
Abstract
PARK2 (PARKIN) is an E3 ubiquitin ligase involved in multiple signaling pathways and cellular processes. Activity of PARK2 is tightly regulated through inter- and intra-molecular interactions. Dysfunction of PARK2 is associated with the progression of parkinsonism. Notably, frequent PARK2 inactivation has been identified in various human cancers. Park2-deficient mice are more susceptible to tumorigenesis, indicating its crucial role as a tumor suppressor. However, biological studies also show that PARK2 possesses both pro-survival and growth suppressive functions. Here, we summarize the genetic lesions of PARK2 in human cancers and discuss the current knowledge of PARK2 in cancer progression. We further highlight future efforts for the study of PARK2 in cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The PARK2 (PARKIN) gene encodes a RING-between-RING-type E3 ubiquitin ligase which serves as a RING/HECT hybrid [1, 2]. The functions of PARK2 have been implicated in protein turnover, stress response, mitochondria homeostasis, xenophagy [3], metabolism, and many other cellular processes regulating cell growth and survival. Genetically, PARK2 status is associated with risk of autosomal recessive juvenile Parkinson's disease (ARJPD), leprosy, typhoid, and paratyphoid fever [4–6].
A growing body of evidence also shows the involvement of somatic PARK2 inactivation in human cancers, albeit the association between PARK2 genotype and cancer susceptibility is still under debate [7]. Park2-deficient mice show increased susceptibility to tumorigenesis. PARK2 depletion promotes the proliferation and tumor formation ability of pancreatic cancer cells [8], whereas ectopic PARK2 reduces the in vitro or in vivo growth of cancer cells of various tissue origin [9–14], strongly suggesting a tumor suppressive role of PARK2. Moreover, PARK2 overexpression inhibits the migration and invasion of multiple cancer cells ([9] and our unpublished data). This review aims to summarize recent advances on structure, regulation, and function of PARK2 and its murine models, with the emphasis on cancer-associated lesions and the potential link between PARK2 inactivation and cancer development.
Expression, structure, and regulation of PARK2
PARK2 is ubiquitously expressed [15]. The transcription of PARK2 can be regulated by N-myc, Max, p53, and ATF4 [16–18], and various environmental stimulations, such as nutrients, growth signals, mitochondrial, and ER stresses [18–22]. PARK2 precursor transcripts can be processed by pre-mRNA splicing factors, TDP-43, and FUS/TLS [23, 24]. Alternative splicing of PARK2 produces multiple tissue-specific variants [15, 25]. Interestingly, an internal in-frame Kozak sequence exists in the full-length PARK2 open reading frame (ORF), which initiates the translation of a special form of PARK2 which lacks the N-terminal ubiquitin-like (UBL) domain.
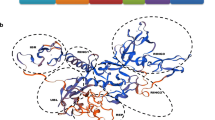
The PARK2 protein is well conserved from nematodes to humans. Full-length PARK2 consists of four important domains: UBL, RING0 (also known as Unique PARKIN domain), RING1, in-between-RING (IBR) domain, and RING2. Additionally, it contains a class II PDZ domain-binding motif towards the C-terminal end [26], and a newly identified Repressor of PARKIN (REP, also known as tether) fragment between IBR and RING2 [27, 28] (Fig. 1a, b). Structural studies reveal an auto-inhibited conformation of PARK2 through complex intra-molecular interactions [27–30]. Briefly, the UBL domain binds to the linker region between IBR and RING2 to stabilize the quaternary structure of PARK2. REP associates with RING1 at the E2 binding site to block E2 recruitment. RING0 intervenes between RING1 and RING2 and buries the catalytic C431, preventing E2-RING2 ubiquitin transfer and subsequent ubiquitin-ester formation (Fig. 1c). Thus, the activation of PARK2 requires massive conformational changes, and the intrinsic auto-inhibition of PARK2 implicates its strict regulation and important function.

