
Abstract
Metabolic cardiomyopathy is an emerging cause of heart failure in patients with obesity, insulin resistance, and diabetes. It is characterized by impaired myocardial metabolic flexibility, intramyocardial triglyceride accumulation, and lipotoxic damage in association with structural and functional alterations of the heart, unrelated to hypertension, coronary artery disease, and other cardiovascular diseases. Oxidative stress plays an important role in the development and progression of metabolic cardiomyopathy. Mitochondria are the most significant sources of reactive oxygen species (ROS) in cardiomyocytes. Disturbances in myocardial substrate metabolism induce mitochondrial adaptation and dysfunction, manifested as a mismatch between mitochondrial fatty acid oxidation and the electron transport chain (ETC) activity, which facilitates ROS production within the ETC components. In addition, non-ETC sources of mitochondrial ROS, such as β-oxidation of fatty acids, may also produce a considerable quantity of ROS in metabolic cardiomyopathy. Augmented ROS production in cardiomyocytes can induce a variety of effects, including the programming of myocardial energy substrate metabolism, modulation of metabolic inflammation, redox modification of ion channels and transporters, and cardiomyocyte apoptosis, ultimately leading to the structural and functional alterations of the heart. Based on the above mechanistic views, the present review summarizes the current understanding of the mechanisms underlying metabolic cardiomyopathy, focusing on the role of oxidative stress.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Obesity represents a major health challenge worldwide due to its escalating prevalence and associated clusters of cardio-metabolic diseases that reduce life quality and expectancy. It is estimated that by 2040, approximately 500 million individuals will be overweight and insulin resistant, and 642 million individuals will develop type 2 diabetes mellitus [1]. Obesity, insulin resistance, and diabetes can inflict numerous harmful effects on the heart. Accumulating evidence demonstrates that people with these conditions are at a higher risk of developing a metabolic cardiomyopathy phenotype, which has become an emerging cause of cardiac dysfunction and heart failure in these individuals [2,3,4]. Metabolic cardiomyopathy represents a chronic metabolic disorder characterized by an intramyocardial accumulation of triglycerides (TGs) and lipotoxic damage in association with structural and functional alterations of the heart, which occur independent of the presence of coronary artery disease, valvular heart disease, hypertension, and other cardiovascular diseases [5, 6]. Of note, the cardiac pathological phenotype of metabolic cardiomyopathy can be manifested as either heart failure with preserved ejection fraction (HFpEF) or heart failure with reduced ejection fraction (HFrEF) [7, 8]. Diabetes-related and obesity-related cardiomyopathies are the most studied subtypes of metabolic cardiomyopathy [9]. Diabetic cardiomyopathy was first reported in postmortem pathological findings in 1972 [10] and further confirmed in a 1974 Framingham Heart Study [11]. It is defined by abnormal myocardial structure and function, beyond that elicited by ischemia or hypertension [12, 13]. Diabetic cardiomyopathy is marked by lipid deposition in cardiomyocytes, reactivation of fetal genes, and left ventricular hypertrophy, which synergistically induce contractile dysfunction [14]. People with obesity, dyslipidemia, and insulin resistance are prone to develop a similar type of cardiomyopathy even in the absence of diabetes, which is often referred to as obesity-related cardiomyopathy [9, 15]. Considering that dysregulated metabolism is the key player in the etiology of these cardiomyopathies and diabetes, insulin resistance, and obesity are frequently overlapped, we group these cardiomyopathies together and collectively refer to them as metabolic cardiomyopathy in the present review, despite that diabetes and obesity may contribute to cardiac structural and functional derangements through several distinct mechanisms [9, 15].
The pathogenesis of metabolic cardiomyopathy is multifactorial, but insulin resistance is believed to be an integral component [5]. An insulin-resistant state leads to increased levels of free fatty acids (FFAs), proinflammatory mediators, and the appearance of certain adipocytokines and hepatokines in the circulation, which can further aggravate systemic insulin resistance and induce cardiac insulin resistance [16]. Cardiac insulin resistance leads to a marked alteration in myocardial energy substrate metabolism due to impaired insulin-regulated metabolic signaling, characterized by decreased glucose uptake and utilization and enhanced fatty acid uptake and β-oxidation [17,18,19]. This change results in impaired myocardial metabolic flexibility and reduced energy production efficiency. Furthermore, when FFA uptake markedly exceeds β-oxidation capacity in cardiomyocytes, excessive FFAs form TG. The deposition of fatty acids and TG in cardiomyocytes induce a phenomenon called cardiac lipotoxicity, which may promote mitochondrial dysfunction, oxidative stress, impaired calcium handling, myocardial cell apoptosis, and metabolic inflammation that ultimately lead to myocardial dysfunction and heart failure [5, 20, 21].
Enhanced oxidative stress in the heart is a common characteristic of metabolic cardiomyopathy [22,23,24]. Oxidative stress reflects a disturbance in the balance between the production of reactive oxygen species (ROS) and the biological system’s capacity to detoxify these reactive intermediates with antioxidants [25]. Depending on the cellular environment and the sources of ROS, ROS can act as signaling molecules that play important regulatory roles in normal physiological processes or modulate maladaptive responses that promote metabolic disorders, inflammation, and cell death [26, 27]. ROS, such as superoxide anion radical (O2•−) and hydrogen peroxide (H2O2), are constantly generated as byproducts of energy substrate metabolism in cardiomyocytes [28]. Cardiac insulin resistance and the resultant alterations in myocardial substrate metabolism can induce mitochondrial dysfunction and facilitate ROS production [19]. Excess generation of various ROS, such as O2•−, H2O2, and hydroxyl radicals, can induce oxidative modification to cellular macromolecules (e.g., lipids, DNA, and proteins) and is considered a crucial mechanism for metabolism-related immune responses and remodeling in the heart [28].
Although the prevalence of metabolic cardiomyopathy will likely grow considerably in the coming decades, its underlying mechanisms are still poorly understood. One major issue is that whether altered energy substrate metabolism is a cause or a consequence of structural and functional derangements in the heart remains elusive. Complicated interactions exist between metabolic alterations and cardiac structure and function, and the precise mechanisms underlying these interactions are not well elucidated [15]. Indeed, the term “metabolic cardiomyopathy” is not a well-established one. Additionally, very few diagnostic and therapeutic tools are currently available for this disease entity [5, 29]. Therefore, further investigation is urgently needed to strengthen the notion of “metabolic cardiomyopathy” and clarify the mechanistic basis underlying this disease. In this review, we summarize the alterations in energy substrate metabolism in metabolic cardiomyopathy and the possible mechanisms underlying the overproduction of ROS in cardiomyocytes. We also summarize the potential effects of ROS on myocardial metabolism, inflammatory responses, calcium regulation, and cell death. Finally, we briefly discuss several challenges facing the development of therapies against metabolic cardiomyopathy.
Myocardial energy substrate metabolism
Myocardial energy substrate metabolism under physiological conditions
The heart has the highest energy demands per gram of any organ. Myocardial ATP is generated at a high rate from the mitochondrial oxidation of diverse energy substrates, such as glucose, fatty acids, and lactate [30]. FFAs (primarily long-chain fatty acids) are the preferred energy substrates [31]. Under normal physiological conditions, ATP production in the myocardium is derived mainly from fatty acid β-oxidation (approximately 70–90%) [32]. Circulating FFAs enter cardiomyocytes via passive diffusion or transport with the help of fatty acid translocase (FAT)/cluster of differentiation (CD)36 [31]. Cytoplasmic long-chain FFAs are converted into long-chain fatty acyl-coenzyme-A (CoA) esters and then transferred into the mitochondria via carnitine palmitoyltransferase (CPT)-1. CPT-1 is a rate-limiting step in fatty acid catabolism, and its activity is inhibited by malonyl-CoA. Malonyl-CoA synthesis is catalyzed by acetyl-CoA carboxylase (ACC), whereas its degradation is catalyzed by malonyl-CoA decarboxylase (MCD) [33,34,35]. The activation of AMP-activated protein kinase (AMPK) induces an inhibitory effect on ACC with a consequent decrease in malonyl-CoA level and an increase in CPT-1 activity [36,37,38]. Once in the mitochondrial matrix, fatty acyl-CoA is broken down via β-oxidation to produce acetyl-CoA, which enters the citric acid cycle to produce nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), the reducing equivalents feeding the electron transport chain (ETC). The oxidation of reducing equivalents by the mitochondrial ETC results in an electrochemical gradient that is necessary for ATP production, but it may also stimulate ROS generation under certain pathological conditions [32, 33].
Although energy conversion in the heart primarily relies on fatty acid oxidation, the heart is metabolically omnivorous and also readily metabolizes glucose, ketone bodies, and amino acids. Approximately 10–30% of the acetyl-CoA used for ATP synthesis is derived from the oxidation of pyruvate, the final product of glycolysis and dehydrogenation [39, 40]. Glucose uptake in cardiomyocytes relies on the action of GLUTs in the cell membrane. The ubiquitous glucose transporter GLUT1 accounts for basal myocardial glucose uptake, while GLUT4 is responsible for insulin or contraction-stimulated glucose uptake [30, 41]. The heart exhibits prominent fuel flexibility under normal conditions and switches between fatty acids and glucose depending on the nutritional status and physical activity, which helps preserve contractile functions in various physiological and pathophysiological conditions [5, 32]. When fatty acid oxidization is enhanced, glucose utilization would be decreased via the Randle cycle [42]. On the other hand, after a high carbohydrate supply, increased insulin levels would induce a shift in cardiac energy substrate toward glucose utilization [31]. Indeed, insulin plays a vital role in metabolic homeostasis of the myocardium. The binding of insulin to the α-subunits of the insulin receptor on the cell surface stimulates the phosphorylation of the insulin receptor β-subunits, which activates the docking protein insulin receptor substrate 1 (IRS1) and subsequently upregulates the downstream phosphatidylinositol 3-kinase (PI3K) and RACβ serine/threonine-protein kinase 2 (AKT2). The activation of PI3K and AKT2 triggers the translocation of GLUT4 and CD36 from the intracellular store to the cell membrane, resulting in enhanced uptake of glucose and FFAs [5, 30]. Increased glucose and FFA uptake promotes mitochondrial oxidative metabolism to produce ATP and supports cardiac contractile function [30].
Myocardial energy substrate metabolism in metabolic cardiomyopathy
Insulin resistance is characterized by decreased insulin sensitivity and suppressed glucose disposal in peripheral tissues, and it is a hallmark of type 2 diabetes, obesity, and metabolic syndrome [43,44,45]. A number of mechanisms associated with systemic metabolic disorders, including increased FFA and ceramide levels, decreased glucose uptake, altered hepatokine profiles (e.g., elevated selenoprotein P and fetuin-A) and adipocytokine profiles (e.g., reduced secretion of adiponectin and enhanced secretion of leptin), augmented release of proinflammatory mediators, and changes in gut microbiota and its metabolites may cause systemic and cardiac insulin resistance [5, 46]. Several clinical studies have provided evidence linking systemic metabolic disorders with cardiac insulin resistance [47,48,49,50]. It is reported that myocardial insulin resistance occurs in about 60% of individuals with type 2 diabetes and is associated with an increased risk of cardiovascular diseases [47]. People with 1-h postload hyperglycemia and impaired glucose tolerance also showed attenuated global myocardial glucose metabolism, indicating that myocardial insulin resistance is an early defect that can already be detectable in subjects with prediabetes [48]. The association between type 2 diabetes and myocardial insulin resistance is independent of coronary artery disease, and myocardial insulin resistance is directly related to whole-body insulin resistance in individuals with type 2 diabetes [49]. It is important to stress that myocardial insulin resistance may act as a response mechanism to fuel overload rather than as a mediator of cardiac dysfunction [51]. Indeed, myocardial insulin resistance may protect the heart from being flooded with excess amounts of energy substrates in disordered metabolic states [52, 53]. This may explain why insulin-sensitizing agents unexpectedly led to adverse cardiovascular events such as the development of heart failure in some clinical studies [51, 54].
Insulin resistance may affect the myocardium via a number of mechanisms [18, 55]. Systemic insulin resistance induces uninhibited lipolysis in adipose tissues, leading to increased circulating FFAs and dramatically enhanced myocardial fatty acid uptake [34, 56, 57]. In the insulin resistance state, FAT/CD36 localization to the sarcolemmal membranes is increased; however, GLUT4 translocation to the plasma membranes is impaired. As a result, the insulin-dependent glucose uptake pathway is attenuated, resulting in a shift away from glycolytic metabolism, with increased reliance on fatty acid β-oxidation for energetic metabolism [58, 59]. This shift leads to metabolic inflexibility of the myocardium. It is well-established that ATP generation from fatty acid oxidation is less efficient than glucose, producing 40% less ATP per oxygen molecule consumed [59]. Disturbed myocardial substrate metabolism and increased left ventricular myocardial oxygen demand have been observed in severely obese patients with normal ejection fraction and without known heart disease [60, 61].
Despite the augmentation in flux via the β-oxidation pathway, excessive fatty acids exceed the mitochondrial respiratory capacity and accumulate in a metabolic low-turnover pool, and the TG content increases [32]. The limited capacity of cardiomyocyte to safely store excessive fatty acids in the TG pool may induce a phenomenon called “cardiac lipotoxicity,” which refers to toxicity resulting from the accumulated lipids and lipid intermediates (e.g., diacylglycerols, ceramides, and lysophosphatidyl choline species) within cardiomyocytes [32, 62]. Lipotoxicity can stimulate cardiomyocyte death via augmented ROS formation and endoplasmic reticulum (ER) stress, leading to structural and functional alterations of the heart. It has been shown that insulin resistance promotes cardiac lipotoxicity in human subjects and murine models [18, 55, 63]. Individuals with type 2 diabetes have increased intramyocardial TG content, which is significantly associated with left ventricular diastolic dysfunction, independent of age, blood pressure, heart rate, body mass index, and visceral fat [64, 65]. In addition, a consistent proportion of patients with nonischemic heart failure exhibit myocardial TG overload and altered expression of genes related to contractile dysfunction [66].
Several animal studies have used transgenic murine models to investigate the possible mechanisms of the effect of cardiac lipotoxicity or myocardial lipids on structural and functional alterations of the heart [18, 63]. Cardiac lipid accumulation, in association with myocardial damages, cell death, and cardiac remodeling and dysfunction, was observed in murine models with a restricted knockout of peroxisome proliferator–activated receptor gamma (PPARγ) in cardiomyocytes and cardiomyocyte overexpression of fatty acid transport proteins, membrane-anchored lipoproteins, acyl Co-A synthetase, diacylglycerol acyltransferase, and PPARα [18, 63]. These toxic effects are generally considered to be triggered by long-chain fatty acids and their derived metabolites, such as diglycerol and ceramide, which can further aggravate insulin resistance [18]. However, it is uncertain whether the findings derived from genetically modified animal models could be easily translated to human pathophysiology.
It is also noteworthy that the serum levels of branched-chain amino acids (BCAAs) are elevated in obesity and insulin resistance. Dysregulated BCAAs play an important role in the etiology of cardiovascular diseases [67]. High levels of serum BCAAs may aggravate myocardial insulin resistance and cardiac contractile dysfunction by inducing mitochondrial dysfunction and upregulating mammalian target of rapamycin (mTOR) [3, 67]. Moreover, the oxidation of BCAAs is crucial for cardiac function [3]. Inhibition of branched-chain α-keto acid dehydrogenase (BCKDH), the essential step for BCAA oxidation, decreases systolic function in the mouse heart, and the use of a pharmacological inhibitor of BCKDH promotes cardiac BCAA degradation and preserves cardiac systolic function [67].
In summary, profound perturbations in energy substrate metabolism occur in metabolic cardiomyopathy, characterized by enhanced fatty acid oxidation and suppressed glucose uptake and utilization (Fig. 1). These changes result in metabolic inflexibility and a reduction in the efficiency of cardiac energy production. Moreover, cardiac lipotoxicity may induce a variety of cellular processes, such as oxidative stress and cardiomyocyte apoptosis, that lead to structural and functional alterations of the heart. In addition, alterations in other energy substrates, such as BCAAs, may also contribute to the development of metabolic cardiomyopathy.

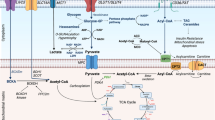
Energy substrate metabolism and mitochondrial reactive oxygen species (ROS) production in cardiomyocyte at physiological or pathophysiological states. Under physiological condition (left panel), the cardiomyocyte is metabolically omnivorous and readily metabolizes various energy substrates such as glucose and free fatty acids (FFAs) for ATP production. In metabolic cardiomyopathy (right panel), cardiac insulin resistance leads to decreased glucose uptake and utilization and enhanced fatty acid (FA) uptake and β-oxidation in cardiomyocytes. When the uptake of FFAs markedly exceeds the β-oxidation capacity, excess FAs form triglyceride (TG) and lipotoxic lipids such as ceramide and diacylglycerol (DAG), leading to cardiac lipotoxicity. FA β-oxidation produces flavin adenine dinucleotide (FADH2) and nicotinamide adenine dinucleotide (NADH), which are the reducing equivalents that feed the electron transport chain (ETC). In well-coupled mitochondria, the electron flow rate through the ETC is limited by the ATP turnover rate. Impaired electron transfer within the ETC facilitates electron leakage from the ETC complexes, resulting in ROS formation. Moreover, enzymes in FA oxidation such as long and very long-chain acyl-CoA dehydrogenase (LCAD\VLCAD) may also produce a considerable quantity of ROS, especially in the context of myocardial metabolic disorders. GLUT, glucose transporter; FAT, fatty acid translocase; CD36, cluster of differentiation 36; PI3K, phosphatidylinositol 3-kinase; FACS, fatty acyl-CoA synthetase; CPT, carnitine palmitoyltransferase; PDH, pyruvate dehydrogenase; ETF, electron transfer flavoprotein; ETFDH, ETF dehydrogenase; ANT, adenine nucleotide translocator
Oxidative stress in metabolic cardiomyopathy
Metabolic cardiomyopathy is closely associated with the presence of oxidative stress. Perturbations in myocardial energy substrate metabolism affect several ROS generators, particularly mitochondria. Augmented ROS production and decreased antioxidant defense mechanisms characterize the myocardium of human subjects and animal models with obesity, insulin resistance, or metabolic syndrome [23, 24].
The most common forms of cardiac ROS include O2•−, H2O2, peroxynitrite (ONOO−), and hydroxyl radical (HO•) [58]. O2•− is a highly reactive molecule generated by a one-electron reduction of oxygen. It is dismutated to H2O2 by superoxide dismutase (SOD), maintaining its concentrations in the picomolar-nanomolar range [68]. Unlike O2•−, H2O2 is relatively more stable and readily crosses lipid membranes. It is a crucial redox signaling molecule that participates in both physiological and pathological processes and exhibits fairly low reactivity and relatively high specificity for molecular targets. H2O2 is decomposed to O2 and H2O by catalase, maintaining its concentration in the nanomolar range [69]. O2•− also rapidly reacts with nitric oxide to generate ONOO−, an oxidant with a relatively short half-life primarily linked to pathology [70]. It can inflict further structural damage via lipid peroxidation and exhibits fairly high reactivity and low specificity for molecular targets [58].
ROS are maintained within a certain range by endogenous antioxidant systems under normal conditions to activate physiological redox signaling, which is necessary for normal cellular functions, such as cell metabolism, proliferation, differentiation, immune defense, and survival [26]. Oxidative stress occurs when ROS generation is elevated and/or the antioxidant mechanisms are impaired. An excess of ROS activates pathological redox signaling that leads to cell damage, particularly when ROS generation shifts to more toxic ROS species [71, 72]. However, identifying the specific ROS species that participate in a signaling event is not easy because one ROS species can be transformed into other species, and their effects on molecular targets might not be noticeably different [73].
Sources of ROS in cardiomyocytes include organelles, such as mitochondria, and enzymes, such as NADPH oxidase (NOX), nitric oxide synthases, and xanthine oxidase [24, 58]. ROS generally accumulate in certain cellular compartments, and each of the organellar compartments has its own redox niche within the cell [74]. Mitochondria are the most quantitatively relevant ROS generators in cardiomyocytes, and these organelles are particularly crucial for ROS produced from energetic metabolism [22, 23]. The ETC is the most studied source of mitochondrial ROS. The monoelectronic reduction of O2 occurs at complexes I, II, and III within the ETC, resulting in the production of O2•− and other derivative species. Of note, accumulating studies demonstrate that other sources of mitochondrial ROS are also quantitatively sizable, such as pyruvate dehydrogenase, glycerol phosphate dehydrogenase, α-ketoglutarate dehydrogenase, and monoaminoxidase [27, 75, 76]. NOX is an important source of cytoplasmic ROS that directly catalyzes the one-electron reduction of O2 to O2•− using NADPH as the electron donor [77]. It is a membrane-bound enzyme complex with seven isoforms (NOX1-5 and DUOX1-2) in mammalian cells, which differ in tissue distribution and ROS production kinetics [78]. NOX2 and NOX4 are considered the major isoforms in myocardium, with NOX2 localized to the cell membrane and NOX4 predominantly to the mitochondria [79, 80]. Increased NOX4 expression and ROS production have been shown in pressure overloaded heart [81]. Nox4 deletion in cardiomyocytes significantly attenuated cardiomyocyte apoptosis, myocardial hypertrophy, and interstitial fibrosis in the presence of pressure overload [81]. In addition, loss of Nox2 could prevent oxidative stress and progression to advanced heart failure in response to pressure overload in mice [82]. Moreover, Ca2+-dependent NOX5 is reported to be a crucial NOX isoform participated in oxidative stress-mediated cardiac hypertrophy [83].
Because mitochondria are the most important cellular source of ROS in metabolic cardiomyopathy due to their key role in myocardial energetic metabolism, the next section focuses on the mechanisms of cardiac ROS overproduction induced by mitochondrial dysfunction under metabolic disorders.
Metabolic alterations induce mitochondrial ROS overproduction
ROS produced from the ETC
The heart is one of the highest energy-demanding organs with extreme dependence on the normal structure and function of mitochondria. Mitochondria are the most important cellular source of ROS in metabolic cardiomyopathy but also suffer ROS-mediated damage [84]. Despite discrepancies, accumulating evidence indicates mitochondrial dysfunction in metabolism-related cardiomyopathy, characterized by alterations in ETC complex activity, oxygen consumption, fatty acid oxidation, and mitochondrial DNA (mtDNA) content. In general, mitochondrial fatty acid oxidation is augmented, or at least preserved, in metabolic cardiomyopathy, but the ETC activity progressively declines with disease progression [2, 3, 19, 22, 23, 58, 59, 85]. The mismatch between mitochondrial fatty acid oxidation and ETC activity induces more substrate-derived reducing equivalents to get into and then leak from the impaired ETC, resulting in the overproduction of oxidants (Fig. 1).
It is reported that FFAs may exert dual effects on mitochondrial ROS production, shown as either an increase or reduction in O2•− formation [73, 86]. O2•− formation from mitochondria oxidizing NAD-dependent substrates dramatically increases in the presence of FFAs, which is related to the partial inactivation of complexes I and III [87,88,89,90]. FFAs can interact with ETC components, thus lowering the rate of forward electron transfer (FET) and promoting O2•− formation, which might be the primary mechanism of the FFA-stimulated production of oxidants in nonphagocytic cells [86]. In addition, FFAs are amphiphilic and may incorporate into mitochondrial inner membranes, which increases membrane fluidity and the chances of electron leakage [91,92,93]. On the other hand, FFAs have mild uncoupling or protonophore effects due to the cyclic movement of their protonated and deprotonated forms across the mitochondrial inner membrane [94]. Therefore, FFAs can act as protonophores and decrease ROS generation in reverse electron transfer (RET) [87, 89, 95]. Notably, a high Δp is required for RET, and only a slight depolarization of the mitochondrial inner membrane by ADP phosphorylation or the mild uncoupling effects of FFAs can abolish the RET-dependent formation of oxidants [96]. This mechanism suggests that the RET-dependent generation of oxidants may have little importance under an in vivo condition, in which mitochondria are actively phosphorylating [73].
ROS produced from non-ETC sources
Several non-ETC sources of ROS exist in mitochondria. For example, several mitochondrial flavoenzymes, including α-ketoglutarate dehydrogenase, pyruvate dehydrogenase, glycerol phosphate, electron transfer flavoprotein (ETF), and ETF-oxidoreductase, are increasingly considered important ROS sources, which may generate a markedly larger quantity of O2•− compared to ETC in certain circumstances [75, 96, 97]. Moreover, a considerable proportion of H2O2 originating from fatty acid oxidation is produced in mitochondrial matrix by flavoproteins upstream of complex III, including short-, medium-, long-, and very long-chain acyl-CoA dehydrogenases (SCAD, MCAD, LCAD, and VLCAD, respectively), which are essential in mitochondrial fatty acid oxidation [98, 99]. Electrons flow from these acyl-CoA dehydrogenases to ETF then ETF dehydrogenase (ETFDH), which transfers electrons to ubiquinone and eventually to complex III. Among the various acyl-CoA substrates, long-chain fatty acid substrates are likely associated with the augmented generation of mitochondrial ROS [98,99,100]. LCAD and VLCAD use long-chain acyl-CoA substrates. Recent studies indicate that VLCAD and LCAD can produce H2O2 directly [99, 101]. It is estimated that VLCAD makes a considerable overall contribution to ROS formation (nmol min−1 mg−1 protein range) due to its continuous oxidase activity [101]. Moreover, recombinant human LCAD is reported to generate H2O2 15-fold faster than VLCAD [98]. Because fatty acid oxidation is the predominant energy source of the heart, VLCAD and LCAD might be important sources of non-ETC oxidants and crucial players in oxidative stress in metabolic cardiomyopathy (Fig. 1). More gain-of-function and loss-of-function studies of fatty acid oxidation–related flavoproteins are needed to elucidate their functional roles in ROS production, contribution to oxidative stress, and regulatory roles in the pathogenic mechanisms of metabolic cardiomyopathy.
