetic diversity for O. rufipogon in China. This result is in good agreement with those from previous allozyme and RAPD studies (Xie 1999; Gao et al. 2000). The Jiangxi populations are the most northern populations of O. rufipogon, and have been under ex situ conservation for many years. It is to be expected, therefore, that the lowest genetic diversity (A = 1.95; P = 70%; H E = 0.274) was observed for this area (P01 and P03) because of their small population sizes and marginal nature.
The present microsatellite data indicate that many populations are under a deficiency of heterozygotes with significantly positive F values (Table 2) and the inbreeding coefficient (f) was significant at seven out of ten loci (Table 3). As pointed out by Roa et al. (2000), heterozygote deficiency can be explained as a result of different factors: unrecognized genetic structure within populations, inbreeding due to consanguineous mating and presence of null alleles, etc. Non-amplifying or null alleles have been highlighted in the literature from human studies (Pemberton et al. 1995). The occurrence of null (non-amplifying) alleles can be the result of divergence in the sequences flanking the microsatellite, undetectable or the unqualified DNA template (Smulders et al. 1997), and thus cause heterozygotes to be mis-scored as homozygous. According to Cho et al. (2000), the null alleles were found in 8–9% of the microsatellite markers in cultivated rice. In the present study, the null alleles were confirmed after several repetitions with different amplification conditions to ensure that no reaction failure existed. As a result, there were totally 27 non-amplifying products and the proportion was as low as 1.14%. Therefore, the null alleles would have little effect on our present results.
Another potential cause of heterozygosity deficits is the unrecognized genetic diversity, but could be excluded in our case because of the hypervariability and higher resolving power of microsatellite markers. Consequently, inbreeding or mating among genetically related individuals to varying extent, may be interpreted as the main reason of the heterozygote deficiency, which was also proposed by previous allozyme and RAPD studies (Xie 1999; Gao et al. 2000). It is of interest to note that two populations (P05 and P45) possessed significantly negative fixation indices (F = –0.4524) but showed low genetic diversity, in particular for population P45 (A = 1.4; P = 40%; H E = 0.164). Xie et al. (2001) showed that the dynamics of sexual vs clonal reproduction in many O. rufipogon populations depended mainly on environmental factors such as external disturbance and water supply. Therefore, in these two populations where human disturbance was observed, the low genetic diversity and negative F values (excess of heterozygotes) are most likely to be attributed to the reproduction by asexual growth of some heterozygotes.
Population differentiation
In the present study, microsatellite data indicate high genetic differentiation among populations with about 50% of the genetic diversity that existed among populations (θ = 0.491; R ST = 0.5199). Xie (1999) and Gao et al. (2000) used allozyme and RAPD markers to investigate more than 20 Chinese wild populations of O. rufipogon collected from similar geographic areas. Based on 22 allozyme loci, Gao et al. (2000) found a moderate genetic differentiation (F ST = 0.310), while Xie (1999) detected higher genetic differentiation (F ST = 0.449) on the basis of AMOVA analysis of 19 RAPD markers. In their comparative studies on an endemic species using allozyme and microsatellite markers, Freville et al. (2001) found that the population differentiation estimated by allozymes was higher (F ST = 0.35) than that estimated by microsatellites (F ST = 0.23). Similar results were also found in many other studies (Streiff et al. 1998; Lemaire et al. 2000). This difference may be attributed to statistical causes related to the discrepancy in allelic richness between allozymes and microsatellites, and differential selection among loci (Streiff et al. 1998; Lemaire et al. 2000). Streiff et al. (1998) pointed out that there is generally a higher variability among allozyme loci than that among microsatellite loci in terms of F ST estimates. In the previous allozyme study on O. rufipogon, a high variance of F ST values among loci (ranging from 0.027 to 0.495) was also observed (Gao et al. 2000), which is in contrast to relatively low variance among microsatellite loci (R ST from 0.220 to 0.773 and θ from 0.346 to 0.753) in the present study. In addition, most populations (14 out of 21) were sampled from Guangxi, and only one population from each of Guangdong, Hainan and Jiangxi were sampled in the allozyme study (Gao et al. 2000). As a consequence, the allozyme data may underestimate the genetic differentiation among populations, and the present microsatellite data is more likely to represent the profile of the population genetic structure of Chinese wild populations of O. rufipogon. The high genetic differentiation among populations is also evidenced by the pairwise tests of differentiation (θ) that were all significant at the 1% nominal level (Table 4).
Moreover, the genetic differentiation between the populations increased with geographical distance (isolation by distance hypothesis). These results suggest that O. rufipogon populations are differentiated by a process of isolation by distance, because the partial Mantel test between a matrix of Weir's pairwise linearized θ and a matrix of geographical distance is significantly positive (r = 0.464, β = 15.58, P = 0.05). The relationships among populations demonstrated by the UPGMA dendrogram parallel this finding (Fig. 2). Specifically, four Hainan (island) populations as a group separated clearly from the other eight mainland populations, a reflection of the physical (sea) barrier between two groups of populations (Figs. 1 and 2).
O. rufipogon is found in swampy areas, pools and ditches which retain soil moisture even in the dry reason (Oka 1988). This species has a typical mixed-mating system and reproduces by both seeds and horizontal stems. The allocation of asexual vs sexual reproduction may vary among populations in different habitats (Oka 1988; Gao et al. 2000; Xie et al. 2001). Given the fact that Chinese O. rufipogon is a predominantly outcrossing perennial (Xie 1999; Gao et al. 2000), the genetic differentiation among populations revealed by the present study is high. To-date, it is well established that the main factors that determine the population genetic structure of plants include the mating and reproductive system, selection or adaptation and the geographic range, among others (Hamrick and Godt 1989). In our case, although it remains difficult to rank them according to their significance, evidence showed that habitat destruction and degradation from decades of agriculture and urbanization throughout the geographic range of O. rufipogon have significantly decreased its populations in China both on scale and in size (Xie 1999; Gao et al. 2000). As a result, the population system of O. rufipogon has become fragmented with the fragmentation of habitats. The isolated and small populations resulted in more or less inbreeding and clonal growth, which in turn increased genetic differentiation among populations.
In summary, the wild populations of O. rufipogon in China maintain moderate diversity and high genetic differentiation among populations. As the most important gene pool for rice breeding and the most endangered wild rice species in China, practical conservation action is urgently needed. With information on the available population genetics, effective conservation management and decision may be easily formulated, whenever ex situ or in situ strategies are undertaken.
References
Akiga H, Yokozeki Y, Inagaki A, Fujimura T (1996) Microsatellite DNA markers for rice chromosomes. Theor Appl Genet 93:1071–1077
Barbier P (1989) Genetic variation and ecotypic differentiation in the wild rice Oryza rufipogon. II. Influence of the mating system and life-history traits on the genetic structure of populations. Jpn J Genet 64:273–285
Chang TT (1984) Conservation of rice genetic resources: luxury or necessity. Science 224:251–256
Chen X, Temnykh S, Xu Y, Cho YG, McCouch SR (1997) Development of a microsatellite framework map providing genome-wide coverage in rice (Oryza sativa L.) Theor Appl Genet 95:553–567
Cho YG, Ishii T, Temnhkh S, Chen X, Lipovich L, McCouch SR, Park WD, Ayres N, Cartinhour S (2000) Diversity of microsatellites derived from genomic libraries and GenBank sequences in rice (Oryza sativa L.). Theor Appl Genet 100:713–722
Collevatti RG, Grattapaglia D, Hay JD (2001) Population genetic structure of the endangered tropical tree species Caryocar brasiliense, based on variability at microsatellite loci. Mol Ecol 10:349–356
Davierwala AP, Chowdari KV, Kumar S, Peddy APK, Ranjekar PK, Gupta VS (2000) Use of three different marker systems to estimate genetic diversity of Indian elite rice varieties. Genetica 108:269–284
Freville H, Justy F, Olivieri I (2001) Comparative allozyme and microsatellite population structure in a narrow endemic plant species, Centaurea corymbosa Pourret (Asteraceae). Mol Ecol 10:879–889
Gao LZ, Ge S, Hong DY (2000) Allozyme variation and population genetic structure of common wild rice Oryza rufipogon Griff. in China. Theor Appl Genet 101:494–502
Garland SH, Lewin L, Abedinia M, Henry R, Blakeney A (1999) The use of microsatellite polymorphisms for the identification of Australian breeding lines of rice (Oryza sativa L.). Euphytica 108:53–63
Ge S, Oliveira GCX, Schaal BA, Gao LZ, Hong DY (1999) RAPD variation within and between natural populations of the wild rice Oryza rufipogon from China and Brazil. Heredity 82:638–644
Goodman SJ (1997) R ST Calc: a collection of computer programs for calculating estimates of genetic differentiation from microsatellite data and determining their significance. Mol Ecol 6:881–885
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (Version 2.9.3). From http://www.unil.ch/izea/softwares/fstat.html
Goudet J, Raymond M, Meeus TD, Rousset F (1996) Testing differentiation in diploid populations. Genetics 144:1933–1940
Guadagnuolo R, Bianchi DS, Felber F (2001) Specific genetic markers for wheat, spelt and four wild relatives: comparison of isozymes, RAPDs and wheat microsatellites. Genome 44:610–621
Hamrick JL, Godt MJW (1989) Allozyme diversity in plant species. In: Brown AHD, Clegg MT, Kahler AL, Weir BS (eds) Plant population genetics, breeding, and genetic resources. Sinauer Associates, Sunderland, pp 43–63
Ishii T, Xu Y, McCouch SR (2001) Nuclear- and chloroplast-microsatellite variation in A-genome species of rice. Genome 44:658–666
Khush GS (1997) Origin, dispersal, cultivation and variation of rice. Plant Mol Biol 35:25–34
Lemaire C, Allegrucci G, Naciri M, Bahri-Sfar L, Kara H, Bonhomme F (2000) Do discrepancies between microsatellite and allozyme variation reveal differential selection between sea and lagoon in the sea bass (Dicentrarchus labrax)? Mol Ecol 9:457–467
McCouch SR, Chen X, Panaud O, Temnhkh S, Xu Y, Cho YG, Huang N, Ishii T, Blair M (1997) Microsatellite marker development, mapping and application in rice genetics and breeding. Plant Mol Biol 35:89–99
Morishima H, Barbier P (1990) Mating system and genetic structure of natural populations in wild rice, Oryza rufipogon. Plant Species Biol 5:31–39
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Ohta T (1982) Linkage disequilibrium due to random drift in finite subdivided populations. Proc Natl Acad Sci USA 79:1940–1944
Oka HI (1988) Origin of cultivated rice. Japan Scientific Societies Press, Tokyo
Olsen KM, Schaal BA (2001) Microsatellite variation in cassava (Manihot esculenta, Euphorbiaceae) and its wild relatives: further evidence for a southern Amazonian origin of domestication. Am J Bot 88:131–142
Olufowote JO, Xu Y, Chen X, Park WD, Beachel HM, Dilay RH, Goto M, McCouch SR (1997) Comparative evaluation of within-cultivar variation of rice (Oryza sativa L.) using microsatellite and RFLP markers. Genome 40:370–378
Page RDM (1996) TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358
Panaud O, Chen X, McCouch SR (1996) Development of microsatellite markers and characterization of simple sequence length polymorphism (SSLP) in rice (Oryza sativa L.). Mol Gen Genet 252:597–607
Pemberton JM, Slate J, Bancroft DR, Barrett JA (1995) Non-amplyfying alleles at microsatellite loci: a caution for parentage and population studies. Mol Ecol 4:249–252
Powell W, Machray GC, Provan J (1996) Polymorphism revealed by simple sequence repeats. Trends Plant Sci 1:215–222
Provan J, Corbett G, McNicol JW, Powell W (1997) Chloroplast DNA variability in wild and cultivated rice (Oryza sativa) revealed by polymorphic chloroplast simple sequence repeats. Genome 40:104–110
Rice WR (1989) Analyzing tables of statistics tests. Evolution 43:223–225
Roa AC, Chavarriaga-Aguirre P, Duque MC, Maya MM, Bonierbale MW, Iglesias C, Tohme J (2000) Cross-species amplication of Cassava (Manihot esculenta) (Euphorbiaceae) microsatellites: allelic polymorphism and degree of relationship. Am J Bot 87:1647–1655
Slaktin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Smulders MJM, Bredemeijer G, Rus-Kortekass W, Arens P, Vosman B (1997) Use of short microsatellites from database sequences to generate polymorphisms among Lycopersicon esculentum cultivars and accessions of other Lycopersicon species. Theor Appl Genet 97:264–272
Streiff R, Labbe T, Bacilieri R, Steinkellner H, Glossl J, Kremer A (1998) Within-population genetic structure in Quercus robur L. and Quercus petraea (Matt.) Liebl. assessed with isozymes and microsatellites. Mol Ecol 7:317–328
Sun CQ, Wang XK, Li ZC, Yoshimura A, Iwata N (2001) Comparison of the genetic diversity of common wild rice (Oryza rufipogon Griff.) and cultivated rice (O. sativa L.) using RFLP markers. Theor Appl Genet 102:157–162
Sun GL, Diaz O, Salomon B, von Bothmer R (1999) Genetic diversity in Elymus caninus as revealed by isozyme, RAPD and microsatellite markers. Genome 42:420–431
Temnykh S, Park WD, Ayres N, Cartinhour S, Hauck N, Lipovich L, Cho YG, Ishii T, McCouch SR (2000) Mapping and genome organization of microsatellite sequences in rice (Oryza sativa L.). Theor Appl Genet 100:697–712
Vaughan DA (1994) The wild relatives of rice: a genetic resources handbook. International Rice Research Institute, Manila, The Philippines
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wright S (1978) Evolution and the genetics of populations, vol. 4. Variability within and among natural populations. University of Chicago Press, Chicago
Wu KS, Tanksley SD (1993) Abundance, polymorphism and genetic mapping of microsatellites in rice. Mol Gen Genet 241:225–235
Xiao J, Grandillo S, Ahn SN, McCouch SR, Tanksley SD (1996) Genes from wild rice improve yield. Nature 384:223–224
Xie ZW (1999) Population genetics and conservation strategies of Oryza rufipogon Griff. in China. PhD dissertation, Institute of Botany, Chinese Academy of Sciences, Beijing, China
Xie ZW, Lu YQ, Ge S, Hong DY, Li FZ (2001) Clonality in wild rice (oryza rufipogon, Poaceae) and its implications for conservation management. Am J Bot 88:1058–1064
Yang GP, Saghai Maroof MA, Xu CG, Zhang QF, Biyashev RM (1994) Comparative analysis of microsatellite DNA polymorphism in landraces and cultivars rice. Theor Appl Genet 245:187–194
Yeh FC, Yang RC, Boyle T (1999) POPGENE version1.32, Microsoft window-base software for population genetic analysis: a quick user's guide. University of Alberta, Center for International Forestry Research, Alberta, Canada
Zhao XP, Kochert G (1992) Characterization and genetic mapping of a short, highly repeated, interspersed DNA sequence from rice (Oryza sativa L.). Theor Appl Genet 231:353–359
Acknowledgements
We thank Dr. Li-zhi Gao, the Institute of Crop Germplasm Resources of the Chinese Academy of Agricultural Sciences, for his technological assistance and providing microsatellite primers. We are also grateful to Dr. Wei Qian for his assistance in the field collections. This research was supported by the National Natural Science Foundation of China (30025005) and the Chinese Academy of Sciences (kscxz-sw-101A).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H.F. Linskens
Rights and permissions
About this article
Cite this article
Zhou, Hf., Xie, Zw. & Ge, S. Microsatellite analysis of genetic diversity and population genetic structure of a wild rice (Oryza rufipogon Griff.) in China. Theor Appl Genet 107, 332–339 (2003). https://doi.org/10.1007/s00122-003-1251-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-003-1251-y