Schematic and spatial illustrations of PARK2 structure. a Functional domains of PARK2 protein. b Structure of full-length PARK2 (PDB 4K95). c Surface representation of full-length PARK2 (remodeling of PDB 4K95) indicating complex intra-molecular interactions and buried catalytic C431
Timely recruitment of substrates and activation are two important aspects to execute the E3 ligase function of PARK2. Phosphorylation (S65), oligomerization, and ligand and/or E2 binding contribute to PARK2 activation [27, 30, 31], whereas the phosphorylations catalyzed by c-Abl (Y143) and Cdk5 (S131) attenuate its activity [32–34] (Fig. 2a). Additionally, phosphorylation of PARK2 may modulate its folding, solubility, and ligand or substrate binding affinity [35–37]. To date, posttranslational modifications and interaction partners of PARK2 have been extensively studied [38]. However, the mechanism of PARK2 activation, how PARK2 transits between active and inactive modes, and what determines the specificity of PARK2 remain largely unclear.

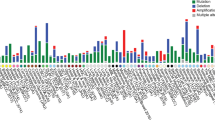
Phosphorylation and cancer-derived recurrent mutations of PARK2. a Sites of PARK2 phosphorylated by various kinases including PINK1, c-Abl, Cdk5, and CK1. b Schematic representation of recurrent mutations of PARK2 in cancer. c Mapping of cancer-derived recurrent mutations onto the PARK2 structure
Inactivation of PARK2 in cancer
Mutation
Mutations of PARK2 occur in both ARJPD and solid tumors. Based on the analysis of recent next generation sequencing data via cBio [39, 40], the frequency of PARK2 mutations is relatively high in cervical cancer (5.6 %), lung squamous cell cancer (5.6 %), colorectal cancer (2.4 ~ 5.6 %), gastric cancer (4.6 %), skin cutaneous melanoma (3.5 %), lung adenocarcinoma (2.7 ~ 3.1 %), and endometrioid cancer (2.1 %). In addition, several cancer cell lines harboring PARK2 mutations have been identified (Supplementary Table 1). Most cancer-derived PARK2 mutations are located at conserved regions (Fig. 2b), and more than 10 % of mutations lead to frame shifts or truncations, suggesting that those mutations may disrupt or abolish the function of PARK2. Notably, several sites mapping to various domains are recurrently mutated, such like A46, T173, T240, P294, P343, Q347, A371, and E395 (Fig. 2b, c). The biological consequences of those mutations need further clarification.
Copy number alterations
Loss of heterozygosity and copy number loss of PARK2 are found in breast cancer [15], clear cell renal cell carcinoma (ccRCC) [41], esophageal adenocarcinoma [42], glioma [12, 43], non-small cell lung cancer [14], lung adenocarcinoma [44], ovarian cancer [15], and pancreatic adenocarcinoma [8] (Table 1). Further analysis based on recent cancer genomic studies reveals that PARK2 deletion is also prevalent in adenoid cystic carcinoma (10 %), skin cutaneous melanoma (3.5 %), ovarian cancer (3.2 %) [39, 40], gastric cancer [45], and triple-negative breast cancer (6 %) [46], suggesting that copy number loss is another leading genomic defect of PARK2.
Promoter hypermethylation
Promoter hypermethylation is a common epigenetic mechanism to alter the gene expression. PARK2 promoter hypermethylation has been found in acute lymphoblastic leukemia (ALL, 26 %), chronic myeloid leukemia (CML, 3 %) [47], and colorectal cancer (4.7 %) [10]. 5-Aza treatment could restore the expression of PARK2 in ALL cell lines with PARK2 promoter aberrant methylation. Interestingly, among 10 samples of CML with lymphoid blast crisis, two showed PARK2 promoter hypermethylation. To date, the function of PARK2 in the pathogenesis of leukemia remains unexplored. Although the frequency of PARK2 promoter hypermethylation is low when compared with mutation or deletion, it may serve as an alternative way to inactivate PARK2.
mRNA/protein aberrant expression
As a result of genomic and epigenetic inactivation, the mRNA expression level of PARK2 is downregulated in a wide spectrum of human malignancies (Table 1). In addition, our unpublished analysis of TCGA dataset supports that the mRNA of PARK2 is significantly lower in ccRCC, bladder urothelial cancer, head and neck squamous cell carcinoma, lung adenocarcinoma, breast cancer, thyroid cancer, and endometrioid cancer compared with corresponding normal tissues [39, 40]. Notably, low transcription of PARK2 correlates with increased lymph node metastasis, higher tumor grade, and worse overall survival in ccRCC [48].
In parallel to mRNA underexpression, PARK2 protein has been shown to be downregulated in a large panel of cancer cell lines [9–13, 15, 49] and primary tumors (Table 1) [8, 13, 48, 50]. In pancreatic cancer, PARK2 expression is negatively correlated with grade and lymph node metastasis [8]. In breast cancer, PARK2 levels can predict the outcome of paclitaxel treatment [51]. Interestingly, stromal PARK2 abundance is remarkably reduced in malignant breast tissues [9], suggesting a potential role of PARK2 in tumor microenvironment.
Aberrant or alternative splicing may also lead to PARK2 abnormal expression. Aberrant transcripts have been identified in ovarian cancer (15 %) [15], colorectal cancer (42 %) [22], and several CML or cancer-derived cell lines [47, 49, 52], which may result in the disruption of PARK2 ORF and protein function.
Together, genetic and epigenetic disruptions of PARK2 are prevalent across human malignancies, suggesting that PARK2 inactivation may be a driving event during neoplastic transformation and progression.
PARK2 and tumorigenesis in animal models
Animal models have helped to investigate the role of PARK2 in tumorigenesis. To date, seven lines of Park2 knockout mice have been generated in an attempt to reproduce Parkinson's disease [53–59]. Generally, Park2 −/− mice develop normally and do not show a severe neurodegeneration phenotype or obvious clinical defects [60].
However, Park2 −/− mice are more susceptible to γ-irradiation-induced tumorigenesis [17]. After irradiation, Park2 is specifically elevated in mouse spleen and thymus in a p53-dependent manner. Park2 −/− mice show significantly shorter γ-irradiation-induced tumor latency compared with wild-type littermates, even though the tumor spectrum is similar (with the predominant type being lymphoma).
Adult Park2 null mice show reduced body weight but enlarged livers compared to wild-type mice [61]. Notably, Park2 −/− mice develop spontaneous hepatocellular carcinoma (HCC) at advanced age [61]. Those tumors histologically recapitulate human HCC with prominent expression of α-fetoprotein and β-catenin. In mouse liver, Park2 is a lipid-responsive gene whose expression facilitates the lipid uptake of hepatocytes and maintains the systematic lipid metabolism [21]. Whether the dysfunction of liver metabolism contributes to the subsequent hepatocellular carcinogenesis in Park2 −/− mice is unclear.
Park2 deficiency also promotes colorectal adenoma development [10]. Park2 +/−; Apc +/min mice show higher incidence (fourfold increase) of adenomas, and earlier onset of intestinal neoplasia compared with Park2 +/+; Apc +/min littermates. The wild-type allele of Park2 is retained in most adenomas derived from Park2 +/−; Apc +/min mice, suggesting that Park2 may be a haploinsufficient tumor suppressor.
Notably, Park2 −/− mice develop liver cancer only at advanced age (72 weeks or older) [61], and Park2 +/−; Apc +/+ mice do not develop intestinal adenoma [10], suggesting that Park2 deficiency alone may not be sufficient to drive rapid neoplastic transformation. Since PARK2 is critical for mitophagy (selective autophagy to degrade damaged mitochondria [62–64]), liver-specific spontaneous tumor formation in Park2 null mice may result from the long-term toxic effect of mitophagy and/or autophagy defects. A similar phenotype is observed in both Becn1 +/− and Atg5 f/f; CAG-Cre mice with their advancing age [65–67].
Involvement of PARK2 in cancer associated signaling pathways
Microtubule organization
Microtubules are critical for diverse cellular processes and have been targeted for cancer therapy for decades. The microtubule filaments are composed of α- and β-tubulin heterodimers. PARK2 co-localizes with microtubules and possesses three independent microtubule/tubulin binding domains, including RING0 (together with linker region between UBL), RING1, and RING2 [68]. PARK2 promotes the polymerization of microtubules, thereby increasing their stabilization in cooperation with paclitaxel treatment, and antagonizing the effect of depolymerizing drugs. In response to microtubule-depolymerizing drugs, PARK2 also suppresses the subsequent activation of microtubule-associated protein kinases (MAPKs) including JNK, ERK, and p38 [69]. Ectopic expression of PARK2 sensitizes breast cancer cell lines to paclitaxel, docetaxel, and epothilone B. Moreover, the PARK2 level correlates with the paclitaxel sensitivity in primary breast cancer cells and predicts the response of paclitaxel treatment in breast cancer [51].
On the other hand, PARK2 also acts as an E3 ligase of α/β-tubulins [70]. Interestingly, all of three microtubule/tubulin binding domains and several E3 ligase-deficient PARK2 mutants are able to rescue the microtubule depolymerizing effect by colchicine [68], suggesting that the microtubule-stabilizing ability of PARK2 is independent of its E3 ligase activity. Further, expression of any one of three domains is sufficient to attenuate the activation of MAPKs upon colchicine and nocodazole treatment [69]. Regarding how PARK2 balances between microtubule stabilization and tubulin degradation, one explanation might be that PARK2 predominantly binds with microtubules and selectively targets misfolded tubulins for proteasomal degradation, similar to the case of DJ-1 [71, 72].
Together, the aforementioned observations suggest that PARK2 is an important regulator of tubulin polymerization and microtubule stability. Of note, ectopically expressed PARK2 suppresses cancer cell migration and invasion in vitro ([9] and our unpublished data). As the dynamics of microtubules have been associated with cell migration [73, 74], PARK2 may negatively regulate cancer cell metastasis through its microtubule-stabilizing activity.
Cell cycle progression
PARK2 appears to play a role in cell cycle progression. A recent study revealed the dynamic subcellular localization of PARK2 during cell cycle progression: in interphase, PARK2 shows perinuclear distribution; in mitotic phase, PARK2 mainly localizes to centrosomes and mitotic spindles; and PARK2 is found at midbody during cytokinesis [8].
Functionally, PARK2 mediates the ubiquitination and degradation of Cyclin E in complex with FBXW7 and Cullin1 [12, 22, 75]. It also downregulates the Cyclin D1 level probably through indirect transcriptional repression ([11] and our unpublished data). Overexpression of PARK2 increases G1-phase arrest and delays mitotic entry [9, 11]. Interestingly, PARK2 upregulates the mRNA level of CDK6 specifically in MCF7 breast cancer cells which leads to the cell cycle arrest and growth suppression [9], suggesting that PARK2 may function in a cell type-specific- or context-dependent manner.
PARK2 depletion increases the cell fraction in S- and G2-M phase [12]. Multiple lines of evidence indicate that PARK2 also regulates centrosome and mitotic spindle partially through interaction with γ-tubulin, a protein with well-established function in nucleation and orientation of microtubules [76–78]. The PARK2/γ-tubulin complexes are physiologically present in the cytosol, and PARK2 is reversibly recruited to the centrosome through HDAC6 and a microtubule-dependent mechanism after proteasome blockage, suggesting a potential role of PARK2 in centrosome function. As centrosomes contribute to the formation of the mitotic spindle, the inactivation of PARK2 in cancer may promote the dysregulation of cell division. Indeed, knockdown of endogenous PARK2 leads to spindle misorientation [8], and the development of multipolar spindles as well as micronucleus [12]. Similarly, cells with exogenous C-terminal truncation of PARK2 display increased ability to bypass the mitotic arrest induced by nocodazole and show a higher frequency of multinucleation [78], suggesting a defect in spindle assembly checkpoint. In addition, PARK2 may help to maintain the bipolar spindle assembly through transcriptional repression of Eg5 [8, 79], hence facilitates the proper chromosome segregation during cell division. Together, PARK2 safeguards the proper mitosis by ensuring the function and organization of centrosome and spindle, and PARK2 loss may contribute to the development of aneuploidy.
Mitochondria homeostasis
Mitochondria are critical for cell metabolism and cell death whose dysfunction contributes directly to cancer development. Increasing amount of evidence indicates that PARK2 is involved in the turnover and function of mitochondria.
Mitochondrial genome
PARK2 binds to mitochondrial DNA (mtDNA), enhances TFAM-mediated mitochondrial transcription, and restores the PGC-1α expression, thereby promoting mitochondria biogenesis [80–82]. Moreover, it protects the mitochondrial genome from reactive oxygen species (ROS)-induced damage and supports mtDNA recovery [81]. Long-term overexpression of PARK2 selectively eliminates mitochondria with deleterious mtDNA mutations, thereby enriching the wild-type mtDNA for normal mitochondrial function [83]. This suggests that PARK2 is important for the maintenance of integrity of the mitochondrial genome, and thus linking PARK2 alterations to tumorigenesis [84–86].
Mitophagy
The role of PARK2 in the induction and progression of mitophagy has been extensively studied, leading to some controversy [62–64]. Generally, mitochondrial stress (depolarization) blocks the inner mitochondrial import of PINK1 and triggers its auto-phosphorylation and stabilization [87–89]. The accumulated PINK1 phosphorylates many substrates including PARK2 at S65, thereby stimulating self-association of PARK2 and then recruiting it to depolarized mitochondrial membrane [31, 90, 91]. Upon activation, PARK2 rapidly catalyzes the ubiquitination of a vast array of mitochondrial proteins, such like FIS1, MFN1/2, RHOT1/2, TOMM70A, and many other substrates [63, 92, 93], and separates mitochondria from the microtubule network [94]. The bulky ubiquitination of mitochondrial proteome subsequently recruits adaptor proteins to connect the autophagy machinery and initiates selective autophagy [95–98]. Ultimately, PARK2-dependent mitophagy selectively degrades damaged mitochondria, thereby maintaining a healthy population of mitochondria.
The function of mitochondria is commonly impaired in cancer [99]. Those mitochondria isolated from the brain of Park2 −/− mice have reduced respiratory capacity [100], suggesting that PARK2 loss undermines the mitochondrial energy production. However, to what extent PARK2 inactivation contributes to the mitochondria impairment in cancer remains uncertain.
Apoptosis pathway
PARK2 alters the intrinsic mitochondrial threshold for cytochrome c release, thereby protecting cells from apoptotic stress [101, 102]. However, the presence of PARK2 in the mitochondria is not sufficient to prevent cytochrome c release, suggesting that the anti-apoptotic function of PARK2 may be indirect, probably mediated through cytosolic factors. Indeed, PARK2 is capable to regulate the activity of several proteins belonging to the pro- and anti-apoptotic BCL-2 family, including BAX, MCL1, and BCL-2 [92, 103–105]. Of note, after apoptosis onset, PARK2 is cleaved by caspase 1 and caspase 8 [106, 107]. However, compared to the well-established protective function in neurons, the role of PARK2 in regulation of cancer cell apoptosis remains elusive. In cancer cells derived from the liver or breast, PARK2 expression augments the apoptotic cell death induced by HDAC inhibitors and microtubule-stabilizing drugs [13, 51]. Park2 −/− hepatocytes are more resistant to anticancer drugs than the wild-type counterpart [61]. Additionally, PARK2 sensitizes Hela cells to TNF-α-induced apoptosis [108]. Together, these observations suggest that PARK2 generally exerts an anti-apoptosis function but it also sensitizes cancer cells to certain stimuli.
Cancer cell metabolism
Warburg effect
Reprogramming energy metabolism is one of the hallmarks of cancer [109]. During malignant transformation, cancer cells switch from mitochondrial respiration to aerobic glycolysis to sustain the bioenergetics and biosynthetic requirement (known as Warburg effect). PARK2 is a p53 target gene and negatively regulates glucose uptake, oxygen consumption, glycolysis, and lactate production, mitigating the Warburg effect [17]. The mechanism underlying the inhibitory activity of PARK2 may be mediated by regulating the mitochondrial function as well as the expression/activity of metabolic enzymes. Proteomic studies have identified many metabolic enzymes which might be regulated by PARK2 [92, 100, 110–112], albeit the functional consequences of most alterations need to be further clarified. As an example, PARK2 positively regulates the expression of PDHA1, which reduces mitochondrial oxidative phosphorylation and promotes glycolysis [17, 100].
Antioxidant defense
Park2 mutant flies or mice show defects in antioxidant defense [100, 113–115]. Consistently, ectopic PARK2 expression reduces the ROS level and increases the glutathione (GSH) level in cells [17, 116], while PARK2 mutants decrease the GSH and elevate the intracellular oxidative damage [117]. Thus, loss of PARK2 may contribute to ROS production during oncogenic transformation, similar to the effect of p53 inactivation. Paradoxically, PARK2 activity may be required for KRAS-driven tumors to maintain mitochondrial quality control and buffer the oxidative stress, since functional mitochondria and mitochondrial ROS generation are essential for the growth of those tumors [118, 119]. In such a context, PARK2 becomes a pro-survival protein in KRAS-transformed cancer cells. On the other hand, excessive ROS modulates the sulfonation, protein folding, and solubility of PARK2, and thus represses its activity [120–123].
PARK2 in the receptor tyrosine kinase pathway
PARK2 interacts with Eps15 and EGFR upon EGF treatment [124]. Thus, loss of PARK2 might accelerate EGFR endocytosis and degradation, and decrease the EGFR-AKT signaling. However, overexpression of PARK2 in glioma cells paradoxically inhibits signaling through AKT/mTOR [11]. Our unpublished data also support the role of PARK2 as a negative regulator of the EGFR-AKT pathway in gliomas, suggesting a differential behavior of PARK2 in cancer cells. Moreover, PARK2 is able to downregulate VEGFR2 in gliomas [11]; thus, it may have a role in suppression of cancer angiogenesis.
Conclusions and future perspectives
As discussed above, although many aspects remain unexplored, recent data highlight the auto-inhibited structure of PARK2 and uncover its important roles in multiple cellular processes relevant to neoplastic transformation and malignant progression, such like cell cycle control, mitochondria homeostasis, and metabolism (Fig. 3). Importantly, advances in cancer genetics reveal frequent inactivation of PARK2 in a broad panel of human cancers. Murine studies further support the tumor suppressive role of PARK2 [10, 17, 61]. However, characterization of the putative roles of PARK2 in cancer still awaits further efforts as outlined below.

Mapping targets and/or pathways associated with PARK2 deficiency to cancer hallmarks defined by Hanahan and Weinberg [109]. MSD, microtubule-stabilizing drug
As an E3 ligase, the substrates of PARK2 involved in tumorigenesis remain largely unknown. Apparently, many lessons can be learned from its role in neuron, including its involvement in key signaling pathways implicated in both neurodegeneration and tumorigenesis, such as NF-κB, Wnt, JNK, and estrogen-related receptor pathways [125–129]. Importantly, transcriptomic and proteomic approaches are required to profile systematically the targets of PARK2 in cancer. In addition, deciphering the functional importance of cancer-associated PARK2 mutations is fertile ground of study.
Moreover, knowledge concerning the regulation of PARK2 needs to be expanded. The transcriptional and posttranslational regulation of PARK2 in cancer is unclear, though both are very likely to be impaired. For example, the association between expression and/or activity of PARK2 and the cellular status of p53, N-myc, and c-Abl in human malignancies has not been determined. How does PARK2 shuttle among different cellular compartments? What coordinates the mitochondrial dependent- and independent-function of PARK2? And to what extent do these dysregulations contribute to cancer?
Additional genetic and in vivo studies, including animal models, are essential to dissect further the function of PARK2 during tumorigenesis. Notably, Park2 deficiency is likely to increase the risk of cancer during exposure to carcinogens or tumor suppressor inactivation [10, 17], suggesting that murine models of Park2 knockout and other oncogenic background may help to clarify its involvement in tumorigenesis. Meanwhile, the role of PARK2 in “mitochondria-addicted” tumors, especially in RAS/RAF-driven tumors needs further study, perhaps by crossing Park2 null mice with Ras/Raf transgenic or knockin mice. Also, generation of Park2-associated tumor models will be a powerful tool to test the in vivo efficacy of small molecules modulating the PARK2 pathway, such as vitamin K2 [130]. Understanding the mechanism of PARK2 activation and function will therefore provide more insights into the development of cancer therapy by targeting the PARK2 pathway.
References
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K et al (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25:302–305
Wenzel DM, Lissounov A, Brzovic PS, Klevit RE (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474:105–108
Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS (2013) The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501:512–516
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Mira MT, Alcais A, Van Thuc N, Moraes MO, Di Flumeri C, Thai VH, Phuong MC, Huong NT, Ba NN, Khoa PX et al (2004) Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 427:636–640
Ali S, Vollaard AM, Widjaja S, Surjadi C, van de Vosse E, van Dissel JT (2006) PARK2/PACRG polymorphisms and susceptibility to typhoid and paratyphoid fever. Clin Exp Immunol 144:425–431
Alcalay RN, Clark LN, Marder KS, Bradley WE (2012) Lack of association between cancer history and PARKIN genotype: a family based study in PARKIN/Parkinson's families. Genes Chromosomes Cancer 51:1109–1113
Sun XD, Liu M, Hao JH, Li DW, Luo YG, Wang XC, Yang YF, Li F, Shui WQ, Chen Q et al (2013) Parkin deficiency contributes to pancreatic tumorigenesis by inducing spindle multipolarity and misorientation. Cell Cycle 12:1133–1141
Tay SP, Yeo CW, Chai C, Chua PJ, Tan HM, Ang AX, Yip DL, Sung JX, Tan PH, Bay BH et al (2010) Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J Biol Chem 285:29231–29238
Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, Luo FJ, Cantley LC, Wyllie AH, Adams DJ et al (2010) PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc Natl Acad Sci U S A 107:15145–15150
Yeo CW, Ng FS, Chai C, Tan JM, Koh GR, Chong YK, Koh LW, Foong CS, Sandanaraj E, Holbrook JD et al (2012) Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Cancer Res 72:2543–2553
Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W et al (2010) Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 42:77–82
Wang F, Denison S, Lai JP, Philips LA, Montoya D, Kock N, Schule B, Klein C, Shridhar V, Roberts LR et al (2004) Parkin gene alterations in hepatocellular carcinoma. Genes Chromosomes Cancer 40:85–96
Picchio MC, Martin ES, Cesari R, Calin GA, Yendamuri S, Kuroki T, Pentimalli F, Sarti M, Yoder K, Kaiser LR et al (2004) Alterations of the tumor suppressor gene Parkin in non-small cell lung cancer. Clin Cancer Res 10:2720–2724
Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, McAdams H, Trapasso F, Drusco A, Shimizu M, Mascillo V et al (2003) Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci U S A 100:5956–5961
West AB, Kapatos G, O'Farrell C, Gonzalez-de-Chavez F, Chiu K, Farrer MJ, Maidment NT (2004) N-myc regulates parkin expression. J Biol Chem 279:28896–28902
Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z (2011) Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci U S A 108:16259–16264
Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D et al (2011) Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ 18:769–782
Klinkenberg M, Gispert S, Dominguez-Bautista JA, Braun I, Auburger G, Jendrach M (2012) Restriction of trophic factors and nutrients induces PARKIN expression. Neurogenetics 13:9–21
Wang HQ, Imai Y, Kataoka A, Takahashi R (2007) Cell type-specific upregulation of parkin in response to ER stress. Antioxid Redox Signal 9:533–542
Kim KY, Stevens MV, Akter MH, Rusk SE, Huang RJ, Cohen A, Noguchi A, Springer D, Bocharov AV, Eggerman TL et al (2011) Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J Clin Invest 121:3701–3712
Ikeuchi K, Marusawa H, Fujiwara M, Matsumoto Y, Endo Y, Watanabe T, Iwai A, Sakai Y, Takahashi R, Chiba T (2009) Attenuation of proteolysis-mediated cyclin E regulation by alternatively spliced parkin in human colorectal cancers. International journal of cancer Journal international du cancer 125:2029–2035
Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C et al (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14:459–468
Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C et al (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15:1488–1497
Thierry-Mieg D, Thierry-Mieg J (2006) AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol 7(Suppl 1):S12 11–14
Fallon L, Moreau F, Croft BG, Labib N, Gu WJ, Fon EA (2002) Parkin and CASK/LIN-2 associate via a PDZ-mediated interaction and are co-localized in lipid rafts and postsynaptic densities in brain. J Biol Chem 277:486–491
Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G et al (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340:1451–1455
Riley BE, Lougheed JC, Callaway K, Velasquez M, Brecht E, Nguyen L, Shaler T, Walker D, Yang Y, Regnstrom K et al (2013) Structure and function of parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun 4:1982
Spratt DE, Martinez-Torres RJ, Noh YJ, Mercier P, Manczyk N, Barber KR, Aguirre JD, Burchell L, Purkiss A, Walden H et al (2013) A molecular explanation for the recessive nature of parkin-linked Parkinson's disease. