
Abstract
Non-steroidal anti-inflammatory drug (NSAID)-associated endoplasmic reticulum (ER) stress (a cyclooxygenase-2-independent mechanism) and consequent autophagic cell death are responsible for NSAID-associated gastric damage. Therefore, alleviating cytotoxicity executed via ER stress and autophagy can be a strategy to prevent NSAID-associated gastric damage. Here, we explored whether genetic or pharmacologic inhibition of autophagy can mitigate NSAID-associated gastric damage in in vitro and in vivo models. To examine the effects of genetic inhibition of NSAID-associated autophagy, we administered indomethacin to RGM1 gastric mucosal cells transfected with shPERK, siLC3B, or shATG5 and microtubule-associated protein light chain 3B knock-out (LC3B−/−) mice. 3-Methyladenine (3-MA) or chloroquine (CQ) was used for pharmacologic inhibition of autophagy in both models. Indomethacin administration increased the expression of ER stress proteins including GRP78, ATF6, and CHOP. Indomethacin provoked the appearance of autophagic vesicles with the increased expression of ATG5 and LC3B-II. Genetic ablation of various ER stress genes significantly attenuated indomethacin-induced autophagy and apoptosis (p < 0.01), whereas knock-down of either ATG5 or LC3B significantly reduced indomethacin-induced cytotoxicity (p < 0.01). Testing each of the genes implicated in ER stress and autophagy showed that indomethacin leads to gastric cell apoptosis through autophagy induction consequent to ER stress. Pharmacological inhibition of autophagy with either 3-MA or CQ in rats or genetic ablation of LC3B in mice all had a significant rescuing effect against indomethacin-associated gastric damage (p < 0.01) and a decrease in molecular markers of autophagic and apoptotic gastric cells. In conclusion, preemptive autophagy inhibition can be a potential strategy to mitigate NSAID-associated gastric damage.
Key messages
-
NSAID administration triggered ER stress and subsequent autophagy.
-
Inhibition of autophagy resulted in attenuated NSAID-associated cytotoxicity.
-
Autophagy inhibitors represent a novel strategy to prevent NSAID-associated gastric damage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are widely prescribed for pain, arthritis, pyretic disease, and even cancer for preventive purposes in spite of their notorious adverse effects in the gastrointestinal (GI) tract such as life-threatening bleeding and perforation [1]. The main pharmacological action of NSAIDs is the inhibition of prostaglandin (PG) synthesis via suppression of cyclooxygenase (COX) activity [2]. However, a decrease in the level of gastroprotective PGE2 is also regarded as a core mechanism responsible for the adverse GI effects. Recent molecular exploration showed that apoptosis mediated by endoplasmic reticulum (ER) stress is another, COX-independent cytotoxic mechanism of NSAIDs [3], that is, autophagic cell death following ER stress leads to NSAID-associated GI toxicity.
The role of autophagy in cancer (causative or preventive) is still debatable. In non-cancer and cancer cells, the answer to “to be or not to be” when autophagy is activated also differs under different circumstances [4, 5]. It was reported that autophagy contributes to cancer cell killing but also that aggressive cancers use autophagy for survival. Regarding the relevance of NSAID-associated cytotoxicity to autophagy, Harada et al. [6] and Narabayashi et al. [7] found that autophagy deficiency diminishes enterocyte damage induced by the NSAID indomethacin; in contrast, Herandez et al. [8] reported that autophagy can protect from aspirin-induced gastrointestinal damage. Therefore, a more clear understanding of the links between NSAID/aspirin-associated gastric damage and autophagy is required. This issue is explored in the current study.
Intense efforts to develop GI-safer NSAIDs resulted in a wide use of selective COX-2 inhibitors (coxibs) or a combination of acid suppressants such as proton pump inhibitors (PPI) in cases of inevitable long-term NSAID administration. However, the use of PPI to rescue NSAID-associated intestinal damages was recently discontinued because of the risk of aggravating outcomes (either dysbiosis [9] or increased intestinal permeability [10]). Therefore, on the basis of our and others’ preliminary data that (1) NSAIDs can induce ER stress and excess apoptosis [3, 11], (2) ER stress is closely linked with autophagy activation [12, 13], (3) autophagy may determine the cell fate (“to be or not to be”) [14, 15], and (4) there are urgent unmet medical needs to develop GI-safer NSAIDs, we put forward a hypothesis that relieving ER stress and consequent autophagic cell death can be a rescuing strategy from NSAID-associated gastric damage. In the current study, we found that either genetic ablation of autophagic genes or pharmacological inhibition of autophagy significantly mitigate indomethacin-induced gastric damage.
Materials and methods
Reagents
LC3B, ATG5, Beclin-1, cleaved caspase-3, phosphor-PERK, PERK, CHOP, phosphor-mTOR, mTOR, phosphor-Akt, Akt from Cell Signaling Technology, PARP-1/2, GRP78, β-actin from Santa Cruz Biotechnology (Santa Cruz, CA), cleaved LC3B from ABGENT (San Diego, CA). All other chemicals and reagents were purchased from Sigma-Aldrich (St. Lois, MO) unless otherwise specified.
Cell culture and cytotoxicity assay
The rat gastric mucosal cells, RGM1, were kindly given by Prof. Hirofumi Matsui (University of Tsukuba, Japan) and were maintained at 37 °C in a humidified atmosphere containing 5% CO2. RGM1 cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% (v/v) fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. 1.0 × 105/ml RGM1 cells were plated and incubated for 24 h after which media was changed with fresh one containing indomethacin or 3-methyladenine (3-MA) or chloroquine (CQ). Cell cytotoxicity was measured by MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] assay.
Annexin V apoptosis assay
Annexin V-FITC apoptosis kit was purchased from BD Biosciences and performed according to manufacturer’s instructions. Briefly, after treatment, media was collected for including death cells and adherent cells were trypsinized for 5 min, then, suspended with previously collected media. After cells were washed, cells were suspended with 1× binding buffer, adjusted to final concentration 1.0 × 106/ml. One hundred μl of cell-binding buffer solution was mixed with 4 μl of anti-Annexin V-FITC and propidium iodide (PI), then solution was incubated 15 min at room temperature in the dark. Reactions were terminated with insertion of 400 μl of binding buffer, then analyzed by FACS Calibur flow cytometer (BD bioscience).
Western blot analysis
This assay was performed as previously described. Briefly, treated cells were washed twice with PBS and then lysed in ice-cold cell lysis buffer (Cell Signaling Technology) containing 1 mM PMSF. After 1 h of incubation, samples were centrifuged at 10,000 g for 10 min. Supernatants were then collected. Proteins in lysates were separated by SDS-PAGE and transferred to PVDF membranes, which were incubated with primary antibodies, washed, incubated with peroxidase-conjugated secondary antibodies, rewashed, and then visualized using an West-zol Plus (Intron biotechnology, Seongnam, Korea).
RNA isolation and RT-PCR
This assay was performed as previously described. After incubation, media was removed by suction and cells were washed with PBS twice. Trizol (Invitrogen) was added to plates, which were then incubated for 10 min at 4 °C. Trizol was harvested and placed in a 1.5-ml tube, and 100-μl chloroform (Merck) was added and gently mixed. After incubation for 10 min in ice, samples were centrifuged at 10,000 g for 30 min. Supernatants were extracted and mixed with 200 μl isopropanol (Merck), and mixtures were incubated at 4 °C for 1 h. After centrifuging at 13,000 g for 30 min, pellets were washed with 70% (v/v) ethanol (Merck). After allowing the ethanol to evaporate completely, pellets were dissolved in 100 μl of DEPC-treated water (Invitrogen). cDNA was prepared using reverse transcriptase originating from Murine-Moloney leukemia virus (Promega), according to the manufacturer’s instructions. PCR was performed over 25 cycles of 94 °C for 20 s, 55 °C for 30 s, and 72 °C for 45 s. Oligonucleotide primers were designed by authors using NCBI/primer-blast except XBP-1. Oligonucleotide primers were purchased from Bioneer (Daejeon, Korea). The sequences of oligonucleotide primers are listed in supplementary table 2.
RNA interference
The expression of LC3B was abolished using target-specific small interference RNA (siRNA) molecules purchased from Bioneer. 100 pM gene-specific or control siRNA was transfected into RGM1 cells using Lipofectamine reagent (Invitrogen) according to the manufacturer’s instructions. The expressions of PERK and ATG5 were abolished using target-specific small hairpin RNA (shRNA) molecules purchased from Santa Cruz Biotechnology and SA biosciences, respectively. One-microgram gene-specific (shATG5, shPERK) or negative control vector (Mock) was transfected into RGM1 cells at 80–90% confluence using Lipofectamine according to the manufacturer’s instructions. The stable cell lines were selected by incubation with media containing 1 μg/ml of puromycin.
Construction of GFP-LC3-expressing RGM1 cells and analysis of GFP-LC3 dots
To obtain stable RGM1 cells expressing GFP-tagged LC3, pCMV-GFP-LC3 vector was purchased from Cell Biolabs. 1.6 μg vector was transfected into RGM1 cells at 80–90% confluence using Lipofectamine reagent according to manufacturer’s instructions. The stable cell lines were selected by incubation with media containing 500 μg/ml of G418. To detect translocation of GFP-LC3 from the cytosolic fraction to autophagic vacuoles, the stable cell lines expressing GFP-LC3 were grown in the Lab-Tek II chamber slide with coverglass (Nalge Nunc) for 24 h, after which cells were treated with indomethacin as well as following chemicals; 3-MA, Compound C, PD98029 (Cell Signaling Technology), SP600125 (Tocris), SB202190 (Tocris), Diphenyleneiodonium chloride (DPI), and rapamycin (LC laboratories). Autofluorescence GFP-LC3 (excitation 488 nm, emission 515 nm) was observed under a confocal microscope (LSM700, Carl Zeiss). LC3-dot-positive cell was determined as more than 5 dots in a cell. LC3-dot-positivity was measured by dividing the number of LC3-dot-positive cells by the number of total cells.
Lysosomal and MDC staining for confocal images
The stable cell lines expressing GFP-LC3 were seeded in the Lab-Tek II chamber slide with coverglass for 24 h, after which cells were treated with indomethacin for 16 h. Cells were stained either with 100-nM Lysotracker Red Lysosomal Probe (Lonza) or with 50-μM monodansylcadaverine (MDC) incubated for 30 min. Cells were washed and fixed with 3.7% formaldehyde at room temperature for 15 min. The fluorescence of Lysotracker Red Lysosomal Probe (excitation 555 nm, emission 570 nm) or MDC (excitation 405 nm, emission 460 nm) was observed under a confocal microscope.
Animal model of indomethacin-induced gastric ulcer
Six-week-old specific-pathogen-free 40 Wistar Kyoto male rats (Orient, Seongnam, Korea) were used for the experiments, fed a sterilized commercial pellet diet (DH Biolink, Eumseong, Korea), given sterile water ad libitum, and housed in an air conditioned room under a 12-h light/dark cycle. Rats were divided into five groups, 8 rats per group, respectively; a non-treated control group (Normal), indomethacin-treated group (Indomethacin), indomethacin plus 3-MA administered group (3-MA), indomethacin plus CQ group (CQ), and indomethacin plus pantoprazole group (Pantoprazole). Animals were handled in an accredited animal facility in accordance with Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International) guidelines of our CACU Institute after IRB approval. Animals were deprived of food, but allowed free access to water 24 h before administration of drugs. Indomethacin, 3-MA, and CQ were all dissolved in 0.5% carboxymethyl cellulose sodium (Tokyo Chemical). Indomethacin was administrated orally to Indomethacin group, but Normal group received vehicle only. At the same time, 25-mg/kg 3-MA was injected by intraperitoneal route to 3-MA group, but Normal group and Indomethacin group were treated with vehicle only. Animals were killed 18 h after drug administration. Stomachs were removed, opened along the greater curvature, and rinsed with phosphate-buffered saline. Isolated tissues were subjected to a histological examination and Western blotting. Pathological lesion index was defined according to the criteria of Szabo et al. [16], all reviewed blindly by two gastroenterology specialists and first author. The details of pathological lesion indexes are shown in supplementary Table 1. The animal experiments were repeated in LC3B−/− mice (purchased from Jackson Lab) as same in the above protocol except sacrifice time, 16 h after indomethacin.
Immunohistochemistry and TdT-mediated biotinylated UTP nick end labeling assay
For immunohistochemical analysis, the sections were deparaffinized and blocked with 2.5% goat serum for 30 min, incubated for 12 h at 4 °C with antibody against cleaved 1:100 LC3B in the presence of 1.0% BSA, and finally incubated for 1 h at room temperature with Alexa Fluor 488 goat anti-rabbit immunoglobulin G in the presence of 1 μg/ml DAPI. The slides were mounted with Prolong Gold anti-fade agent (Invitrogen) and inspected using a confocal microscopy. TdT-mediated biotinylated UTP nick end labeling (TUNEL) assay kit was purchased and performed according to manufacturer’s instruction (Promega).
Statistical analysis
The data are presented as means ± standard deviations (SD). The Tukey’s test or the Student’s t test for unpaired results was used to evaluate differences between more than three groups or between two groups, respectively. Differences were considered to be significant for values of p < 0.05.
Results
Indomethacin induced cytotoxicity of gastric epithelial cells through ER stress and subsequent autophagy
When RGM1 gastric mucosal cells were exposed to either 500-μM indomethacin for various periods of time or various concentrations of indomethacin for up to 24 h, their viability decreased in a dose-dependent and time-dependent manner (Fig. 1a). Flow cytometry analysis using Annexin V-FITC was performed to see whether indomethacin-induced cytotoxicity was associated with apoptosis. Annexin V absorbance significantly increased after 16 h of indomethacin treatment (Fig. 1a), showing that indomethacin administration is associated with significant cytotoxicity in normal gastric mucosal cells via apoptotic cell death. Western blotting showed the increased levels of the cleaved forms of caspase-3 and poly (ADP-ribose) polymerase (PARP) after 16 h of indomethacin treatment, indicating the induction of apoptosis. (Fig. 1b). Mammalian ER stress response is mediated by the following three transmembrane proteins: namely inositol-requiring ER-to-nucleus signal kinase 1 (IRE1), which is both a protein kinase and an endonuclease, protein kinase R-like ER kinase/pancreatic eukaryotic translocation initiation factor 2 kinase (PERK/PEK), and activating transcription factor 6 (ATF6). The PERK/eIF2 pathway induces the expression of glucose-regulated protein of 78 kDa (GRP78) and DNA-damage-inducible transcript 3 (DDIT3, also known as C/EBP-homologous transcription factor, CHOP) [17]. Therefore, we checked changes in the expression of these genes after indomethacin administration. As shown in Fig. 1c, the ratio of phospho-PERK to total PERK increased in response to indomethacin treatment, although not as much as after treatment with a well-known ER stress activator, thapsigargin [18]. The protein levels of GRP78 and CHOP were also increased in indomethacin-treated cells (Fig. 1c). The ER stress-related transcription factors X-box-binding protein-1 (XBP-1) and ATF6 increase CHOP mRNA expression. XBP-1 mRNA splicing by IRE1 converts it into a potent activator of CHOP transcription. The levels of GRP78, CHOP, ATF6, and XBP-1 mRNAs were increased in indomethacin-treated cells in comparison with vehicle-treated cells (Fig. 1c). All of these findings consistently suggest that indomethacin induced apoptosis through ER stress and autophagy induced by indomethacin led to cell death. To check whether indomethacin triggers autophagy, we examined dot formation by GFP-tagged LC3 in indomethacin-treated pCMV-GFP-LC3-transfected RGM1 cells. As shown in Fig. 1d, indomethacin significantly increased GFP-LC3 dot formation in a time-dependent manner, with a maximum at ~16 h (p < 0.01). More than 90% of GFP-LC3 dots were co-localized with Lysotracker Red-stained lysosomes, indicating that indomethacin induced autolysosome formation. To further confirm the formation of autophagosomes surrounded by a double membrane, indomethacin-treated RGM1 cells were stained with monodansylcadaverine (MDC). Confocal imaging showed that indomethacin significantly increased MDC dot formation in pCMV-GFP-LC3-transfected RGM1 cells; MDC dots were co-localized with GFP-LC3 dots (Fig. 1e). When autophagy is activated, LC3B is transformed into LC3B-II, which binds to phosphatidylethanolamine. LC3B-II is visible as band 2 kDa below that of LC3B-I (the inactive form). In response to indomethacin, the levels of the LC3B-II protein and ATG5 protein, a crucial positive regulator of autophagy [19], clearly increased in a dose-dependent manner (Fig. 1e). To determine whether the signaling pathway involved in indomethacin-induced autophagy is related to class III phosphoinositide-3-kinase (PI3K), AMP-activated protein kinase (AMPK), mitogen-activated protein kinases (MAPKs), oxidative stress, or mechanistic target of rapamycin (mTOR), we examined the effects of 3-MA (class III PI3K inhibitor), compound C (AMPK inhibitor), PD98029 (MEK-1 inhibitor), SP600125 (JNK inhibitor), SB202190 (p38 inhibitor), diphenyleneiodonium chloride (DPI; NOX inhibitor), and rapamycin (mTOR inhibitor) on indomethacin-induced autophagosome formation (Fig. 1f). 3-MA and compound C co-administered with indomethacin significantly blocked GFP-LC3 dot formation, which was reduced to a nearly basal level, whereas rapamycin treatment alone efficiently increased GFP-LC3 dot formation. After cell incubation with different doses of indomethacin for different periods of time, significant increases in LC3B-II and ATG5 were noted on a Western blot (p < 0.01, Fig. 1g). The mTOR pathway plays a central role in the initiation and maturation of autophagosomes and in signaling upstream of AKT. Upon administration of 500-μM indomethacin, the ratio of phospho-mTOR to total mTOR was initially increased until 8 h, but then gradually decreased to less than 10% of the pre-treatment level at 16 h, indicating inactivation of the mTOR pathway; similarly, the AKT pathway, which is a crucial activator of mTOR, was also significantly blocked by indomethacin treatment (p < 0.05 for both kinases, Fig. 1h). From the above experiments, we conclude that the mTOR/AKT pathway is important upstream of indomethacin-induced autophagy.

Indomethacin induced cytotoxicity through ER stress and autophagy. a RGM1 cells treated with 500-μM indomethacin for 0–24 h and indomethacin (0–1000 μM) were subjected to MTT assay for 16 h. Bars represent mean ± SD (n = 3). *p < 0.05. Floating and adherent cells were collected and analyzed by flow cytometry after annexin V-FITC and propidium iodide (PI) staining. x-axis, annexin V; y-axis, PI. b Western blots for PARP and cleaved caspase-3. RGM1 cells were treated with indomethacin (500 μM) for various periods of time as indicated. c RT-PCR for ER stress genes and a Western blot for ER stress proteins. RGM1 cells were treated as in a. Treatment with thapsigargin (#Thp, 2 μM) for 6 h was used as a positive control for the induction of ER stress. d RGM1 cells transfected with pCMV-GFP-LC3 were treated with DMSO or 500-μM indomethacin for 16 h, followed by incubation with LysoTracker Red for 30 min. Left part, confocal microscopy (×200). Cells were treated with indomethacin for different periods of time and the number of GFP-LC3 dot-positive cells was divided by the total cell number. Right part, the percentages of GFP-LC3 dot-positive cells. Bars represent mean ± SD (n = 3). e DMSO- or indomethacin-treated GFP-LC3-expressing cells were incubated with 50 μM MDC for 30 min. Confocal microscopy images (×800) are shown. f GFP-LC3-expressing RGM1 cells were treated with inhibitors of various signaling pathways: 3-MA (10 μM), Compound C (10 μM), PD98029 (PD, 10 μM), SP600125 (SP, 10 μM), SB202190 (SB, 10 μM), diphenyleneiodonium chloride (DPI, 10 μM), and rapamycin (2 μM) in the absence or presence of 500-μM indomethacin for 16 h. The percentages of GFP-LC3 dot-positive cells were determined. Bars represent mean ± SD (n = 3). g. Western blot for LC3B after different doses of indomethacin, in which rapamycin was compared with professional LC3B II inducer and ATG5 expression was measured after different times of 500-μM indomethacin. Rpm* stands for rapamycin. Indomethacin administration significantly increased either LC3B II or ATG5. h. Western blot for p-mTOR, mTOR, p-Akt, and AKT after different times of 500-μM indomethacin. Upon indomethacin administration, the ratio of phospho-mTOR to total mTOR was initially increased until 8 h, after which it gradually decreased to less than 10% of the pre-treatment level at 16 h (p < 0.05) and in the same manner, the AKT pathway, which is a crucial activator of mTOR, was significantly blocked by 500-mM indomethacin treatment after 16 h (p < 0.05)
Knock-down of the PERK, LC3B, and ATG5 genes mitigated indomethacin-associated gastric epithelial cell cytotoxicity
Since several studies have demonstrated that ER stress induced by hypoxia, thapsigargin, tunicamycin, amino acid deprivation, or expanded polyglutamine aggregation can induce autophagy [15], it was necessary to check whether indomethacin-induced ER stress precedes autophagy and resulting cell death. To test this hypothesis, we knocked down PERK with a small hairpin RNA (shPERK). The levels of ER stress-related mRNAs such as GRP78, CHOP, and XBP-1 induced by indomethacin were lower in shPERK-transfected cells than in mock-transfected cells (Fig. 2a). PERK knock-down also significantly attenuated the levels of ATG5 and LC3B-II and increased the level of phospho-mTOR, confirming that ER stress might be involved in indomethacin-induced autophagy. LC3B knock-down significantly reduced indomethacin-induced caspase-3 cleavage and apoptosis (p < 0.01, Fig. 2b, c), suggesting that indomethacin cytotoxicity is associated with autophagic cell death. To further confirm that autophagy consequent to indomethacin-induced ER stress promotes apoptosis rather than rescues gastric epithelial cells from apoptosis, we knocked down the ATG5 gene using shATG5. As shown in Fig. 2d, e, indomethacin-induced caspase-3 cleavage and resulting apoptosis were significantly decreased in shATG5-transfected cells in comparison with mock-transfected cells (p < 0.01). All of these findings consistently showed that indomethacin-induced ER stress precedes autophagy and that autophagy after indomethacin-induced ER stress induces cell death rather than promotes cell survival. To discriminate between necroptosis and apoptosis after indomethacin treatment, we measured the changes in RIPK1, a key signaling molecule in the programmed necrosis pathway, in RGM1 cells treated with indomethacin alone or a combination of indomethacin and chloroquine (CQ, an autophagy inhibitor), and did not find any significant differences between the two groups (Supplementary Fig. 1a). Scanning electron microscopy revealed that indomethacin treatment led to numerous cell surface blebs, i.e., morphology distinct from that expected for necroptosis or necrosis (Supplementary Fig. 1b).

Influence of knocking out PERK, LC3B, or ATG5 on indomethacin-induced autophagy and apoptosis. a RGM1 cells stably transfected with the shPERK vector (shPERK) and empty vector (mock) were treated with 500-μM indomethacin for 16 h, and cell lysates were immunoblotted with anti-PERK, anti-phospho-mTOR, anti-mTOR, anti-ATG5, anti-LC3B II, and anti-β-actin antibodies. b Target-specific siRNA for LC3B (siLC3B) or a negative control RNA (mock) was transfected into RGM1 cells for 24 h. The expression of LC3B mRNA was checked by RT-PCR (upper part) and the cleavage of caspase-3 (lower part) was compared between mock-transfected and siLC3B-transfected cells. The level of cleaved caspase-3 was significantly decreased in cells transfected with siLC3B. Bars represent mean ± SD (n = 3). c Mock-transfected cells and siLC3B-transfected cells were treated with indomethacin (500 μM) for 16 h and analyzed by flow cytometry for annexin V-FITC and PI staining. The relative levels of the cleaved form of caspase-3 and β-actin were quantified by densitometry, and the ratio of cleaved caspase-3 to β-actin was calculated. Bars represent mean ± SD (n = 3). d Autophagy inhibition in shATG5-transfected RGM1. Cells were treated with indomethacin and LC3B and the cleaved form of caspase-3 were analyzed by Western blotting. Bars represent mean ± SD (n = 3). e Mock-transfected cells and shATG5-transfected cells were treated with indomethacin (500 μM) for 16 h and analyzed by flow cytometry for annexin V-FITC and PI staining
Pharmacologic inhibition of autophagy with 3-MA or CQ significantly decreased indomethacin-associated gastric epithelial cytotoxicity
On the basis of the above findings that genetic inhibition of autophagy significantly alleviated indomethacin-associated gastric epithelial cytotoxicity, we assessed the effects of pharmacological inhibition of autophagy with either 3-MA or CQ on indomethacin-associated cytotoxicity. Indomethacin significantly increased apoptosis as measured by the levels of Annexin V and cleaved caspase-3 (both p < 0.01), but co-treatment with 3-MA and indomethacin significantly decreased apoptosis (p < 0.05, Fig. 3a). Similarly, CQ significantly attenuated indomethacin-induced apoptosis (p < 0.05, Fig. 3a, d). As shown in Fig. 3b, d, indomethacin significantly increased, whereas either 3-MA or CQ significantly decreased the levels of LC3B-II, ATG5, cleaved caspase-3, and cleaved PARP (all p < 0.05). Confocal observation of GFP-LC3 and Lysotracker Red showed that indomethacin induced significant autophagosome formation (p < 0.005), whereas 3-MA and CQ significantly inhibited autophagy (p < 0.05, Fig. 3c).

Autophagy inhibition by 3-MA or CQ. a RGM1 cells were co-treated with indomethacin (500 μM) and 3-MA (10 μM) for 16 h, after which an Annexin V staining was performed. Cells were stained with annexin V-FITC and PI and analyzed by flow cytometry. Bars represent mean ± SD (n = 3). b Western blot for cleaved caspase-3 after indomethacin alone and a combination of indomethacin and 3-MA. RGM1 cells were exposed to 3-MA and indomethacin for 16 h and soluble protein extracts were subjected Western blotting with the indicated antibodies. The ratios of cleaved caspase-3 to β-actin for each treatment were quantified by densitometry. Bars represent mean ± SD (n = 3; right). c Confocal imaging of GFP-LC3 and Lysotracker Red after 3-MA and CQ. d Flow cytometry after CQ treatment during indomethacin administration. e RGM1 cells were exposed to CQ and indomethacin for 16 h and soluble protein extracts were subjected to SDS-PAGE and Western blotting with the indicated antibodies. β-actin was used as a loading control (left). The ratio of cleaved caspase-3 to β-actin were quantified by densitometry. Bars represent mean ± SD (n = 3)
Genetic ablation of the LC3B gene in mice significantly mitigated gastric damage induced by indomethacin
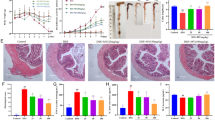
Since the above in vitro experiments consistently showed that autophagy inhibition can be a rescue strategy from NSAID-induced cytotoxicity, we next compared indomethacin-associated gastric damage in both wild-type (WT) and LC3B knock-out (LC3B−/−) mice. Administration of indomethacin (20 mg/kg) for 16 h provoked significant gastric mucosal damage including multiple linear ulcerations, multiple hemorrhagic streaks, and profuse bleeding (Fig. 4a). On microscopic evaluation, indomethacin induced overt ulceration surrounded by necrotic debris, mucosal erosion and hemorrhages, inflammatory cell infiltration, and submucosal edema (Fig. 4a). However, indomethacin-induced gastric damage was significantly attenuated in LC3B−/− mice in comparison with WT mice (p < 0.01, Fig. 4a). Indomethacin-associated gastric damage was significantly associated with apoptosis (p < 0.001, Fig. 4b), but apoptosis was significantly inhibited in LC3B−/− mice (p < 0.001, Fig. 4b). These data suggest that apoptosis is a key mechanism involved in indomethacin-associated gastric damage.

Influence of genetic ablation of autophagy on indomethacin-associated gastric damages. a An indomethacin-induced gastric ulcer in LC3B−/− mice and wild-type (WT) littermates. A representative image, gross lesion index and pathologic lesion index of WT littermates and LC3B−/− mice. Bars represent mean ± SD (n = 8). b Representative images of TUNEL staining performed in WT and LC3B−/− mice before and after indomethacin administration. Apoptotic index (AI) is shown for each group. c Western blot analysis of LC3B, ATG5, caspase-3, Beclin-1, PERK, p-PERK, and β-actin. Bars represent mean ± SD (n = 6)
Indomethacin significantly increased the levels of LC3B and ATG5 as well as cleavage of caspase-3 in stomach homogenates of WT mice (p < 0.01), but LC3B and Atg5 expression was significantly lower in LC3B−/− mice even after indomethacin administration (p < 0.01, Fig. 4c). After indomethacin administration, the expression of PERK and Beclin-1, proteins involved in ER stress, was significantly lower in the stomachs of LC3B−/− mice than in those of WT mice, confirming the above in vitro results and indicating that genetic ablation of autophagy protects mice from indomethacin-associated gastric damage.
Pharmacologic inhibition of autophagy with either 3-MA or CQ significantly rescued from indomethacin-associated gastric damage
Since genetic inhibition of autophagy showed a significant rescuing effect against indomethacin-induced gastric damage (Fig. 4), we checked whether the autophagy inhibitors 3-MA or CQ can mitigate indomethacin-associated gastric damage in Sprague-Dawley rats. As shown in Fig. 5a, rats were divided into four groups: untreated, indomethacin alone (50 mg/kg instead of 20 mg/kg in the above mice model, but the same time points up to 16 h), indomethacin + 3-MA, and indomethacin + CQ (n = 8 per group). Indomethacin treatment resulted in significant gastric damage including overt gastric ulcers, bleeding, and edematous and erythematous gastric mucosa (p < 0.01, Fig. 5b). However, co-treatment with 3-MA or CQ and indomethacin significantly reduced the gross lesion index and pathologic lesion index in comparison with indomethacin treatment alone (p < 0.01, Fig. 5b). The preventive efficacy of 3-MA or CQ was similar or superior to that of the proton pump inhibitor pantoprazole (data not shown). To confirm that autophagy and apoptosis are responsible for indomethacin-associated gastric damage in the rat model and autophagy inhibition contributes to mitigation of indomethacin-induced gastric ulcers, we performed Western blotting for LC3B and caspase-3, TUNEL assay, and immunohistochemical staining with LC3B antibody. We found significant activation of LC3B and caspase-3 after indomethacin treatment (p < 0.05, Fig. 5c). The number of TUNEL-positive cells was dramatically increased around ulcers and in the ulcer niches, and the level of LC3B-II was significantly increased at a location similar to that of TUNEL staining. However, TUNEL positivity and LC3B expression were significantly decreased even at ulcer sites in the 3-MA- and CQ-treated groups (Fig. 5d). From the data shown in Figs. 4 and 5, we conclude that either genetic ablation of autophagy genes or pharmacological inhibition of autophagy significantly mitigates indomethacin-associated gastric damage (Fig. 6).

Pharmacological inhibition of autophagy on indomethacin-associated gastric damages. a Efficacy of the autophagy inhibitors 3-MA or CQ against indomethacin-induced gastric ulcers in rats. Schematic protocol of the indomethacin-induced gastric ulcer rat model. Rats were killed 18 h after the completion of each treatment. n = 8 SD rats/group. b Representative gross features of the stomach in different groups. The pathologic lesion index of each group was calculated. Bars represent mean ± SD (n = 8). c Tissue lysates of each group were immunoblotted with anti-LC3B, anti-cleaved caspase-3, and anti-β-actin antibodies. Bars represent mean ± SD (n = 6). d Sections of gastric tissues were subjected to H&E staining and TUNEL assay. Confocal images of immunohistochemical staining with LC3B are shown

Schematic summary. The current study provides new hope that inhibiting autophagy can be a potential strategy to alleviate NSAID-induced gastric damage
Discussion
The current study has clearly demonstrated that NSAID-induced ER stress and consequent autophagy can evoke apoptotic cell death in gastric epithelial cells, leading to NSAID-associated gastric damage. Accordingly, autophagy inhibition efficiently rescued gastric cells or gastric wall from NSAID-associated gastric damage. In addition to in vitro evidence that apoptotic cytotoxicity after NSAID treatment is regulated by autophagy inhibition, our in vivo animal model consistently showed that inhibition of autophagy either by genetic means in LC3B knock-out mice or by pharmacological means using CQ and 3-MA in rats significantly mitigated indomethacin-associated gastric damage. Similar to PPI, CQ achieved significant rescuing efficacy against indomethacin-induced gastric damage, which strongly supports the necessity of well-designed clinical trials for its future clinical applications (Fig. 6). Though it has been reported [8] that aspirin-induced GI damage is associated with inhibition of epithelial cell autophagy, our findings support the findings by Harada et al. [6] and Narabayashi et al. [7] that autophagy inhibition can diminish indomethacin-induced intestinal cell damage. A possible explanation of the discrepancy between studies is that we documented the rescuing action of autophagy inhibition in stomach and gastric cells (RGM1), while other studies assessed intestinal damage and used IEC-6 enterocytes. Furthermore, we explored in detail the effects of genetic and pharmacological inhibition of autophagy in both in vitro cell models and in vivo animal models, and our data agree with the results of Harada et al. [6] and Narabayashi et al. [7]. Although Harada et al. used ATG5 conditional knock-out rats, whereas our experimental model was LC3B knock-out mice, both studies reached the same conclusion.
Disturbances in the normal functions of the ER lead to the evolutionarily conserved stress response, which is initially aimed at compensating for the incurred damage. However, if the ER protein-folding capacity is overwhelmed or dysfunction is prolonged, ER stress eventually triggers cell death [20, 21]. The ER stress response can protect cells under mild stresses but can kill cells through apoptosis under severe stress [22]. Similarly, autophagy might be regarded as a defensive and salvaging process following the ER stress response. However, excessive autophagy under prolonged NSAID administration is never protective and can lead to autophagic cell death.
In quiescent cells, the level of autophagy remains usually low when LC3, the homolog of yeast atg8, is present diffusely in the cytoplasm. However, when autophagy needs to be activated following cell stress, e.g., NSAID administration and resulting ER stress, LC3 is transformed into LC3-II (Fig. 1) [19, 23]. Although pharmacological or genetic inhibition of autophagy may sensitize cells to apoptosis in response to chemotherapeutic agents or radiation [24, 25], autophagy itself can mediate or trigger apoptosis under different conditions through ER stress [26, 27]. Tsutsumi et al. [3] first examined the association between the ER stress response and NSAID-induced apoptosis and found that exposure of gastric mucosal cells to indomethacin induces GRP78, CHOP, ATF6, and XBP-1 and causes NSAID cytotoxicity regardless of the NSAID used. Chen et al. [28] found that the cytotoxic effect of different pharmacological agents that affect COX-2 inhibitory potency is closely related to the ability of these agents to trigger ER stress. Therefore, ER stress seems to be a COX-independent mechanism responsible for NSAID-induced gastric mucosal cytotoxicity (Fig. 1) [29–31]. Though basal levels of autophagy ensure the physiological turnover of old and damaged organelles and massive accumulation of autophagic vacuoles may represent either an alternative pathway of cell death or an ultimate attempt to survive [32], activation of the autophagic pathway beyond a certain threshold may promote cell death by causing the collapse of cellular functions, so called type II cell death. Autophagy is a lysosomal degradation pathway essential for survival, differentiation, development, and homeostasis, but in the current setting such as NSAID administration, the self-cannibalistic and apoptotic functions of autophagy may be deleterious and play a role in NSAID-induced gastric damage. From the current study, the facts that shPERK-transfected cells reduced the expression of ATG5 and increased the ratio of phospho-mTOR to total mTOR in comparison with mock-transfected cells (Fig. 2) suggest that ER stress might be an upstream regulator of autophagy induction after indomethacin treatment.
Though CQs have been used for treatment of malaria for over 500 years and have also been known to have disease-modifying properties in systemic lupus erythematosus [33] for over 50 years, newer iterations of this class of anti-inflammatory agents have been used to treat autoimmune diseases and various inflammatory diseases by exploiting Toll-like receptor-associated mechanisms. In the current study, we have demonstrated that CQ is an autophagy inhibitor and that CQ can be used to mitigate NSAID-associated cytotoxicity. Some researchers consider CQ as an anti-cancer drug as old drugs are being investigated for their ability to target cancer metabolism [34, 35]. In oncology, anti-cancer clinical trials are underway that use CQ to sensitize cancer cells to chemotherapy independently of autophagy [36] and CQ is repositioned to eliminate cancer stem cells in premalignant lesions [37]. Our study might be the first to suggest CQ as a potential preventive agent against NSAID-associated gastric damage. Among autophagy inhibitors, in the current study, we have also shown the preventive action of 3-MA but not bafilomycin A1. Although bafilomycin A1 is a late-stage autophagy inhibitor, our preliminary study showed that bafilomycin A1 as well as pantoprazole (a PPI) showed significant cytotoxicity [38].
In conclusion, our study showed that since NSAID-induced ER stress and subsequent autophagy seem to induce cell death in gastric epithelial cells, optimal modulation of autophagy can be a good opportunity to mitigate NSAID-associated gastric damage. After well-designed, extensive large-scale clinical studies, an autophagy inhibitor alone or a combination of an NSAID and an autophagy inhibitor can become a novel therapeutic strategy to protect the stomach from NSAID-associated damage.
References
Graham DY, Chan FK (2008) NSAIDs, risks, and gastroprotective strategies: current status and future. Gastroenterology 134:1240–1246
Vane JR (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231:232–235
Tsutsumi S, Gotoh T, Tomisato W, Mima S, Hoshino T, Hwang HJ, Takenaka H, Tsuchiya T, Mori M, Mizushima T (2004) Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ 11:1009–1016
Chen N, Karantza-Wadsworth V (2009) Role and regulation of autophagy in cancer. Biochim Biophys Acta 1793:1516–1523
Chen HY, White E (2011) Role of autophagy in cancer prevention. Cancer Prev Res (Phila) 4:973–983
Harada S, Nakagawa T, Yokoe S, Edogawa S, Takeuchi T, Inoue T, Higuchi K, Asahi M (2015) Autophagy deficiency diminishes indomethacin-induced intestinal epithelial cell damage through activation of the ERK/Nrf2/HO-1 pathway. J Pharmacol Exp Ther 355:353–361
Narabayashi K, Ito Y, Eid N, Maemura K, Inoue T, Takeuchi T, Otsuki Y, Higuchi K (2015) Indomethacin suppresses LAMP-2 expression and induces lipophagy and lipoapoptosis in rat enterocytes via the ER stress pathway. J Gastroenterol 50:541–554
Hernandez C, Barrachina MD, Vallecillo-Hernandez J, Alvarez A, Ortiz-Masia D, Cosin-Roger J, Esplugues JV, Calatayud S (2016) Aspirin-induced gastrointestinal damage is associated with an inhibition of epithelial cell autophagy. J Gastroenterol 51:691–701
Wallace JL, Syer S, Denou E, de Palma G, Vong L, McKnight W, Jury J, Bolla M, Bercik P, Collins SM et al (2011) Proton pump inhibitors exacerbate NSAID-induced small intestinal injury by inducing dysbiosis. Gastroenterology 141:1314–1322 1322 e1311-1315
Mullin JM, Valenzano MC, Whitby M, Lurie D, Schmidt JD, Jain V, Tully O, Kearney K, Lazowick D, Mercogliano G et al (2008) Esomeprazole induces upper gastrointestinal tract transmucosal permeability increase. Aliment Pharmacol Ther 28:1317–1325
Marciniak SJ, Ron D (2006) Endoplasmic reticulum stress signaling in disease. Physiol Rev 86:1133–1149
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K et al (2006) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26:9220–9231
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T (2007) ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 14:230–239
Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H (2009) Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann Surg Oncol 16:761–771
Li J, Hou N, Faried A, Tsutsumi S, Kuwano H (2010) Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur J Cancer 46:1900–1909
Szabo S, Trier JS, Brown A, Schnoor J, Homan HD, Bradford JC (1985) A quantitative method for assessing the extent of experimental gastric erosions and ulcers. J Pharmacol Methods 13:59–66
Kaufman RJ (2002) Orchestrating the unfolded protein response in health and disease. J Clin Invest 110:1389–1398
Treiman M, Caspersen C, Christensen SB (1998) A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca(2+)-ATPases. Trends Pharmacol Sci 19:131–135
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728
Xu C, Bailly-Maitre B, Reed JC (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115:2656–2664
Lai E, Teodoro T, Volchuk A (2007) Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology (Bethesda) 22:193–201
Wu J, Kaufman RJ (2006) From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ 13:374–384
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ et al (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36:585–595
Kondo Y, Kanzawa T, Sawaya R, Kondo S (2005) The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 5:726–734
Moretti L, Attia A, Kim KW, Lu B (2007) Crosstalk between Bak/Bax and mTOR signaling regulates radiation-induced autophagy. Autophagy 3:142–144
Guillon-Munos A, van Bemmelen MX, Clarke PG (2006) Autophagy can be a killer even in apoptosis-competent cells. Autophagy 2:140–142
Xue L, Fletcher GC, Tolkovsky AM (1999) Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol Cell Neurosci 14:180–198
Chen ST, Thomas S, Gaffney KJ, Louie SG, Petasis NA, Schonthal AH (2010) Cytotoxic effects of celecoxib on Raji lymphoma cells correlate with aggravated endoplasmic reticulum stress but not with inhibition of cyclooxygenase-2. Leuk Res 34:250–253
Kardosh A, Golden EB, Pyrko P, Uddin J, Hofman FM, Chen TC, Louie SG, Petasis NA, Schonthal AH (2008) Aggravated endoplasmic reticulum stress as a basis for enhanced glioblastoma cell killing by bortezomib in combination with celecoxib or its non-coxib analogue, 2,5-dimethyl-celecoxib. Cancer Res 68:843–851
Pae HO, Jeong SO, Jeong GS, Kim KM, Kim HS, Kim SA, Kim YC, Kang SD, Kim BN, Chung HT (2007) Curcumin induces pro-apoptotic endoplasmic reticulum stress in human leukemia HL-60 cells. Biochem Biophys Res Commun 353:1040–1045
Chinta SJ, Poksay KS, Kaundinya G, Hart M, Bredesen DE, Andersen JK, Rao RV (2009) Endoplasmic reticulum stress-induced cell death in dopaminergic cells: effect of resveratrol. J Mol Neurosci 39:157–168
Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G (2008) To die or not to die: that is the autophagic question. Curr Mol Med 8:78–91
Wallace DJ, Gudsoorkar VS, Weisman MH, Venuturupalli SR (2012) New insights into mechanisms of therapeutic effects of antimalarial agents in SLE. Nat Rev Rheumatol 8:522–533
Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E (2011) Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 17:654–666
Yang ZJ, Chee CE, Huang S, Sinicrope FA (2011) The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 10:1533–1541
Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A (2012) Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 8:200–212
Vazquez-Martin A, Lopez-Bonetc E, Cufi S, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA (2011) Repositioning chloroquine and metformin to eliminate cancer stem cell traits in pre-malignant lesions. Drug Resist Updat 14:212–223
Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J et al (2012) Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem 287:12455–12468
Acknowledgements
The current study was granted by The Korean Society of Upper GI Disease and Helicobacter, by a National Research Foundation grant of Korea government (2010-0002052), and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2014R1A1A2058732).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Chan Young Ock and Jong-Min Park contributed equally
Electronic supplementary material
ESM 1
(PDF 529 kb)
Rights and permissions
About this article

Cite this article
Ock, C.Y., Park, JM., Han, YM. et al. Genetic ablation or pharmacologic inhibition of autophagy mitigated NSAID-associated gastric damages. J Mol Med 95, 405–416 (2017). https://doi.org/10.1007/s00109-016-1491-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-016-1491-3