Oxidative damage to the mitochondria
Mitochondria-originated ROS can modulate cellular redox-sensitive signalings via reversible modification of critical elements in signal transduction to exert multiple physiological functions [75]. Conversely, ROS at a high concentration can lead to non-specific damages to intracellular macromolecules, which generally produce more reactive intermediates and initiate a chain of damage amplification [73]. Indeed, fatty acid–induced ROS generation may trigger both oxidative damages to the mitochondria and mitochondrial uncoupling that leads to impaired ATP production, indicating a pathophysiological association between oxidative stress and mitochondrial dysfunction [102, 103].
Mitochondria have their own DNA that is circular, double-stranded, 16,569 base-paired DNA encoding thirteen subunits of mammalian respiratory complexes, 22 tRNAs, and 2 rRNAs [104]. mtDNA shows a higher sensitivity to oxidative damages compared to nuclear DNA. Several human and animal studies have suggested that mtDNA may accumulate significantly more oxidized bases relative to nuclear DNA [105]. This is because mtDNA is closer to ROS-producing respiratory chains and it lacks the protection of histone-like proteins. H2O2 and O2•− synthesized in the mitochondrial inner membrane can readily react with proximal mtDNA and cause the accumulation of point mutations and/or deletions and reduced DNA copy number, impairing mitochondrial functions [105, 106]. Mitochondria possess well-defined DNA repair mechanisms quite similar to those of the nucleus, such as base excision repair, mismatch repair, single-strand break repair, and homology recombination–dependent repair. Several mitochondrial repair mechanisms have been reported to improve mitochondrial dysfunction and cardiac structural and functional abnormality [107,108,109,110]. However, the DNA repairing capacity in the mitochondria appears to be less powerful than that in the nucleus [107, 111]. Moreover, it is suggested that oxidative stress could directly or indirectly impair the mtDNA repair pathways, resulting in an increased number of mtDNA mutations [110, 111]. Nevertheless, the mechanisms of mtDNA repair and their role in contributing to mitochondrial dysfunction in cardiomyocytes require further investigation.
Oxidative damage to mtDNA leads to a decrease in mitochondrial transcript and protein levels and impaired ETC function and ATP formation, which further trigger mitochondrial ROS production and mtDNA mutations to form a vicious cycle of damage amplification [112,113,114]. Several studies have indicated that elevated oxidative mtDNA damages are involved in the development of cardiomyopathy and heart failure [115,116,117]. The coexistence of ROS-mutated mtDNA and wild-type mtDNA in the same mitochondrion is called mtDNA heteroplasmy [118, 119]. As the percentage of mutant mtDNA rises above certain thresholds (60–70%), mitochondrial homeostatic mechanisms are disturbed, resulting in impaired mitochondrial bioenergetic capacity, reduced mitophagy, and imbalance of mitochondrial fusion and fission events. Dysfunctional mitochondria accumulated in cardiomyocytes further lead to impaired ATP generation, cell atrophy, or death, and also interfere with certain metabolic mechanisms and signaling pathways, which contributes to the development of cardiomyopathy [118]. Indeed, the increase in mtDNA heteroplasmy not only causes mitochondrial dysfunction but also induces broad alterations in gene expression [120,121,122], as mitochondria serve as critical intracellular signaling hubs modulating nuclear gene expression at both transcriptional and epigenetic levels. To date, over 400 mtDNA mutations have been identified to be associated with human diseases such as heart failure [119]. In this context, developing mitochondrial therapies to deal with mtDNA heteroplasmy has received growing attention for the treatment of cardiovascular diseases [118].
ROS not only affect mtDNA but also lipids and proteins. Accumulation of fatty acids and lipid-derived metabolites in the internal mitochondrial membrane may increase lipid peroxidation. Lipid peroxidation products, such as 4-hydroxynonenal (4-HNE), 4-hydroxy hexenal, and malondialdehyde (MDA), are involved in the development of metabolic cardiomyopathy [123]. There is evidence that lipid peroxidation products may damage mitochondrial membranes, suppress cyclooxygenase (COX), and activate uncoupling protein (UCP)-2, leading to ETC dysfunction and exacerbation of ROS production [73, 123]. One of the primary phospholipids of the mitochondrial inner membranes, cardiolipin, is also prone to modification by oxidants [124]. Oxidative modification of cardiolipin can impair mitochondrial membrane fluidity, reduce ETC activity, and induce mitochondrial permeability transition pore (mPTP) opening, resulting in profound consequences on mitochondrial function [124]. Moreover, changes in the mitochondrial proteome and related mitochondrial dysfunctions are involved in cardiac diseases [84]. Several lines of evidence showed an underlying connection between cardiac oxidative stress and alterations of the mitochondrial proteome [125, 126]. Significant cardiac mitochondrial proteome remodeling has been observed in diabetic mice in response to transverse aortic constriction, which could be significantly attenuated by mitochondrial catalase–mediated scavenging of mitochondrial ROS [126]. However, the association between oxidative stress and alteration of cardiac mitochondrial proteome needs to be further studied with an in-depth analysis of sequencing technology.
Consequences of oxidative stress in metabolic cardiomyopathy
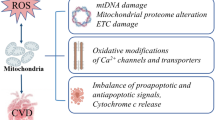
ROS are important regulators of multiple intracellular responses via redox signaling pathways. However, the role of ROS in physiology or pathology depends on their type, concentration, and the site of formation [75]. Low concentrations of ROS primarily participate in physiological processes, such as cell proliferation and differentiation and excitation–contraction coupling (ECC). In contrast, high concentrations of ROS induce oxidative stress–regulated signaling events and modify the structure and function of critical molecules in cardiomyocytes, which further worsens myocardial metabolism, induces dysfunctions in mitochondria and ion channels and transporters, and triggers inflammation and cardiomyocyte apoptosis (Fig. 2). These changes ultimately result in impaired cardiac remodeling and heart failure [127].

Consequences of oxidative stress in metabolic cardiomyopathy. Augmented reactive oxygen species (ROS) production in cardiomyocytes induces a variety of effects, including modulation of key regulators in myocardial energy substrate metabolism, alterations in insulin sensitivity, modulation of metabolic inflammation, redox modification of calcium channels and transporters, and mitochondrial dysfunction as well as cardiomyocyte apoptosis, which ultimately lead to structural and functional alterations of the heart. AMPK, AMP-activated protein kinase; PPAR, peroxisome proliferator–activated receptor; TLR, Toll-like receptor; NLRP3, NLR family pyrin domain–containing 3; JNK, c-Jun N-terminal kinase; ASK1, apoptosis signal-regulating kinase 1; NF-κB, nuclear factor-kappa B; SERCA, sarcoplasmic reticulum Ca2+-adenosine triphosphatase; RYR, ryanodine receptor; mPTP, mitochondrial permeability transition pore
Effects of ROS on myocardial energy substrate metabolism
Modulation of key regulators in energy substrate metabolism
A coordinated network of nuclear receptors finely regulates the enzymes involved in cardiac energy substrate metabolism [32]. Emerging evidence suggests that some of these nuclear receptors may act as redox sensors, and ROS exposure can affect the expression and activity of several nuclear receptors and critical enzymes in different cultured cell models [128]. PPARs, including PPARα, PPARβ/δ, and PPARγ, are a nuclear receptor superfamily implicated in the regulation of metabolism [129, 130]. PPARα is highly expressed in the myocardium and regulates the expression of many enzymes responsible for various processes in metabolism [131]. H2O2 exposure can downregulate the expression of PPARα and its downstream target genes CPT-1 and ACOX [132]. Moreover, H2O2 and lipid peroxidation products, such as 4-HNE, can suppress PPARγ expression, which promotes insulin sensitivity and fatty acid oxidation [133, 134]. AMPK, the key metabolic sensor and regulator in myocardial energetic metabolism, has recently emerged as a redox sensor that contributes to cardiac physiology maintenance and disease progression prevention [135]. Both mitochondrial and cytoplasmic ROS have been shown to promote AMPK activity [136]. H2O2 could directly modulate AMPK activity and its downstream metabolic pathways via oxidative modification of the AMPKα subunit and s-glutathionylation of the α- and β-subunits of AMPK [137]. AMPK activation attenuates oxidative stress by acting on the expression of prooxidant and antioxidant genes, thereby protecting the heart from injuries [135].
Regulation of insulin signaling
A number of studies have been conducted to directly link ROS-mediated signaling to the development of cardiac insulin resistance. For instance, cardiac insulin resistance in mice with Glut4 deletion was associated with upregulation of NOX1 and NOX2 [138]. Moreover, myocardial overexpression of the antioxidants catalase and metallothionein showed that antioxidants play a critical role in the signaling and contractile dysregulation associated with insulin resistance [139, 140]. Although oxidative stress has long been considered a critical element in insulin resistance, inconsistent results of the association between ROS and insulin sensitivity are reported [141, 142]. Recent studies showed that approaches promoting H2O2 elimination provided greater protection against insulin resistance than those targeting O2•− [141, 143, 144]. Indeed, H2O2 is considered to act as a second messenger in signal transduction, whereas O2•− seems to act simply as a precursor of H2O2 rather than an important second messenger [145]. It is now recognized that H2O2 can induce a stimulatory or an inhibitory effect on insulin signaling, depending on its concentration and production site relative to different components of the insulin signaling pathway [142]. Several signaling pathways, such as the c-Jun N-terminal kinase (JNK) pathway, were proposed as a mechanistic link between H2O2 production and insulin resistance [146]. JNK catalyzes the phosphorylation of IRS-1 at serine residues and blocks signal transduction downstream of IRS-1. High concentrations of H2O2 (≥ 5 μM) activate the protein tyrosine phosphatases PTP-1B and JNK1 in vitro and suppress insulin-stimulated phosphorylation of IRS and Akt [146].
The role of ROS in metabolic inflammation
Metabolic stress–induced inflammation is deeply involved in the development and progression of metabolic cardiomyopathy [5]. Notably, metabolic inflammation is primarily modulated by the innate immune system [5]. Cardiac innate immune signaling plays an important role in the regulation of several critical pathophysiological processes, such as chronic inflammation, fibrogenesis, insulin resistance, and redox imbalance [147,148,149,150,151,152,153,154]. Therefore, cardiac innate immune signaling may serve as a driving force in the development of metabolism-related cardiomyopathy [155,156,157,158].
Accumulating evidence indicates that innate immune signaling and redox signaling interact widely, but the molecular mechanisms underlying these interactions are not well understood [73]. ROS have been shown to participate in innate immune signaling by interacting with several critical components in signal transduction, including pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain–like receptors (NLRs), intracellular kinases, such as JNK and apoptosis signal-regulating kinase 1 (ASK1), and transcription factors, such as nuclear factor-kappa B (NF-κB) [73]. For example, studies have shown that O2•− promotes inflammation via TLR4 [159], and TLR2 participates in the mechanism of H2O2-induced cytotoxicity on cardiomyocytes [160]. In addition, both mitochondrial and NOX-originated ROS can stimulate the formation of the NLR family pyrin domain–containing 3 (NLRP3) inflammasome [161], and SOD mimetics and catalase can inhibit NLRP3 activation [162]. NLRP3 inflammasome plays a crucial role in the pathophysiology of cardiac dysfunction by activating caspase-1, which subsequently cleaves pro-interleukin (IL)-1β or pro-IL-18 to their mature forms [5]. Indeed, the crystal structure of NLRP3 suggests a high sensitivity to alterations in redox status [163]. However, whether the priming step of NLRP3 inflammasome activation requires ROS participation needs to be further studied. Moreover, ROS can activate the JNK signaling pathway by suppressing the activity of mitogen-activated protein kinase (MAPK) phosphatase, and in turn, the activated JNK can promote mitochondrial ROS formation via SAB, thereby forming a positive feedback loop [164]. Furthermore, ASK1 is a redox-sensitive intracellular kinase that may play a critical role in oxidative stress–induced effects in the heart [127]. Mice with Ask1 deletion exhibit smaller increase in left ventricular end-diastolic and end-systolic size, smaller reduction in fractional shortening, and a lower cell apoptosis level than wild-type mice after thoracic transverse aortic constriction or coronary artery ligation [165]. Although ROS can activate ASK1 and its downstream signaling pathways, such as the p38 and JNK pathways, by preventing the binding of TrxR to ASK1 [166, 167], ROS can also inhibit ASK1 activity by activating phosphatase 5 (PP5) [168]. In addition, ROS and their secondary products, such as oxidized phospholipids and 4-HNE, can directly or indirectly activate NF-κB [169, 170], which is a ROS-sensitive transcription factor that modulates the expression of a wide range of proinflammatory and prooxidant genes in the heart [73].
Recent evidence demonstrates that adaptive immunity is an emerging player in the progression of cardio-metabolic diseases. T1 and T17 cells are implicated in the development and progression of heart failure, which contribute to the induction of specific pathological phenotypes of mononuclear cells and sustained pathological chronic inflammation [171]. Mitochondrial dysfunction in T cells has been shown to contribute to severe cardiovascular complications [172]. Moreover, mechanisms underlying B lymphocyte activation including the production of proinflammatory chemokines, cytokines, and cardiac autoantibodies are also involved in the pathogenesis of heart failure [171]. Thus, both unbalanced T and B cell pathways in the adaptive immune network contribute to cardiomyocyte death and cardiac remodeling and dysfunction [171]. Oxidative stress is associated with the activation, differentiation, and survival of T and B cells [173]. Moderate levels of ROS are necessary for T cell activation [173]. The activation of TCR induces mitochondrial ROS production, which promotes T cell activation by modulating IL2 and IL4 expression [174]. However, excessive ROS production could downregulate NF-кB phosphorylation and suppress T cell activity [175]. In addition, ROS can regulate T cell differentiation and thereby modulate the generation of cytokines by polarized T cell subsets [173]. For example, in vitro administration of H2O2 decreased IFNγ secretion of activated Th1 and promoted IL4 production of activated Th2 [176]. Mice with mutated p45phox or gp91phox showed blocked Treg induction and T cell suppression, indicating NOX-derived ROS are crucial for Treg differentiation and function [177]. Similarly, BCR stimulation induces rapid ROS generation in primary resting murine B cells, and ROS produced by both the NOX and mitochondria are participated in B cell activation [173, 178]. NOX2-derived ROS are mainly engaged in the early stage of B cell activation and mitochondrial respiration-derived ROS are at a later stage [178].
Collectively, ROS actively participate in signal transduction in both innate and adaptive immune responses and are engaged in some feedback loops that are part of the extensive interactions between immune signaling and redox signaling to form a fine-tuned network in the development of metabolism-related cardiomyopathy.
Oxidative modifications of ion channels and transporters
Increased ROS formation can severely affect cardiomyocyte electrophysiology. Altered Ca2+ handling is a hallmark of the contractile dysfunction observed in patients with heart failure [5]. Oxidative stress may contribute to cardiac dysfunction in the context of obesity and insulin resistance by affecting Ca2+ handling [179]. Accumulating evidence indicates that increased ROS formation under myocardial metabolic disorders may induce Ca2+ mishandling via redox modulation of critical proteins implicated in this process, such as sarcoplasmic reticulum Ca2+-adenosine triphosphatase 2a (SERCA2a) and ryanodine receptor 2 (RyR2) [84, 180]. The sulfhydryl groups of the cysteine residues of these ion channels and ion pumps are critical targets for oxidative modification [84, 181]. Studies in obese and insulin-resistant animal models showed augmented formation of ROS and oxidative damage products, such as protein carbonyl and lipid peroxidation, in the heart and ventricular myocytes in association with altered activity of Ca2+-handling proteins and impaired cardiac relaxation and contraction [179, 182,183,184]. ROS-related posttranslational modifications can induce diverse functional results according to the type of ion channels, pumps, and other transporters [84]. Oxidative modification of SERCA2a leads to prolonged Ca2+ transients and slower SERCA2a-mediated Ca2+ reuptake [179]. In ob/ob mice, augmented irreversible carbonyl oxidation and decreased activity of SERCA2a have been observed, which were associated with impaired relaxation of the heart [182]. In addition, ventricular myocytes isolated from rats fed sucrose showed enhanced oxidation and decreased activity of SERCA2a, which could be reversed with the antioxidant N-acetylcysteine (NAC) [185]. Moreover, RyR is a redox-sensitive sarcoplasmic reticulum Ca2+ release channel located in the inner mitochondrial membrane that can be hyperactivated by the redox modification of thiol groups, which results in calcium leakage and altered calcium kinetics [84]. Furthermore, ROS also increase the influx of Ca2+ via L-type calcium channels and reverse the function of the Na+/Ca2+ exchanger (NCX), which results in Ca2+ influx and Na+ efflux [180]. Therefore, oxidative stress contributes to cardiac dysfunction via the oxidative modification of ion channels, pumps, or other transporter types, leading to alterations in their activities and eventually impaired ECC [84].
Oxidative stress–induced cardiomyocyte apoptosis
Cardiomyocyte apoptosis is a crucial contributor to hypertrophic remodeling and cardiac dysfunction [186]. Oxidative stress is a major risk factor for triggering cardiomyocyte apoptosis via the modulation of downstream signaling pathways [187]. Previous studies found that exposure of human cardiac progenitor cells (CPCs) to H2O2 triggered apoptosis by activating the JNK signaling pathway [188]. Furthermore, ROS production contributed to palmitate-induced apoptosis in human CPCs, and pretreatment with antioxidant NAC in human CPCs attenuated both palmitate-induced ROS production and apoptosis [189]. The mechanisms involved in ROS-induced mitochondria-dependent apoptosis in cardiomyocytes may include the activation of proapoptotic signaling pathways (e.g., the JNK, p38, and ASK1 pathways), suppression of antiapoptotic signaling pathways (e.g., the PI3K/AKT pathway and the ERK1/2 pathway), and immediate ROS-triggered effects on mitochondria that induce the release of cytochrome c [84]. Alterations in the mPTP and membrane potentials are key early signals of oxidative stress responses. Oxidative stress and ATP consumption lead to a long opening of mPTP in the mitochondrial intima and reduction in the proton gradient and potential energy, which cause mitochondrial swelling and the release of large quantities of cytochrome c and apoptosis-inducing factors. These events trigger caspase-dependent and caspase-independent cascade apoptosis reactions, ultimately contributing to the development and progression of cardiac diseases [84]. Moreover, the ROS-induced activation of mPTP opening can lead to alterations in intra- and intermitochondrial redox environments and facilitate the release of ROS, a phenomenon termed “ROS-induced ROS release” (RIRR), which may result in different outcomes depending on the levels of ROS, ranging from cell injury or even cell and organismal death [96]. Maintaining mitochondrial integrity and blocking the expression of apoptotic genetic programs could be a viable therapeutic strategy for cardiomyocyte apoptosis [84].
Challenges in the development of therapies to treat metabolic cardiomyopathy
Several therapies aimed at inhibiting oxidative stress or augmenting antioxidant defenses improved cardiac dysfunction related to metabolism in animal models [3]. These strategies included (1) natural antioxidant compounds, such as vitamins, polyphenols, and flavonoids, (2) synthetic antioxidant agents that selectively target mitochondria, such as SOD mimetics, coenzyme Q10 and its analogs, and mitochondria-targeted peptides, and (3) gene transfer therapy [24]. Despite the promising results in preclinical models, data from large-scale clinical trials with antioxidative therapies for cardiovascular diseases are disappointing due to the lack of efficacy and undesired adverse effects [3, 180]. Thus, there are still many challenges in the development of successful antioxidative therapies for the treatment of metabolism-related cardiomyopathy. Differences in the sources, spatial distribution, local concentrations, and targets of ROS complicate the selection of drugs to target oxidative stress in cardiomyocytes effectively. In addition, maintenance of the ROS levels needed for physiological processes in the heart is a major challenge. Further treatments should inhibit oxidative stress and meanwhile not severely affect general redox homeostasis. Moreover, determination of the cell and tissue specificity of antioxidant drugs is also a difficulty. Antioxidant agents in the future should specifically target and counteract cardiomyocyte injuries with little harm to normal cardiac cells. Another challenge for the development of therapeutic agents is the poor selectivity and permeability of the cell membranes. Advances in drug delivery technology, such as the use of polymeric nanoparticles as drug delivery systems, may be a viable solution [190, 191].
The selection of appropriate in vivo and in vitro models is also vital for the translational study of metabolic cardiomyopathy. Several animal models, including diet-induced, genetic, or models with a combination of more than one intervention, have been widely applied in the mechanistic studies of cardiomyopathy related to metabolism [3]. Although animal models are indispensable for the preclinical evaluation of potential drugs, they are largely incompatible with high-throughput drug discovery due to the cost and the length of time involved in model generation and validation. Furthermore, concerns have been raised about the interspecies differences in clinical manifestation, pathophysiology, and disease severity [192]. The logical approach would thereby be to conduct research on primary cardiomyocytes obtained directly from human heart tissue. Nevertheless, it is impractical to perform meaningful long-term investigational and interventional studies on such cells due to their limited proliferative ability, lifespan, and availability. Fortunately, recent advances in human induced pluripotent stem cell (iPSC) technologies have provided a promising tool for modeling cardiomyopathy via human heart tissue in a dish [192]. As summarized in recent reviews [192,193,194], human iPSC-derived cardiomyocytes (iPSC-CMs) have been applied as a platform to study a wide range of cardiac disorders related to metabolism and oxidative stress. For instance, Venkatesh et al. identified LONP1 protease as a negative regulator of mitochondrial fatty acid oxidation by using iPSC-CMs and proteomics [195]. SCO2 deficiency in iPSC-CMs led to cardiomyopathy with abnormalities in mitochondrial morphology and function [196]. PRKAG2-mutated iPSC-CMs showed marked metabolic perturbation, characterized by increased AMPK activity, excessive glycogen deposition, and enhanced fatty acid oxidation [197]. In iPSC-CMs derived from patients with Barth syndrome, excessive ROS production was detected, and ROS scavenging by small molecules improved mitochondrial dysfunction [198]. Thus, iPSC-CMs have the potential to become a key translational asset for drug discovery in metabolic cardiomyopathy [193]. A major limitation of iPSC-CMs in metabolic study is their immature phenotype, as iPSC-CMs often resemble fetal cardiomyocytes in structure and function, such as a metabolic preference for glucose utilization [199]. Several new maturation protocols have shown promising results to promote the metabolic and functional maturation of iPSC-CMs, making them applicable to recapitulate adult-like metabolic phenotypes and model cardiomyopathy related to metabolic disorders [192].
Concluding remarks and future perspectives
Metabolic cardiomyopathy is characterized by impaired myocardial metabolic flexibility, intramyocardial TG accumulation, and lipotoxic damage in association with structural and functional alterations of the heart. Oxidative stress actively participates in the pathogenesis of metabolic cardiomyopathy. Mitochondria are the most significant ROS sources in cardiomyocytes, and metabolic cardiomyopathy is associated with mitochondrial dysfunction, which manifests as the mismatch between mitochondrial fatty acid oxidation and ETC activity. The ETC is the most well-studied site of mitochondrial ROS production, but non-ETC sources of ROS in the mitochondria, such as fatty acid β-oxidation, might also produce a considerable quantity of ROS in the context of myocardial metabolic disorders. The consequences of augmented cardiac ROS generation include the reprogramming of myocardial energy substrate metabolism, alterations in insulin sensitivity, regulation of metabolic inflammation, redox modification of ion channels and transporters, and cardiomyocyte apoptosis. These effects suggest that oxidative stress plays a critical role in the pathogenesis of metabolic cardiomyopathy.
In this context, treatments of metabolic cardiomyopathy such as antioxidative therapies have gained much attention. However, many challenges must be conquered for successful translation from the laboratory to the clinic. Indeed, there are still many unanswered questions, such as (1) whether altered energy substrate metabolism is a cause or a consequence of cardiac structural and functional abnormities? (2) What are the central metabolic events that drive the transition from metabolic disorders to cardiac injuries? How metabolic intermediates and oxidative stress are linked with immune responses in the heart? How to identify the most key molecules in the progression of metabolic cardiomyopathy and how to determine their cell-specific functions? (3) How to identify specific agents that selectively target pathogenic pathways (e.g., metabolic, oxidative, or immunological pathways) in cardiomyocytes while maintaining their normal biological functions? (4) Why in individuals with similar metabolic disorders (e.g., diabetes, insulin resistance, or obesity), some develop metabolic cardiomyopathy while others develop atherosclerotic cardiovascular diseases? (5) How to identify individuals with a higher risk of metabolic cardiomyopathy at an early stage? How to optimize patient management based on risk stratification?
To overcome these daunting challenges, systems-based multi-omic analyses, in combination with recent advances in iPSC technology and clinically relevant animal models, would be useful to gain an in-depth understanding of the underlying mechanisms linking metabolic disorders, oxidative stress, and immune responses in the heart. In addition, novel organelle-targeted probes and antioxidant compounds would be useful to clarify the contribution of compartment-specific oxidants and their effects on critical intracellular processes. It is important to stress that a formal definition for metabolic cardiomyopathy as a distinct clinical entity remains vague. Most clinical studies to date have focused on atherosclerotic cardiovascular events, but not on cardiomyopathy related to myocardial metabolic disturbances. Thus, more large-scale epidemiological studies and basic researches with clinically relevant animal models should be conducted to strengthen the notion of “metabolic cardiomyopathy,” provide solid evidence for a causal relationship between systemic metabolic disorders and cardiomyopathy, and increase the awareness of this disease entity among physicians, biomedical researchers, and the public. Finally, there are no approved drugs for the treatment of metabolic cardiomyopathy. Further clinical trials with adequate duration and power are required to assess the long-term efficacy and safety of potential treatment options for this disease.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH et al (2017) IDF Diabetes Atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract 128:40–50. https://doi.org/10.1016/j.diabres.2017.03.024
Czibik G, d’Humieres T, Derumeaux G (2021) When does too much energy become a danger to the heart? Eur Heart J. https://doi.org/10.1093/eurheartj/ehab801
Ren J, Wu NN, Wang S, Sowers JR, Zhang Y (2021) Obesity cardiomyopathy: evidence, mechanisms, and therapeutic implications. Physiol Rev 101(4):1745–1807. https://doi.org/10.1152/physrev.00030.2020
Zhou J, Bai L, Zhang XJ, Li H, Cai J (2021) Nonalcoholic fatty liver disease and cardiac remodeling risk: pathophysiological mechanisms and clinical implications. Hepatology 74(5):2839–2847. https://doi.org/10.1002/hep.32072
Nishida K, Otsu K (2017) Inflammation and metabolic cardiomyopathy. Cardiovasc Res 113(4):389–398. https://doi.org/10.1093/cvr/cvx012
Maack C, Murphy E (2017) Metabolic cardiomyopathies - fighting the next epidemic. Cardiovasc Res 113(4):367–369. https://doi.org/10.1093/cvr/cvx022
Lavie CJ, Alpert MA, Arena R, Mehra MR, Milani RV, Ventura HO (2013) Impact of obesity and the obesity paradox on prevalence and prognosis in heart failure. JACC Heart Fail 1(2):93–102. https://doi.org/10.1016/j.jchf.2013.01.006
Mishra S, Kass DA (2021) Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 18(6):400–423. https://doi.org/10.1038/s41569-020-00480-6
Li J, Li J, Chen Y, Hu W, Gong X, Qiu H et al (2022) The role of mitochondria in metabolic syndrome-associated cardiomyopathy. Oxid Med Cell Longev 2022:9196232. https://doi.org/10.1155/2022/9196232
Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A (1972) New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol 30(6):595–602. https://doi.org/10.1016/0002-9149(72)90595-4
Kannel WB, Hjortland M, Castelli WP (1974) Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 34(1):29–34. https://doi.org/10.1016/0002-9149(74)90089-7
Jia G, Hill MA, Sowers JR (2018) Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res 122(4):624–638. https://doi.org/10.1161/CIRCRESAHA.117.311586
Bugger H, Abel ED (2014) Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57(4):660–671. https://doi.org/10.1007/s00125-014-3171-6
Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M et al (2012) Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest 122(3):1109–1118. https://doi.org/10.1172/JCI60329
Nakamura M, Sadoshima J (2020) Cardiomyopathy in obesity, insulin resistance and diabetes. J Physiol 598(14):2977–2993. https://doi.org/10.1113/JP276747
Mandavia CH, Aroor AR, Demarco VG, Sowers JR (2013) Molecular and metabolic mechanisms of cardiac dysfunction in diabetes. Life Sci 92(11):601–608. https://doi.org/10.1016/j.lfs.2012.10.028
Cai J, Zhang XJ, Ji YX, Zhang P, She ZG, Li H (2020) Nonalcoholic fatty liver disease pandemic fuels the upsurge in cardiovascular diseases. Circ Res 126(5):679–704. https://doi.org/10.1161/CIRCRESAHA.119.316337
Alpert MA, Karthikeyan K, Abdullah O, Ghadban R (2018) Obesity and cardiac remodeling in adults: mechanisms and clinical implications. Prog Cardiovasc Dis 61(2):114–123. https://doi.org/10.1016/j.pcad.2018.07.012
Jia G, DeMarco VG, Sowers JR (2016) Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol 12(3):144–153. https://doi.org/10.1038/nrendo.2015.216
Schilling JD, Machkovech HM, Kim AH, Schwendener R, Schaffer JE (2012) Macrophages modulate cardiac function in lipotoxic cardiomyopathy. Am J Physiol Heart Circ Physiol 303(11):H1366–H1373. https://doi.org/10.1152/ajpheart.00111.2012
Chen Z, Liu J, Zhou F, Li H, Zhang XJ, She ZG et al (2021) Nonalcoholic fatty liver disease: an emerging driver of cardiac arrhythmia. Circ Res 128(11):1747–1765. https://doi.org/10.1161/CIRCRESAHA.121.319059
Bugger H, Abel ED (2008) Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci (Lond) 114(3):195–210. https://doi.org/10.1042/CS20070166
Nicolson GL (2007) Metabolic syndrome and mitochondrial function: molecular replacement and antioxidant supplements to prevent membrane peroxidation and restore mitochondrial function. J Cell Biochem 100(6):1352–1369. https://doi.org/10.1002/jcb.21247
Ilkun O, Boudina S (2013) Cardiac dysfunction and oxidative stress in the metabolic syndrome: an update on antioxidant therapies. Curr Pharm Des 19(27):4806–4817. https://doi.org/10.2174/1381612811319270003
Forman HJ, Zhang H (2021) Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov 20(9):689–709. https://doi.org/10.1038/s41573-021-00233-1
Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK (2018) Reactive oxygen species in metabolic and inflammatory signaling. Circ Res 122(6):877–902. https://doi.org/10.1161/CIRCRESAHA.117.311401
van der Vliet A, Janssen-Heininger YMW, Anathy V (2018) Oxidative stress in chronic lung disease: from mitochondrial dysfunction to dysregulated redox signaling. Mol Aspects Med 63:59–69. https://doi.org/10.1016/j.mam.2018.08.001
Shah AK, Bhullar SK, Elimban V, Dhalla NS (2021) Oxidative stress as a mechanism for functional alterations in cardiac hypertrophy and heart failure. Antioxidants (Basel) 10(6). https://doi.org/10.3390/antiox10060931
Costantino S, Akhmedov A, Melina G, Mohammed SA, Othman A, Ambrosini S et al (2019) Obesity-induced activation of JunD promotes myocardial lipid accumulation and metabolic cardiomyopathy. Eur Heart J 40(12):997–1008. https://doi.org/10.1093/eurheartj/ehy903
Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L (2020) Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol 17(9):585–607. https://doi.org/10.1038/s41569-020-0339-2
Palomer X, Salvado L, Barroso E, Vazquez-Carrera M (2013) An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int J Cardiol 168(4):3160–3172. https://doi.org/10.1016/j.ijcard.2013.07.150
Sletten AC, Peterson LR, Schaffer JE (2018) Manifestations and mechanisms of myocardial lipotoxicity in obesity. J Intern Med 284(5):478–491. https://doi.org/10.1111/joim.12728
Roul D, Recchia FA (2015) Metabolic alterations induce oxidative stress in diabetic and failing hearts: different pathways, same outcome. Antioxid Redox Signal 22(17):1502–1514. https://doi.org/10.1089/ars.2015.6311
Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z et al (2004) Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 109(18):2191–2196. https://doi.org/10.1161/01.CIR.0000127959.28627.F8
Chen Z, Yu Y, Cai J, Li H (2019) Emerging molecular targets for treatment of nonalcoholic fatty liver disease. Trends Endocrinol Metab 30(12):903–914. https://doi.org/10.1016/j.tem.2019.08.006
Folmes CD, Lopaschuk GD (2007) Role of malonyl-CoA in heart disease and the hypothalamic control of obesity. Cardiovasc Res 73(2):278–287. https://doi.org/10.1016/j.cardiores.2006.10.008
Zhang XJ, Cai J, Li H (2021) Targeting ACC for NASH resolution. Trends Mol Med. https://doi.org/10.1016/j.molmed.2021.11.002
Jian C, Fu J, Cheng X, Shen LJ, Ji YX, Wang X et al (2020) Low-dose sorafenib acts as a mitochondrial uncoupler and ameliorates nonalcoholic steatohepatitis. Cell Metab 31(5):892–908 e11. https://doi.org/10.1016/j.cmet.2020.04.011
Stanley WC, Lopaschuk GD, Hall JL, McCormack JG (1997) Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions Cardiovasc Res 33(2):243–257. https://doi.org/10.1016/s0008-6363(96)00245-3
Wisneski JA, Gertz EW, Neese RA, Gruenke LD, Morris DL, Craig JC (1985) Metabolic fate of extracted glucose in normal human myocardium. J Clin Invest 76(5):1819–1827. https://doi.org/10.1172/JCI112174
Davey KA, Garlick PB, Warley A, Southworth R (2007) Immunogold labeling study of the distribution of GLUT-1 and GLUT-4 in cardiac tissue following stimulation by insulin or ischemia. Am J Physiol Heart Circ Physiol 292(4):H2009–H2019. https://doi.org/10.1152/ajpheart.00663.2006
Randle PJ, Garland PB, Hales CN, Newsholme EA (1963) The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1(7285):785–9. https://doi.org/10.1016/s0140-6736(63)91500-9
Luo P, Wang PX, Li ZZ, Zhang XJ, Jiang X, Gong J et al (2016) Hepatic oncostatin M receptor beta regulates obesity-induced steatosis and insulin resistance. Am J Pathol 186(5):1278–1292. https://doi.org/10.1016/j.ajpath.2015.12.028
Wang XA, Deng S, Jiang D, Zhang R, Zhang S, Zhong J et al (2013) CARD3 deficiency exacerbates diet-induced obesity, hepatosteatosis, and insulin resistance in male mice. Endocrinology 154(2):685–697. https://doi.org/10.1210/en.2012-1911
Yan FJ, Zhang XJ, Wang WX, Ji YX, Wang PX, Yang Y et al (2017) The E3 ligase tripartite motif 8 targets TAK1 to promote insulin resistance and steatohepatitis. Hepatology 65(5):1492–1511. https://doi.org/10.1002/hep.28971
Zhao YC, Zhao GJ, Chen Z, She ZG, Cai J, Li H (2020) Nonalcoholic fatty liver disease: an emerging driver of hypertension. Hypertension 75(2):275–284. https://doi.org/10.1161/HYPERTENSIONAHA.119.13419
Herance JR, Martin-Saladich Q, Velasquez MA, Hernandez C, Aparicio C, Ramirez-Serra C et al (2022) Identification of myocardial insulin resistance by using liver tests: a simple approach for clinical practice. Int J Mol Sci 23(15). https://doi.org/10.3390/ijms23158783
Succurro E, Pedace E, Andreozzi F, Papa A, Vizza P, Fiorentino TV et al (2020) Reduction in global myocardial glucose metabolism in subjects with 1-hour postload hyperglycemia and impaired glucose tolerance. Diabetes Care 43(3):669–676. https://doi.org/10.2337/dc19-1975
Iozzo P, Chareonthaitawee P, Dutka D, Betteridge DJ, Ferrannini E, Camici PG (2002) Independent association of type 2 diabetes and coronary artery disease with myocardial insulin resistance. Diabetes 51(10):3020–3024. https://doi.org/10.2337/diabetes.51.10.3020
Hu L, Qiu C, Wang X, Xu M, Shao X, Wang Y (2018) The association between diabetes mellitus and reduction in myocardial glucose uptake: a population-based (18)F-FDG PET/CT study. BMC Cardiovasc Disord 18(1):203. https://doi.org/10.1186/s12872-018-0943-9
Taegtmeyer H, Beauloye C, Harmancey R, Hue L (2013) Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. Am J Physiol Heart Circ Physiol 305(12):H1693–H1697. https://doi.org/10.1152/ajpheart.00854.2012
Harmancey R, Wilson CR, Taegtmeyer H (2008) Adaptation and maladaptation of the heart in obesity. Hypertension 52(2):181–187. https://doi.org/10.1161/HYPERTENSIONAHA.108.110031
Hue L, Taegtmeyer H (2009) The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 297(3):E578–E591. https://doi.org/10.1152/ajpendo.00093.2009
Nissen SE, Wolski K (2007) Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 356(24):2457–2471. https://doi.org/10.1056/NEJMoa072761
Peterson LR (2006) Obesity and insulin resistance: effects on cardiac structure, function, and substrate metabolism. Curr Hypertens Rep 8(6):451–456. https://doi.org/10.1007/s11906-006-0022-y
Lopaschuk GD, Folmes CD, Stanley WC (2007) Cardiac energy metabolism in obesity. Circ Res 101(4):335–347. https://doi.org/10.1161/CIRCRESAHA.107.150417
Herrero P, Peterson LR, McGill JB, Matthew S, Lesniak D, Dence C et al (2006) Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol 47(3):598–604. https://doi.org/10.1016/j.jacc.2005.09.030
Mellor KM, Ritchie RH, Delbridge LM (2010) Reactive oxygen species and insulin-resistant cardiomyopathy. Clin Exp Pharmacol Physiol 37(2):222–228. https://doi.org/10.1111/j.1440-1681.2009.05274.x
Witteles RM, Fowler MB (2008) Insulin-resistant cardiomyopathy clinical evidence, mechanisms, and treatment options. J Am Coll Cardiol 51(2):93–102. https://doi.org/10.1016/j.jacc.2007.10.021
Grymyr LMD, Nadirpour S, Gerdts E, Nedrebo BG, Hjertaas JJ, Matre K et al (2021) Left ventricular myocardial oxygen demand and subclinical dysfunction in patients with severe obesity referred for bariatric surgery. Nutr Metab Cardiovasc Dis 31(2):666–674. https://doi.org/10.1016/j.numecd.2020.10.009
Hannukainen JC, Lautamaki R, Parkka J, Strandberg M, Saunavaara V, Hurme S et al (2018) Reversibility of myocardial metabolism and remodelling in morbidly obese patients 6 months after bariatric surgery. Diabetes Obes Metab 20(4):963–973. https://doi.org/10.1111/dom.13183
Szczepaniak LS, Dobbins RL, Metzger GJ, Sartoni-D’Ambrosia G, Arbique D, Vongpatanasin W et al (2003) Myocardial triglycerides and systolic function in humans: in vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn Reson Med 49(3):417–423. https://doi.org/10.1002/mrm.10372
Wende AR, Symons JD, Abel ED (2012) Mechanisms of lipotoxicity in the cardiovascular system. Curr Hypertens Rep 14(6):517–531. https://doi.org/10.1007/s11906-012-0307-2
Rijzewijk LJ, van der Meer RW, Smit JW, Diamant M, Bax JJ, Hammer S et al (2008) Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol 52(22):1793–1799. https://doi.org/10.1016/j.jacc.2008.07.062
Ng AC, Delgado V, Bertini M, van der Meer RW, Rijzewijk LJ, Hooi Ewe S et al (2010) Myocardial steatosis and biventricular strain and strain rate imaging in patients with type 2 diabetes mellitus. Circulation 122(24):2538–2544. https://doi.org/10.1161/CIRCULATIONAHA.110.955542
Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K et al (2004) Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18(14):1692–1700. https://doi.org/10.1096/fj.04-2263com
Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z et al (2016) Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 133(21):2038–2049. https://doi.org/10.1161/CIRCULATIONAHA.115.020226
Beyer W, Imlay J, Fridovich I (1991) Superoxide dismutases. Prog Nucleic Acid Res Mol Biol 40:221–253. https://doi.org/10.1016/s0079-6603(08)60843-0
Oshino N, Chance B, Sies H, Bucher T (1973) The role of H 2 O 2 generation in perfused rat liver and the reaction of catalase compound I and hydrogen donors. Arch Biochem Biophys 154(1):117–131. https://doi.org/10.1016/0003-9861(73)90040-4
White CR, Brock TA, Chang LY, Crapo J, Briscoe P, Ku D et al (1994) Superoxide and peroxynitrite in atherosclerosis. Proc Natl Acad Sci USA 91(3):1044–1048. https://doi.org/10.1073/pnas.91.3.1044
Zhang L, Wang X, Cueto R, Effi C, Zhang Y, Tan H et al (2019) Biochemical basis and metabolic interplay of redox regulation. Redox Biol 26:101284. https://doi.org/10.1016/j.redox.2019.101284
Campbell EL, Colgan SP (2019) Control and dysregulation of redox signalling in the gastrointestinal tract. Nat Rev Gastroenterol Hepatol 16(2):106–120. https://doi.org/10.1038/s41575-018-0079-5
Chen Z, Tian R, She Z, Cai J, Li H (2020) Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med 152:116–141. https://doi.org/10.1016/j.freeradbiomed.2020.02.025
Naviaux RK (2012) Oxidative shielding or oxidative stress? J Pharmacol Exp Ther 342(3):608–618. https://doi.org/10.1124/jpet.112.192120
Figueira TR, Barros MH, Camargo AA, Castilho RF, Ferreira JC, Kowaltowski AJ et al (2013) Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid Redox Signal 18(16):2029–2074. https://doi.org/10.1089/ars.2012.4729
Vercesi AE, Castilho RF, Kowaltowski AJ, de Oliveira HCF, de Souza-Pinto NC, Figueira TR et al (2018) Mitochondrial calcium transport and the redox nature of the calcium-induced membrane permeability transition. Free Radic Biol Med 129:1–24. https://doi.org/10.1016/j.freeradbiomed.2018.08.034
Nabeebaccus A, Zhang M, Shah AM (2011) NADPH oxidases and cardiac remodelling. Heart Fail Rev 16(1):5–12. https://doi.org/10.1007/s10741-010-9186-2
Zhang M, Perino A, Ghigo A, Hirsch E, Shah AM (2013) NADPH oxidases in heart failure: poachers or gamekeepers? Antioxid Redox Signal 18(9):1024–1041. https://doi.org/10.1089/ars.2012.4550
Kayama Y, Raaz U, Jagger A, Adam M, Schellinger IN, Sakamoto M et al (2015) Diabetic cardiovascular disease induced by oxidative stress. Int J Mol Sci 16(10):25234–25263. https://doi.org/10.3390/ijms161025234
Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J (2010) Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res 106(7):1253–1264. https://doi.org/10.1161/CIRCRESAHA.109.213116
Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J (2010) NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA 107(35):15565–15570. https://doi.org/10.1073/pnas.1002178107
Parajuli N, Patel VB, Wang W, Basu R, Oudit GY (2014) Loss of NOX2 (gp91phox) prevents oxidative stress and progression to advanced heart failure. Clin Sci (Lond) 127(5):331–340. https://doi.org/10.1042/CS20130787
Zhao GJ, Zhao CL, Ouyang S, Deng KQ, Zhu L, Montezano AC et al (2020) Ca(2+)-dependent NOX5 (NADPH oxidase 5) exaggerates cardiac hypertrophy through reactive oxygen species production. Hypertension 76(3):827–838. https://doi.org/10.1161/HYPERTENSIONAHA.120.15558
Dhalla NS, Shah AK, Tappia PS (2020) Role of oxidative stress in metabolic and subcellular abnormalities in diabetic cardiomyopathy. Int J Mol Sci 21(7). https://doi.org/10.3390/ijms21072413
Saotome M, Ikoma T, Hasan P, Maekawa Y (2019) Cardiac insulin resistance in heart failure: the role of mitochondrial dynamics. Int J Mol Sci 20(14). https://doi.org/10.3390/ijms20143552
Schonfeld P, Wojtczak L (2008) Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic Biol Med 45(3):231–241. https://doi.org/10.1016/j.freeradbiomed.2008.04.029
Schonfeld P, Wojtczak L (2007) Fatty acids decrease mitochondrial generation of reactive oxygen species at the reverse electron transport but increase it at the forward transport. Biochim Biophys Acta 1767(8):1032–1040. https://doi.org/10.1016/j.bbabio.2007.04.005
Cocco T, Di Paola M, Papa S, Lorusso M (1999) Arachidonic acid interaction with the mitochondrial electron transport chain promotes reactive oxygen species generation. Free Radic Biol Med 27(1–2):51–59. https://doi.org/10.1016/s0891-5849(99)00034-9
Loskovich MV, Grivennikova VG, Cecchini G, Vinogradov AD (2005) Inhibitory effect of palmitate on the mitochondrial NADH: ubiquinone oxidoreductase (complex I) as related to the active-de-active enzyme transition. Biochem J 387(Pt 3):677–683. https://doi.org/10.1042/BJ20041703
Schonfeld P, Reiser G (2006) Rotenone-like action of the branched-chain phytanic acid induces oxidative stress in mitochondria. J Biol Chem 281(11):7136–7142. https://doi.org/10.1074/jbc.M513198200
Stillwell W, Jenski LJ, Crump FT, Ehringer W (1997) Effect of docosahexaenoic acid on mouse mitochondrial membrane properties. Lipids 32(5):497–506. https://doi.org/10.1007/s11745-997-0064-6
Schonfeld P, Struy H (1999) Refsum disease diagnostic marker phytanic acid alters the physical state of membrane proteins of liver mitochondria. FEBS Lett 457(2):179–183. https://doi.org/10.1016/s0014-5793(99)01009-1
Gille L, Nohl H (2001) The ubiquinol/bc1 redox couple regulates mitochondrial oxygen radical formation. Arch Biochem Biophys 388(1):34–38. https://doi.org/10.1006/abbi.2000.2257
Skulachev VP (1991) Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS Lett 294(3):158–162. https://doi.org/10.1016/0014-5793(91)80658-p
Korshunov SS, Korkina OV, Ruuge EK, Skulachev VP, Starkov AA (1998) Fatty acids as natural uncouplers preventing generation of O2- and H2O2 by mitochondria in the resting state. FEBS Lett 435(2–3):215–8. https://doi.org/10.1016/s0014-5793(98)01073-4
Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94(3):909–950. https://doi.org/10.1152/physrev.00026.2013
Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE (2009) Mitochondria and reactive oxygen species. Free Radic Biol Med 47(4):333–343. https://doi.org/10.1016/j.freeradbiomed.2009.05.004
Zhang Y, Bharathi SS, Beck ME, Goetzman ES (2019) The fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase can be a source of mitochondrial hydrogen peroxide. Redox Biol 26:101253. https://doi.org/10.1016/j.redox.2019.101253
Cardoso AR, Kakimoto PA, Kowaltowski AJ (2013) Diet-sensitive sources of reactive oxygen species in liver mitochondria: role of very long chain acyl-CoA dehydrogenases. PLoS ONE 8(10):e77088. https://doi.org/10.1371/journal.pone.0077088
Montgomery MK, Osborne B, Brown SH, Small L, Mitchell TW, Cooney GJ et al (2013) Contrasting metabolic effects of medium- versus long-chain fatty acids in skeletal muscle. J Lipid Res 54(12):3322–3333. https://doi.org/10.1194/jlr.M040451
Kakimoto PA, Tamaki FK, Cardoso AR, Marana SR, Kowaltowski AJ (2015) H2O2 release from the very long chain acyl-CoA dehydrogenase. Redox Biol 4:375–380. https://doi.org/10.1016/j.redox.2015.02.003
Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED (2005) Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 112(17):2686–2695. https://doi.org/10.1161/CIRCULATIONAHA.105.554360
Boudina S, Bugger H, Sena S, O’Neill BT, Zaha VG, Ilkun O et al (2009) Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 119(9):1272–1283. https://doi.org/10.1161/CIRCULATIONAHA.108.792101
Simoes ICM, Fontes A, Pinton P, Zischka H, Wieckowski MR (2018) Mitochondria in non-alcoholic fatty liver disease. Int J Biochem Cell Biol 95:93–99. https://doi.org/10.1016/j.biocel.2017.12.019
Muftuoglu M, Mori MP, de Souza-Pinto NC (2014) Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion 17:164–181. https://doi.org/10.1016/j.mito.2014.03.007
Kawahara H, Fukura M, Tsuchishima M, Takase S (2007) Mutation of mitochondrial DNA in livers from patients with alcoholic hepatitis and nonalcoholic steatohepatitis. Alcohol Clin Exp Res 31(1 Suppl):S54-60. https://doi.org/10.1111/j.1530-0277.2006.00287.x
Marin-Garcia J (2016) Mitochondrial DNA repair: a novel therapeutic target for heart failure. Heart Fail Rev 21(5):475–487. https://doi.org/10.1007/s10741-016-9543-x
Bradley JM, Li Z, Organ CL, Polhemus DJ, Otsuka H, Islam KN et al (2018) A novel mtDNA repair fusion protein attenuates maladaptive remodeling and preserves cardiac function in heart failure. Am J Physiol Heart Circ Physiol 314(2):H311–H321. https://doi.org/10.1152/ajpheart.00515.2017
Cividini F, Scott BT, Dai A, Han W, Suarez J, Diaz-Juarez J et al (2016) O-GlcNAcylation of 8-oxoguanine DNA glycosylase (Ogg1) impairs oxidative mitochondrial DNA lesion repair in diabetic hearts. J Biol Chem 291(51):26515–26528. https://doi.org/10.1074/jbc.M116.754481
Gredilla R, Sanchez-Roman I, Gomez A, Lopez-Torres M, Barja G (2020) Mitochondrial base excision repair positively correlates with longevity in the liver and heart of mammals. Geroscience 42(2):653–665. https://doi.org/10.1007/s11357-020-00158-4
Lee SR, Han J (2017) Mitochondrial mutations in cardiac disorders. Adv Exp Med Biol 982:81–111. https://doi.org/10.1007/978-3-319-55330-6_5
Pohjoismaki JL, Goffart S, Tyynismaa H, Willcox S, Ide T, Kang D et al (2009) Human heart mitochondrial DNA is organized in complex catenated networks containing abundant four-way junctions and replication forks. J Biol Chem 284(32):21446–21457. https://doi.org/10.1074/jbc.M109.016600
Miller FJ, Rosenfeldt FL, Zhang C, Linnane AW, Nagley P (2003) Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res 31(11):e61. https://doi.org/10.1093/nar/gng060
Frahm T, Mohamed SA, Bruse P, Gemund C, Oehmichen M, Meissner C (2005) Lack of age-related increase of mitochondrial DNA amount in brain, skeletal muscle and human heart. Mech Ageing Dev 126(11):1192–1200. https://doi.org/10.1016/j.mad.2005.06.008
Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P et al (1999) Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet 21(1):133–137. https://doi.org/10.1038/5089
Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J et al (2000) Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc Natl Acad Sci USA 97(7):3467–3472. https://doi.org/10.1073/pnas.97.7.3467
Marin-Garcia J, Goldenthal MJ, Moe GW (2001) Mitochondrial pathology in cardiac failure. Cardiovasc Res 49(1):17–26. https://doi.org/10.1016/s0008-6363(00)00241-8
Elorza AA, Soffia JP (2021) mtDNA heteroplasmy at the core of aging-associated heart failure. An integrative view of OXPHOS and mitochondrial life cycle in cardiac mitochondrial physiology. Front Cell Dev Biol 9:625020. https://doi.org/10.3389/fcell.2021.625020
Li M, Schonberg A, Schaefer M, Schroeder R, Nasidze I, Stoneking M (2010) Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am J Hum Genet 87(2):237–249. https://doi.org/10.1016/j.ajhg.2010.07.014
Lakshmanan LN, Yee Z, Ng LF, Gunawan R, Halliwell B, Gruber J (2018) Clonal expansion of mitochondrial DNA deletions is a private mechanism of aging in long-lived animals. Aging Cell 17(5):e12814. https://doi.org/10.1111/acel.12814
Tuppen HA, Blakely EL, Turnbull DM, Taylor RW (2010) Mitochondrial DNA mutations and human disease. Biochim Biophys Acta 1797(2):113–128. https://doi.org/10.1016/j.bbabio.2009.09.005
Li H, Slone J, Fei L, Huang T (2019) Mitochondrial DNA variants and common diseases: a mathematical model for the diversity of age-related mtDNA mutations. Cells 8(6). https://doi.org/10.3390/cells8060608
Martinelli I, Tomassoni D, Moruzzi M, Roy P, Cifani C, Amenta F et al (2020) Cardiovascular changes related to metabolic syndrome: evidence in obese zucker rats. Int J Mol Sci 21(6). https://doi.org/10.3390/ijms21062035
Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y et al (2010) Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab 12(2):154–165. https://doi.org/10.1016/j.cmet.2010.07.003
Tappia PS, Adameova A, Dhalla NS (2018) Attenuation of diabetes-induced cardiac and subcellular defects by sulphur-containing amino acids. Curr Med Chem 25(3):336–345. https://doi.org/10.2174/0929867324666170705115207
Wilson AJ, Gill EK, Abudalo RA, Edgar KS, Watson CJ, Grieve DJ (2018) Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart 104(4):293–299. https://doi.org/10.1136/heartjnl-2017-311448
D’Oria R, Schipani R, Leonardini A, Natalicchio A, Perrini S, Cignarelli A et al (2020) The role of oxidative stress in cardiac disease: from physiological response to injury factor. Oxid Med Cell Longev 2020:5732956. https://doi.org/10.1155/2020/5732956
Quijano C, Trujillo M, Castro L, Trostchansky A (2016) Interplay between oxidant species and energy metabolism. Redox Biol 8:28–42. https://doi.org/10.1016/j.redox.2015.11.010
Zhu LH, Wang A, Luo P, Wang X, Jiang DS, Deng W et al (2014) Mindin/Spondin 2 inhibits hepatic steatosis, insulin resistance, and obesity via interaction with peroxisome proliferator-activated receptor alpha in mice. J Hepatol 60(5):1046–1054. https://doi.org/10.1016/j.jhep.2014.01.011
Tong J, Han CJ, Zhang JZ, He WZ, Zhao GJ, Cheng X et al (2019) Hepatic interferon regulatory factor 6 alleviates liver steatosis and metabolic disorder by transcriptionally suppressing peroxisome proliferator-activated receptor gamma in mice. Hepatology 69(6):2471–2488. https://doi.org/10.1002/hep.30559
Li S, Yang B, Du Y, Lin Y, Liu J, Huang S et al (2018) Targeting PPARalpha for the treatment and understanding of cardiovascular diseases. Cell Physiol Biochem 51(6):2760–2775. https://doi.org/10.1159/000495969
Li JL, Wang QY, Luan HY, Kang ZC, Wang CB (2012) Effects of L-carnitine against oxidative stress in human hepatocytes: involvement of peroxisome proliferator-activated receptor alpha. J Biomed Sci 19:32. https://doi.org/10.1186/1423-0127-19-32
Blanquicett C, Kang BY, Ritzenthaler JD, Jones DP, Hart CM (2010) Oxidative stress modulates PPAR gamma in vascular endothelial cells. Free Radic Biol Med 48(12):1618–1625. https://doi.org/10.1016/j.freeradbiomed.2010.03.007
Pizzimenti S, Laurora S, Briatore F, Ferretti C, Dianzani MU, Barrera G (2002) Synergistic effect of 4-hydroxynonenal and PPAR ligands in controlling human leukemic cell growth and differentiation. Free Radic Biol Med 32(3):233–245. https://doi.org/10.1016/s0891-5849(01)00798-5
Marino A, Hausenloy DJ, Andreadou I, Horman S, Bertrand L, Beauloye C (2021) AMP-activated protein kinase: a remarkable contributor to preserve a healthy heart against ROS injury. Free Radic Biol Med 166:238–254. https://doi.org/10.1016/j.freeradbiomed.2021.02.047
Cardaci S, Filomeni G, Ciriolo MR (2012) Redox implications of AMPK-mediated signal transduction beyond energetic clues. J Cell Sci 125(Pt 9):2115–2125. https://doi.org/10.1242/jcs.095216
Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E (2010) Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem 285(43):33154–33164. https://doi.org/10.1074/jbc.M110.143685
Ritchie RH, Quinn JM, Cao AH, Drummond GR, Kaye DM, Favaloro JM et al (2007) The antioxidant tempol inhibits cardiac hypertrophy in the insulin-resistant GLUT4-deficient mouse in vivo. J Mol Cell Cardiol 42(6):1119–1128. https://doi.org/10.1016/j.yjmcc.2007.03.900
Fang CX, Dong F, Ren BH, Epstein PN, Ren J (2005) Metallothionein alleviates cardiac contractile dysfunction induced by insulin resistance: role of Akt phosphorylation, PTB1B. PPARgamma and c-Jun Diabetologia 48(11):2412–2421. https://doi.org/10.1007/s00125-005-1940-y
Turdi S, Li Q, Lopez FL, Ren J (2007) Catalase alleviates cardiomyocyte dysfunction in diabetes: role of Akt, Forkhead transcriptional factor and silent information regulator 2. Life Sci 81(11):895–905. https://doi.org/10.1016/j.lfs.2007.07.029
Di Meo S, Iossa S, Venditti P (2017) Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J Endocrinol 233(1):R15–R42. https://doi.org/10.1530/JOE-16-0598
Rindler PM, Crewe CL, Fernandes J, Kinter M, Szweda LI (2013) Redox regulation of insulin sensitivity due to enhanced fatty acid utilization in the mitochondria. Am J Physiol Heart Circ Physiol 305(5):H634–H643. https://doi.org/10.1152/ajpheart.00799.2012
Boden MJ, Brandon AE, Tid-Ang JD, Preston E, Wilks D, Stuart E et al (2012) Overexpression of manganese superoxide dismutase ameliorates high-fat diet-induced insulin resistance in rat skeletal muscle. Am J Physiol Endocrinol Metab 303(6):E798-805. https://doi.org/10.1152/ajpendo.00577.2011
Lark DS, Kang L, Lustig ME, Bonner JS, James FD, Neufer PD et al (2015) Enhanced mitochondrial superoxide scavenging does not improve muscle insulin action in the high fat-fed mouse. PLoS ONE 10(5):e0126732. https://doi.org/10.1371/journal.pone.0126732
Forman HJ, Maiorino M, Ursini F (2010) Signaling functions of reactive oxygen species. Biochemistry 49(5):835–842. https://doi.org/10.1021/bi9020378
Iwakami S, Misu H, Takeda T, Sugimori M, Matsugo S, Kaneko S et al (2011) Concentration-dependent dual effects of hydrogen peroxide on insulin signal transduction in H4IIEC hepatocytes. PLoS ONE 6(11):e27401. https://doi.org/10.1371/journal.pone.0027401
Cai J, Xu M, Zhang X, Li H (2019) Innate immune signaling in nonalcoholic fatty liver disease and cardiovascular diseases. Annu Rev Pathol 14:153–184. https://doi.org/10.1146/annurev-pathmechdis-012418-013003
Deng KQ, Zhao GN, Wang Z, Fang J, Jiang Z, Gong J et al (2018) Targeting transmembrane BAX inhibitor motif containing 1 alleviates pathological cardiac hypertrophy. Circulation 137(14):1486–1504. https://doi.org/10.1161/CIRCULATIONAHA.117.031659
Ji YX, Zhang P, Zhang XJ, Zhao YC, Deng KQ, Jiang X et al (2016) The ubiquitin E3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via TAK1-dependent signalling. Nat Commun 7:11267. https://doi.org/10.1038/ncomms11267
Chen L, Huang J, Ji YX, Mei F, Wang PX, Deng KQ et al (2017) Tripartite motif 8 contributes to pathological cardiac hypertrophy through enhancing transforming growth factor beta-activated kinase 1-dependent signaling pathways. Hypertension 69(2):249–258. https://doi.org/10.1161/HYPERTENSIONAHA.116.07741
Zhang Y, Li H (2017) Reprogramming interferon regulatory factor signaling in cardiometabolic diseases. Physiology (Bethesda) 32(3):210–223. https://doi.org/10.1152/physiol.00038.2016
Zhang XJ, Zhang P, Li H (2015) Interferon regulatory factor signalings in cardiometabolic diseases. Hypertension 66(2):222–247. https://doi.org/10.1161/HYPERTENSIONAHA.115.04898
Zhang XJ, Liu X, Hu M, Zhao GJ, Sun D, Cheng X et al (2021) Pharmacological inhibition of arachidonate 12-lipoxygenase ameliorates myocardial ischemia-reperfusion injury in multiple species. Cell Metab 33(10):2059–75 e10. https://doi.org/10.1016/j.cmet.2021.08.014
Deng KQ, Wang A, Ji YX, Zhang XJ, Fang J, Zhang Y et al (2016) Suppressor of IKKvarepsilon is an essential negative regulator of pathological cardiac hypertrophy. Nat Commun 7:11432. https://doi.org/10.1038/ncomms11432
Xu M, Liu PP, Li H (2019) Innate immune signaling and its role in metabolic and cardiovascular diseases. Physiol Rev 99(1):893–948. https://doi.org/10.1152/physrev.00065.2017
Zhang Y, Zhang XJ, Wang PX, Zhang P, Li H (2017) Reprogramming innate immune signaling in cardiometabolic disease. Hypertension 69(5):747–760. https://doi.org/10.1161/HYPERTENSIONAHA.116.08192
Zhang Y, Zhang XJ, Li H (2017) Targeting interferon regulatory factor for cardiometabolic diseases: opportunities and challenges. Curr Drug Targets 18(15):1754–1778. https://doi.org/10.2174/1389450116666150804110412
Wang W, Zhang Y, Yang L, Li H (2017) The innate immune signaling in cancer and cardiometabolic diseases: friends or foes? Cancer Lett 387:46–60. https://doi.org/10.1016/j.canlet.2016.06.004
Al-Khafaji AB, Tohme S, Yazdani HO, Miller D, Huang H, Tsung A (2016) Superoxide induces neutrophil extracellular trap formation in a TLR-4 and NOX-dependent mechanism. Mol Med 22:621–631. https://doi.org/10.2119/molmed.2016.00054
Frantz S, Kelly RA, Bourcier T (2001) Role of TLR-2 in the activation of nuclear factor kappaB by oxidative stress in cardiac myocytes. J Biol Chem 276(7):5197–5203. https://doi.org/10.1074/jbc.M009160200
Doridot L, Jeljeli M, Chene C, Batteux F (2019) Implication of oxidative stress in the pathogenesis of systemic sclerosis via inflammation, autoimmunity and fibrosis. Redox Biol 25:101122. https://doi.org/10.1016/j.redox.2019.101122
Abais JM, Xia M, Li G, Gehr TW, Boini KM, Li PL (2014) Contribution of endogenously produced reactive oxygen species to the activation of podocyte NLRP3 inflammasomes in hyperhomocysteinemia. Free Radic Biol Med 67:211–220. https://doi.org/10.1016/j.freeradbiomed.2013.10.009
Bae JY, Park HH (2011) Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J Biol Chem 286(45):39528–39536. https://doi.org/10.1074/jbc.M111.278812
Win S, Than TA, Fernandez-Checa JC, Kaplowitz N (2014) JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis 5:e989. https://doi.org/10.1038/cddis.2013.522
Yamaguchi O, Higuchi Y, Hirotani S, Kashiwase K, Nakayama H, Hikoso S et al (2003) Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proc Natl Acad Sci USA 100(26):15883–15888. https://doi.org/10.1073/pnas.2136717100
Noguchi T, Takeda K, Matsuzawa A, Saegusa K, Nakano H, Gohda J et al (2005) Recruitment of tumor necrosis factor receptor-associated factor family proteins to apoptosis signal-regulating kinase 1 signalosome is essential for oxidative stress-induced cell death. J Biol Chem 280(44):37033–37040. https://doi.org/10.1074/jbc.M506771200
Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y et al (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17(9):2596–2606. https://doi.org/10.1093/emboj/17.9.2596
Sekine Y, Hatanaka R, Watanabe T, Sono N, Iemura S, Natsume T et al (2012) The Kelch repeat protein KLHDC10 regulates oxidative stress-induced ASK1 activation by suppressing PP5. Mol Cell 48(5):692–704. https://doi.org/10.1016/j.molcel.2012.09.018
Yadav UC, Ramana KV (2013) Regulation of NF-kappaB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid Med Cell Longev 2013:690545. https://doi.org/10.1155/2013/690545
Furnkranz A, Leitinger N (2004) Regulation of inflammatory responses by oxidized phospholipids: structure-function relationships. Curr Pharm Des 10(8):915–921. https://doi.org/10.2174/1381612043452929
Sanchez-Trujillo L, Vazquez-Garza E, Castillo EC, Garcia-Rivas G, Torre-Amione G (2017) Role of adaptive immunity in the development and progression of heart failure: new evidence. Arch Med Res 48(1):1–11. https://doi.org/10.1016/j.arcmed.2016.12.008
Desdin-Mico G, Soto-Heredero G, Aranda JF, Oller J, Carrasco E, Gabande-Rodriguez E et al (2020) T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 368(6497):1371–1376. https://doi.org/10.1126/science.aax0860
Sun L, Wang X, Saredy J, Yuan Z, Yang X, Wang H (2020) Innate-adaptive immunity interplay and redox regulation in immune response. Redox Biol 37:101759. https://doi.org/10.1016/j.redox.2020.101759
Kaminski MM, Sauer SW, Klemke CD, Suss D, Okun JG, Krammer PH et al (2010) Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: mechanism of ciprofloxacin-mediated immunosuppression. J Immunol 184(9):4827–4841. https://doi.org/10.4049/jimmunol.0901662
Chen X, Song M, Zhang B, Zhang Y (2016) Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid Med Cell Longev 2016:1580967. https://doi.org/10.1155/2016/1580967
Frossi B, De Carli M, Piemonte M, Pucillo C (2008) Oxidative microenvironment exerts an opposite regulatory effect on cytokine production by Th1 and Th2 cells. Mol Immunol 45(1):58–64. https://doi.org/10.1016/j.molimm.2007.05.008
Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM et al (2010) Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci USA 107(41):17686–17691. https://doi.org/10.1073/pnas.1012016107
Wheeler ML, Defranco AL (2012) Prolonged production of reactive oxygen species in response to B cell receptor stimulation promotes B cell activation and proliferation. J Immunol 189(9):4405–4416. https://doi.org/10.4049/jimmunol.1201433
Carvajal K, Balderas-Villalobos J, Bello-Sanchez MD, Phillips-Farfan B, Molina-Munoz T, Aldana-Quintero H et al (2014) Ca(2+) mishandling and cardiac dysfunction in obesity and insulin resistance: role of oxidative stress. Cell Calcium 56(5):408–415. https://doi.org/10.1016/j.ceca.2014.08.003
van der Pol A, van Gilst WH, Voors AA, van der Meer P (2019) Treating oxidative stress in heart failure: past, present and future. Eur J Heart Fail 21(4):425–435. https://doi.org/10.1002/ejhf.1320
Lancel S, Qin F, Lennon SL, Zhang J, Tong X, Mazzini MJ et al (2010) Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ Res 107(2):228–232. https://doi.org/10.1161/CIRCRESAHA.110.217570
Li SY, Yang X, Ceylan-Isik AF, Du M, Sreejayan N, Ren J (2006) Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch. Diabetologia 49(6):1434–1446. https://doi.org/10.1007/s00125-006-0229-0
Dong F, Fang CX, Yang X, Zhang X, Lopez FL, Ren J (2006) Cardiac overexpression of catalase rescues cardiac contractile dysfunction induced by insulin resistance: Role of oxidative stress, protein carbonyl formation and insulin sensitivity. Diabetologia 49(6):1421–1433. https://doi.org/10.1007/s00125-006-0230-7
Balderas-Villalobos J, Molina-Munoz T, Mailloux-Salinas P, Bravo G, Carvajal K, Gomez-Viquez NL (2013) Oxidative stress in cardiomyocytes contributes to decreased SERCA2a activity in rats with metabolic syndrome. Am J Physiol Heart Circ Physiol 305(9):H1344–H1353. https://doi.org/10.1152/ajpheart.00211.2013
Gomez-Viquez NL, Balderas-Villalobos J, Bello-Sanchez MD, Mayorga-Luna M, Mailloux-Salinas P, Garcia-Castaneda M et al (2021) Oxidative stress in early metabolic syndrome impairs cardiac RyR2 and SERCA2a activity and modifies the interplay of these proteins during Ca(2+) waves. Arch Physiol Biochem 1–13. https://doi.org/10.1080/13813455.2021.1895224
Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B et al (2001) Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res 89(3):279–286. https://doi.org/10.1161/hh1501.094115
Barlaka E, Gorbe A, Gaspar R, Paloczi J, Ferdinandy P, Lazou A (2015) Activation of PPARbeta/delta protects cardiac myocytes from oxidative stress-induced apoptosis by suppressing generation of reactive oxygen/nitrogen species and expression of matrix metalloproteinases. Pharmacol Res 95–96:102–110. https://doi.org/10.1016/j.phrs.2015.03.008
Laviola L, Leonardini A, Melchiorre M, Orlando MR, Peschechera A, Bortone A et al (2012) Glucagon-like peptide-1 counteracts oxidative stress-dependent apoptosis of human cardiac progenitor cells by inhibiting the activation of the c-Jun N-terminal protein kinase signaling pathway. Endocrinology 153(12):5770–5781. https://doi.org/10.1210/en.2012-1461
Leonardini A, D’Oria R, Incalza MA, Caccioppoli C, Andrulli Buccheri V, Cignarelli A et al (2017) GLP-1 receptor activation inhibits palmitate-induced apoptosis via ceramide in human cardiac progenitor cells. J Clin Endocrinol Metab 102(11):4136–4147. https://doi.org/10.1210/jc.2017-00970
Rana S, Datta K, Reddy TL, Chatterjee E, Sen P, Pal-Bhadra M et al (2015) A spatio-temporal cardiomyocyte targeted vector system for efficient delivery of therapeutic payloads to regress cardiac hypertrophy abating bystander effect. J Control Release 200:167–178. https://doi.org/10.1016/j.jconrel.2015.01.008
Zhao X, Luo W, Hu J, Zuo L, Wang J, Hu R et al (2018) Cardiomyocyte-targeted and 17beta-estradiol-loaded acoustic nanoprobes as a theranostic platform for cardiac hypertrophy. J Nanobiotechnology 16(1):36. https://doi.org/10.1186/s12951-018-0360-3
Ramachandra CJA, Chua J, Cong S, Kp MMJ, Shim W, Wu JC et al (2021) Human-induced pluripotent stem cells for modelling metabolic perturbations and impaired bioenergetics underlying cardiomyopathies. Cardiovasc Res 117(3):694–711. https://doi.org/10.1093/cvr/cvaa125
Caudal A, Ren L, Tu C, Wu JC (2022) Human induced pluripotent stem cells for studying mitochondrial diseases in the heart. FEBS Lett 596(14):1735–1745. https://doi.org/10.1002/1873-3468.14444
Vuckovic S, Dinani R, Nollet EE, Kuster DWD, Buikema JW, Houtkooper RH et al (2022) Characterization of cardiac metabolism in iPSC-derived cardiomyocytes: lessons from maturation and disease modeling. Stem Cell Res Ther 13(1):332. https://doi.org/10.1186/s13287-022-03021-9
Venkatesh S, Baljinnyam E, Tong M, Kashihara T, Yan L, Liu T et al (2021) Proteomic analysis of mitochondrial biogenesis in cardiomyocytes differentiated from human induced pluripotent stem cells. Am J Physiol Regul Integr Comp Physiol 320(4):R547–R562. https://doi.org/10.1152/ajpregu.00207.2020
Hallas T, Eisen B, Shemer Y, Ben Jehuda R, Mekies LN, Naor S et al (2018) Investigating the cardiac pathology of SCO2-mediated hypertrophic cardiomyopathy using patients induced pluripotent stem cell-derived cardiomyocytes. J Cell Mol Med 22(2):913–925. https://doi.org/10.1111/jcmm.13392
Zhan Y, Sun X, Li B, Cai H, Xu C, Liang Q et al (2018) Establishment of a PRKAG2 cardiac syndrome disease model and mechanism study using human induced pluripotent stem cells. J Mol Cell Cardiol 117:49–61. https://doi.org/10.1016/j.yjmcc.2018.02.007
Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A et al (2014) Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med 20(6):616–623. https://doi.org/10.1038/nm.3545
Wu JC, Garg P, Yoshida Y, Yamanaka S, Gepstein L, Hulot JS et al (2019) Towards precision medicine with human iPSCs for cardiac channelopathies. Circ Res 125(6):653–658. https://doi.org/10.1161/CIRCRESAHA.119.315209
Funding
This work was supported by grants from the Hubei Province Innovation Platform Construction Project (20204201117303072238), Wuhan Science and Technology Planning Project (2020021105012439), the National Science Foundation of China (82000386, 81870171), and the Excellent Doctoral Program of Zhongnan Hospital of Wuhan University (ZNYB2019001).
Author information
Authors and Affiliations
Contributions
All authors contributed toward this work. Conceptualization: Huo-Ping Li, Zhibing Lu, and Hongliang Li; literature search and analysis: Ze Chen, Zhao-Xia Jin, Ruyan Li, Ke-Qiong Deng, Yan-Xiao Ji, and Fang Lei; writing of the first draft of the manuscript: Ze Chen and Zhao-Xia Jin; review and editing: Jingjing Cai, Ruyan Li, Huo-Ping Li, Zhibing Lu, and Hongliang Li. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
All the authors give their consent for participation.
Consent for publication
All the authors give their consent for publication.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, Z., Jin, ZX., Cai, J. et al. Energy substrate metabolism and oxidative stress in metabolic cardiomyopathy. J Mol Med 100, 1721–1739 (2022). https://doi.org/10.1007/s00109-022-02269-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-022-02269-1