
Abstract
Autophagy, a fundamental cellular catabolic process, is involved in the development of numerous diseases including cancer. Autophagy seems to have an ambivalent impact on tumor development. While increasing evidence indicates a cytoprotective role for autophagy that can contribute to resistance against chemotherapy and even against the adverse, hypoxic environment of established tumors, relatively few publications focus on the role of autophagy in early tumorigenesis. However, the consensus is that autophagy is inhibitory for the genesis of tumors. To understand this apparent contradiction, more detailed information about the roles of the individual participants in autophagy is needed. This review will address this topic with respect to autophagy-related protein 5 (ATG5), which in several lines of investigation has been ascribed special significance in the autophagic pathway. Furthermore, it was recently shown that an ATG5 deficiency in melanocytes interferes with oncogene-induced senescence, thus promoting melanoma tumorigenesis. Similarly, an ATG5 deficiency resulted in tumors of the lung and liver in experimental mouse models. Taken together, these findings indicate that ATG5 and the autophagy to which it contributes are essential gatekeepers restricting early tumorigenesis in multiple tissues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
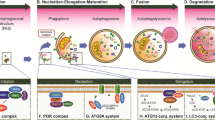
Autophagy is a highly conserved catabolic process which ensures cell survival and homeostasis under unfavorable conditions. By sequestering supernumerary or toxic, misfolded proteins and malfunctioning organelles in double-membrane vesicles called autophagosomes which then fuse with lysosomes, cells are able to supply themselves with building blocks needed for energy and new protein synthesis [1, 2]. Autophagy is highly dependent on the availability of the so-called autophagy-related proteins (ATGs) (Fig. 1). Among others, ATG5 is recognized as a key player needed for the formation of autophagosomes which is also important because of its potential contribution to apoptosis.

ATG5 is one of the key players during autophagosome elongation. Starting from the autophagosome formation site, the autophagosome is elongated, sealed around neighboring targets, and then fused with a lysosome to allow the degradation of the engulfed contents. Two ubiquitin-like conjugation systems are involved in the expansion of autophagosome: ATG5-ATG12 and LC3-PE
The onset of autophagy is regulated by the mammalian target of rapamycin complex 1 (mTORC1), a cellular metabolic regulatory node responding to the availability of nutrients and to growth factor stimulation [3]. As a serine/threonine protein kinase, mTORC1 maintains an inhibitory phosphorylation on the ATG1/unc-51-like kinase, (Ulk)1/2. Following mTORC1 inactivation, for example, owing to starvation, Ulk1/2 initiates autophagy by phosphorylating ATG13 and the focal adhesion kinase family-interacting protein of 200 kD (FIP200), stimulating the localization of the complex of all three to a site on the endoplasmatic reticulum, thus beginning the process of autophagosome formation [3]. The migration of a second complex containing vacuolar protein sorting-34 (Vps34), a class III phosphatidylinositol 3-kinase, together with ATG14 ligand and Beclin 1 to this site [4, 5] allows the local production of the phosphatidylinositol 3-phosphate (PI3P) which then recruits PI3P-binding proteins, such as WD-repeat protein interacting with phosphoinositides (WIPI) and their interaction partners, finally activating the elongation of the incipient autophagosome [6–8].
For this, two ubiquitin-like conjugation systems are involved, producing the two conjugates needed: ATG5-ATG12 and microtubule-associated protein 1 light chain 3—phosphatidylethanolamine (LC3-PE) [9, 10]. ATG12 is activated by the E1-like enzyme, ATG7, to form a thioester bond between the C-terminal Gly 186 of ATG12 and the Cys 507 of ATG7 [11]. Activated ATG12 is transferred to ATG10, an E2-like enzyme, and finally conjugated to ATG5 at Lys 149 through an isopeptide bond [12]. The ATG5 substituent then interacts with ATG16L leading to the formation of a multimeric complex localizing to the developing autophagosome membrane [13]. Thereafter, after removal of its C-terminal Arg 117 by the proteolytic activity of ATG4, LC3 is first activated by ATG7. Activated LC3 is then transferred to the E2-like enzyme ATG3 and finally attached to PE through an amide bond [14] (Fig. 1). ATG12-ATG5-ATG16L is mainly located on the outer surface of the developing autophagosome and is dissociated from the membrane once the autophagosome is complete [14]. The autophagosome with its enclosed targets is transported to the lysosome and, with the help of syntaxin 17, undergoes fusion [15]. Following the subsequent acidification of the phagolysosome, the contents undergo degradation by a variety of hydrolytic enzymes.
ATG5 is essential for autophagy, and its absence in knockout mice causes neonatal lethality [16]. This is probably because, in liver, ATG5 is required for a special form of autophagy called lipophagy, in which lipid droplets are partially engulfed and processed by autophagosomes [17, 18]. Furthermore, in the context of autophagy, ATG5 is essential for the maintenance and differentiation of adult stem cells [19, 20], but there is also evidence for functions carried out by ATG5 beyond any role in autophagy [9, 21, 22]. ATG5 is cleaved by calpain to produce a fragment translocating to the mitochondria and inducing apoptosis [21]. Using a neutrophil model of spontaneous apoptosis, these authors demonstrated that calpain-mediated cleavage of ATG5 produces a 24-kDa amino-terminal apoptogenic fragment. The inhibition of calpain blocked neutrophil apoptosis and also the appearance of the 24-kDa fragment. Interestingly, the transduction of cells with a construct expressing the 24-kDa ATG5 amino-terminal fragment resulted in rapid apoptosis; cells expressing high levels of BCL-2, however, elicited only little death [21]. The truncated 24-kDa ATG5 protein caused cytochrome c release from mitochondria and caspase-3 activation. It was found complexed with BCL-XL in apoptotic Jurkat and HeLa cells as well as in neutrophils, but not in non-apoptotic cells [21].
DNA damage caused, for example, by etoposide or cisplatin, induces ATG5 expression. ATG5 subsequently translocates to the nucleus where it interacts with survivin to induce cell cycle arrest followed by mitotic catastrophe [23]. Surprisingly, the same phenotype was also observed in cells overexpressing ATG5 ectopically without any DNA-damaging drug treatment [23]. Furthermore, the nuclear effects of ATG5, its binding to survivin with resulting disturbances in cytokinesis leading to mitotic catastrophe, occur independent of autophagy since ATG5 causes cell cycle arrest and mitotic catastrophe even when autophagy is blocked by 3-methyladenine inhibition. A mutant, ATG5K130R, which is unable to associate with ATG12, hence, is incapable of supporting autophagy, still induces the nuclear effects including the complex with survivin [23]. The mechanism linking ATG5 overexpression and the DNA damage response is the ability of ATG5 itself to induce p53 up-regulation and activation [24].
The contradictory effects of ATG5 and autophagy on cell death, namely by either blocking or promoting anticancer drug-induced cytotoxicity, have been shown in diverse types of cancers [25–30]. In contrast to this large body of literature reporting the involvement of ATG5 in response to anticancer drugs, little is known about the role of ATG5 in tumorigenesis. In the following, we will address this point with respect to the formation of liver and lung carcinomas and melanomas.
The role of ATG5 in tumorigenesis
Liver tumorigenesis
Since a systemic deletion of Atg5 leads to neonatal lethality [16], a mosaic deletion of Atg5 was achieved by crossing Atg5 flox/flox mice with CAG-Cre transgenic mice in which the Cre recombinase is ubiquitously expressed. In contrast to the expected overall deletion of Atg5, the resulting Atg5 flox/flox;CAG-Cre mice showed an incomplete deletion which was only observed in the liver, heart, skeletal muscle, and brain [31]. Because the incompletely deleted phenotype is reproducible and the mice are viable for more than 19 months, Takamura et al. used this mouse model to investigate the role of ATG5 in the tissues where Atg5 was knocked out [32]. Within 6 months after birth, severe hepatomegaly was observed in Atg5 flox/flox;CAG-Cre mice. By 6–9 months, multiple small tumors were visible; the number and size of which increased with age. All of the mice examined (n = 17) showed multiple tumors in their livers [32]. Interestingly, there was no evidence of tumor formation in other Atg5-deficient organs [32].
The histological examination of Atg5-deficient liver tumors at a later stage (month 19) showed a slight lymphocytic infiltration and intracellular vacuolation with necrotic hepatocytes. No obvious malignancy-associated signs, such as abnormal nuclear morphology or local invasion, were detected, even at the 19th month, suggesting a benign adenoma [32]. The vacuoles were shown to be lipid droplets by Oil-red O staining [32]. The formation of numerous lipid droplets in the liver has also been observed in p73 knockout mice, a phenomenon explainable by the fact that p73 is essential for Atg5 transactivation. Hence, p73−/− mice also exhibit a deficiency for autophagy [18].
Since autophagy is a dynamic degradation process, impaired autophagic activity can be monitored by an accumulation of ubiquitinated protein aggregates and an increased level of p62, a selective substrate in autophagy, responsible for aggregating cargo proteins to be enclosed in the autophagosome [33, 34]. To confirm the autophagy deficiency of Atg5 flox/flox;CAG-Cre adenoma cells, immunostaining of ubiquitin and p62 was performed. Indeed, more than 80 % of the hepatocytes in the tumor areas showed greater ubiquitin aggregates than those in the non-tumor regions [32]. Similarly, p62-positive dots were more often (nearly 80 %) seen in the tumor areas than those in non-tumor areas. These data suggested that a lack of autophagy was probably the main cause of tumor formation in the liver of Atg5 flox/flox;CAG-Cre mice [32]. In addition, Atg5-deficient hepatocytes had a growth advantage over normal cells in vivo as evidenced by the frequency of the proliferation marker, Ki-67 [32]. The histological examination of the liver of Atg5 flox/flox;CAG-Cre mice by electron microscopy showed abnormally enlarged mitochondria in the hepatocytes, suggestive of increased oxidative stresses in these cells. The immunostaining of 8-hydroxydeoxyguanosine (8-OHdG), an indicator of the oxidative stress [35], showed that most hepatocytes were positive for 8-OHdG, especially in the tumor areas in Atg5 flox/flox;CAG-Cre mice. Furthermore, a small fraction of the hepatocytes were positive for phospho-histone H2A.X in the nuclei (approximately 2 %) of Atg5 flox/flox;CAG-Cre mice as compared with the control mice (approximately 0.4 %), suggesting an induction of the DNA damage response in these mice [32]. Taken together, these observations indicate that an autophagy deficiency leads to increased oxidative stress and the induction of the DNA damage response often implicated in tumorigenesis [36].
To explore the mechanism and chronologic development of pathological abnormalities in Atg5-deficient hepatocytes, hepatocyte-specific Atg5 knockout mice were generated by crossing Atg5 flox/flox mice with albumin Cre mice (Hep-Atg5 knockout (KO)). In agreement with previous findings [32], these mice also developed liver adenomas as early as 9 months [37]. An impaired autophagic activity was documented by undetectable levels of LC3-II and increased amounts of p62 in primary cultures of Atg5-deficient hepatocytes [37]. A higher proliferation rate of the tumor cells and the presence of abnormal, swollen mitochondria were observed in the livers of these mice as well [37]. Interestingly, Atg5-deficient hepatocytes showed increased expression of the proapoptotic protein, BAX, whereas levels of the FLICE-like protein (FLIP), which inhibits caspase-8 activation declined [37]. Primary cultures of these hepatocytes exhibited the induction of apoptosis as shown by increased caspase-3 activity and PARP cleavage [37]. The histological examination of liver sections from the Hep-Atg5 KO mice showed infiltration of neutrophils and macrophages. Accordingly, the secretion of inflammatory cytokines was also elevated in the liver of Hep-Atg5 KO mice. Additionally, liver fibrosis was also evidenced by the up-regulation of profibrotic genes, including collagen type 1, connective tissue growth factor, transforming growth factor β1, and α-smooth muscle actin [37]. Based on previous publications, showing that the loss of autophagy in livers leads to the accumulation of p62 [32] and that p62 activates Nrf2 by the non-canonical p62-keap1-Nrf2 pathway [38], the possible involvement of Nrf2 in the pathogenesis of liver tumors was considered. In human hepatocellular carcinomas, the increased levels of p62 and the activation of Nrf2 have also been described [39]. Indeed, the deletion of Nrf-2 in Hep-Atg5 KO mice markedly abolished these tumor-associated pathological features; no liver tumors were observed in Atg5 flox/flox;Alb Cre +/Nrf2−/− double knockout mice [37]. This work has improved our current understanding of the contribution of Nrf2, a transcription factor regulating the expression of cytoprotective genes and hepatic detoxification enzymes [40, 41], in the pathogenesis of liver cancer (Fig. 2).

An ATG5 deficiency promotes early tumorigenesis in the liver, lung, and melanocytes. Insufficient ATG5 and/or autophagic dysfunction result in features associated with tumorigenesis, such as inflammation, accelerated proliferation, induction of oxidative stresses, and a DNA damage response in the liver. In the lung, deficient ATG5 and the resulting inhibition of autophagy lead to immunosuppression by infiltration of tumor cells by Tregs and an up-regulation of ENTPD1/CD39, an ecto-ATPase that converts the immunostimulatory ATP to immunosuppressive adenosine. In the skin, reduced ATG5 expression delays the onset of OIS, leading to an uncontrolled proliferation of melanocytes
Lung tumorigenesis
A dual role for ATG5 and autophagy in tumorigenesis has been evidenced by the study of KRasG12D-driven lung cancer mouse models [42]. In a KRas;Atg5 flox/flox mouse model, where the Cre recombinase can activate the KRas G12D oncogene in pneumocytes [43] leading to a stepwise progression from epithelial hyperplasia to benign adenoma and finally adenocarcinoma of the lung, Rao et al. studied the role of ATG5 in lung tumorigenesis by depleting Atg5 using the adenoviral delivery of the Cre recombinase [42]. They found a prolonged overall survival of autophagy-deficient KRas;Atg5 flox/flox mice. However, tumor initiation in these mice was accelerated. As early as 2–6 weeks after Atg5 depletion, enhanced tumor foci were observed in KRas;Atg5 flox/flox mice compared with KRas;Atg5 flox/+ controls. By 8 weeks after Atg5 depletion, however, the overall tumor burden values for autophagy-competent and autophagy-deficient mice were comparable. Interestingly, the autophagy-deficient KRas;Atg5 flox/flox mice, which showed increased tumor formation at early stages (until 8 weeks after Atg5 depletion), exhibited reduced tumor progression from adenoma to adenocarcinoma of the lung as compared to the autophagy-competent KRas;Atg5 flox/+ controls [42]. Mechanistically, p53 was found to be an essential tumor suppressor in the progression from adenoma to adenocarcinoma in the lung [42]. However, the depletion of p53 did not affect the increased tumor initiation in the KRas;Atg5 flox/flox mice, suggesting that p53-independent mechanisms are involved in promoting early tumorigenesis in autophagy-deficient KRas;Atg5 flox/flox mice. Indeed, it could be shown that increased infiltration by FOXP3-positive T regulatory cells (Tregs) correlated with the accelerated tumor initiation in autophagy-deficient KRas;Atg5 flox/flox mice. The depletion of the infiltrated Tregs slowed down the onset of the oncogenesis in these mice. Furthermore, it was shown that the ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1) or CD39, an ecto-ATPase that converts the immunostimulatory extracellular ATP to immunosuppressive adenosine [44], was induced in KRas;Atg5 flox/flox mice [45]. The pharmacological inhibition of ENTPD1 or the blockage of adenosinergic receptors reduced the infiltration of lung adenoma by Tregs and slowed down the tumor initiation to a level comparable with the controls, providing a link between autophagy deficiency and the inhibition of anticancer immunosurveillance [45] (Fig. 2).
Melanoma tumorigenesis
Consistent with a tumor-suppressive function of ATG5 in early tumorigenesis in the KRasG12D-driven lung cancer mouse model [42], ATG5 expression was found strongly reduced in a large cohort of primary melanomas (n = 194) as compared to benign nevi (n = 150) [46, 47]. Follow-up on 158 primary melanoma patients showed that patients with low levels of ATG5 experienced shorter progression-free survival times than those with high levels of ATG5 [46], indicating a prognostic value of ATG5 in primary melanoma. A reduced level of basal autophagy was also detected in primary melanomas as evidenced by low levels of LC3 II and accumulation of p62 [46]. On the other hand, no obvious difference in Beclin 1 expression was observed between these cohorts of melanoma and nevi patients [46]. Promoter methylation of the Atg5 gene was shown to be one of the possible mechanisms leading to a down-regulation of ATG5 in primary melanomas [46]. The enforced expression of ATG5 in melanoma cell lines restored the reduced basal level of autophagy and sensitized these cells toward autophagy inducers as evidenced by LC3 lipidation [46]. Furthermore, the overexpression of ATG5 reduced the colony-forming ability of melanoma cells and induced senescence as shown by the positivity of senescence-associated β-galactosidase [46]. More importantly, reducing ATG5 expression in mutated BRAF-transduced normal human melanocytes accelerated the proliferation of melanocytes by interfering with oncogene-induced senescence (OIS) [46]. This study, in particular the data obtained from the in vitro model of melanoma tumorigenesis, suggests that a tumor-suppressive function of ATG5 in melanoma tumor initiation is obtained through its promotion of OIS.
Conclusion and perspectives
Unlike apoptosis, the role of autophagy in cell death is two sided, either promoting or preventing death, leading to a highly complex function of autophagy in tumorigenesis. Based on recent publications reporting a role of ATG5 in tumorigenesis, it seems that ATG5 and autophagy suppress early tumorigenesis in liver and lung epithelial cells, as well as in melanocytes. These studies also provide evidence that the function of ATG5 in tumor formation may be stage- and cell/tissue-type dependent. Although the overall survival of the autophagy-deficient KRas;Atg5 flox/flox mice was longer, the initiation of lung adenoma was earlier than the autophagy-competent KRas;Atg5 flox/+ controls. Furthermore, in autophagy-competent KRas;Atg5 flox/+ mice, lesions progress faster from adenoma to adenocarcinoma of the lung than in the autophagy-deficient KRas;Atg5 flox/flox mice. Similarly, ATG5 was found to be down-regulated in primary melanomas as compared with benign nevi. Lowering ATG5 expression accelerated the proliferation of mutant BRAF-transformed melanocytes as a consequence of bypassing, or at least delaying, OIS. These data provide clinically relevant evidence that ATG5 could suppress the early transition from benign to malignant tumors in melanocytes. Given that Atg5 is deficient in the liver, heart, skeletal muscle, and brain in Atg5 flox/flox;CAG-Cre mice, tumor foci were only observed in the liver, but not in other organs where Atg5 was also depleted, thus raising the notion that the tumor-suppressive function of ATG5 and of autophagy may be organ/tissue specific. Since autophagy has been reported to be up-regulated in multiple cancer cells in response to chemotherapeutic drugs, the inhibition of autophagy is being considered as a supplement to current anticancer therapies [23, 48]. However, the basal level of autophagy in different types of cancers must be evaluated, and the stage of the targeted tumors must be considered. Future work on the role of ATG5 and autophagy in the formation and progression of other types of cancer should be carried out to improve our current understanding of the function of ATG5 and autophagy in tumorigenesis.
References
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147(4):728–741
Kim J, Huang WP, Stromhaug PE, Klionsky DJ (2002) Convergence of multiple autophagy and cytoplasm to vacuole targeting components to a perivacuolar membrane compartment prior to de novo vesicle formation. J Biol Chem 277(1):763–773
Yang Z, Klionsky DJ (2010) Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22(2):124–131
Obara K, Noda T, Niimi K, Ohsumi Y (2008) Transport of phosphatidylinositol 3-phosphate into the vacuole via autophagic membranes in Saccharomyces cerevisiae. Genes Cells 13(6):537–547
Russell RC, Yuan HX, Guan KL (2014) Autophagy regulation by nutrient signaling. Cell Res 24(1):42–57
Reggiori F, Tucker KA, Stromhaug PE, Klionsky DJ (2004) The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell 6(1):79–90
Lu Q, Yang P, Huang X, Hu W, Guo B, Wu F, Lin L, Kovács AL, Yu L, Zhang H (2011) The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell 21(2):343–357
Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA (2012) Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 23(10):1860–1873
Yousefi S, Simon HU (2007) Apoptosis regulation by autophagy gene 5. Crit Rev Oncol Hematol 63(3):241–244
Geng J, Klionsky DJ (2008) The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modification: beyond the usual suspects’ review series. EMBO Rep 9(9):859–864
Tanida I, Mizushima N, Kiyooka M, Ohsumi M, Ueno T, Ohsumi Y, Kominami E (1999) Apg7p/Cvt2p: a novel protein-activating enzyme essential for autophagy. Mol Biol Cell 10(5):1367–1379
Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y (1998) A protein conjugation system essential for autophagy. Nature 395(6700):395–398
Mizushima N, Noda T, Ohsumi Y (1999) Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J 18(14):3888–3896
Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M et al (2000) A ubiquitin-like system mediates protein lipidation. Nature 408(6811):488–492
Itakura E, Kishi-Itakura C, Mizushima N (2012) The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151(6):1256–1269
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432(7020):1032–1036
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458(7242):1131–1135
He Z, Liu H, Agostini M, Yousefi S, Perren A, Tschan MP, Mak TW, Melino G, Simon HU (2013) p73 regulates autophagy and hepatocellular lipid metabolism through a transcriptional activation of the ATG5 gene. Cell Death Differ 20(10):1415–1424
Salemi S, Yousefi S, Constantinescu MA, Fey MF, Simon HU (2012) Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res 22(2):432–435
Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegué E (2013) FOXO3A directs a protective autophagy program in hematopoietic stem cells. Nature 494(7437):323–327
Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU (2006) Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 8(10):1124–1132
Codogno P, Meijer AJ (2006) Atg5: more than an autophagy factor. Nat Cell Biol 8(10):1045–1047
Maskey D, Yousefi S, Schmid I, Zlobec I, Perren A, Friis R, Simon HU (2013) ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun 4:2130
Simon HU, Yousefi S, Schmid I, Friis R (2014) ATG5 can regulate p53 expression and activation. Cell Death Dis 5:e1339
Liu H, He Z, Simon HU (2013) Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin Cancer Biol 23(5):352–360
Wei MF, Chen MW, Chen KC, Lou PJ, Lin SY, Hung SC, Hsiao M, Yao CJ, Shieh MJ (2014) Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy 10(7):1179–1192
Kumar D, Shankar S, Srivastava RK (2014) Rottlerin induces autophagy and apoptosis in prostate cancer stem cells via PI3K/Akt/mTOR signaling pathway. Cancer Lett 343(2):179–189
Fan J, Zeng X, Li Y, Wang S, Wang Z, Sun Y, Gao H, Zhang G, Feng M, Ju D (2013) Autophagy plays a critical role in ChLym-1-induced cytotoxicity of non-Hodgkin’s lymphoma cells. PLoS One 8(8):e72478
Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Corominas-Faja B, Cuyàs E, López-Bonet E, Martin-Castillo B, Joven J, Menendez JA (2013) The anti-malarial chloroquine overcomes primary resistance and restores sensitivity to trastuzumab in HER2-positive breast cancer. Sci Rep 3:2469
Pan X, Liu D, Wang J, Zhang X, Yan M, Zhang D, Zhang J, Liu W (2013) Peneciraistin C induces caspase-independent autophagic cell death through mitochondrial-derived reactive oxygen species production in lung cancer cells. Cancer Sci 104(11):1476–1482
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441(7095):885–889
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N (2011) Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25(8):795–800
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y et al (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169(3):425–434
Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171(4):603–614
Cadet J, Douki T, Gasparutto D, Ravanat JL (2003) Oxidative damage to DNA: formation, measurement and biochemical features. Mutat Res 531(1–2):5–23
Dizdaroglu M (2012) Oxidatively induced DNA damage: mechanisms, repair and disease. Cancer Lett 327(1–2):26–47
Ni HM, Woolbright BL, Williams J, Copple B, Cui W, Luyendyk JP, Jaeschke H, Ding WX (2014) Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol 61(3):617–625
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou Y-S, Ueno I , Sakamoto A, Tong KI et al (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12(3):213–223
Komatsu M (2011) Potential role of p62 in tumor development. Autophagy 7(9):1088–1090
Shin SM, Yang JH, Ki SH (2013) Role of the Nrf2-ARE pathway in liver diseases. Oxidative Med Cell Longev 2013:763257
Vomhof-Dekrey EE, Picklo MJ Sr (2012) The Nrf2-antioxidant response element pathway: a target for regulating energy metabolism. J Nutr Biochem 23(10):1201–1206
Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, Sykacek P, Frank L, Schramek D, Komnenovic V et al (2014) A dual role for autophagy in a murine model of lung cancer. Nat Commun 5:3056
Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T (2001) Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410(6832):1111–1116
Roberts V, Stagg J, Dwyer KM (2014) The role of ectonucleotidases CD39 and CD73 and adenosine signaling in solid organ transplantation. Front Immunol 5:64
Rao S, Yang H, Penninger JM, Kroemer G (2014) Autophagy in non-small cell lung carcinogenesis: a positive regulator of antitumor immunosurveillance. Autophagy 10(3):529–531
Liu H, He Z, von Rütte T, Yousefi S, Hunger RE, Simon HU (2013) Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med 5(202):202ra123
Liu H, He Z, Simon HU (2014) Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy 10(2):372–373
Tschan MP, Simon HU (2010) The role of autophagy in anticancer therapy: promises and uncertainties. J Intern Med 268(5):410–418
Acknowledgments
Work in the laboratory of HUS is funded by grants from the Swiss National Science Foundation (310030-146181) and Swiss Cancer League (3099-02-2013).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Liu, H., He, Z. & Simon, HU. Protective role of autophagy and autophagy-related protein 5 in early tumorigenesis. J Mol Med 93, 159–164 (2015). https://doi.org/10.1007/s00109-014-1241-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-014-1241-3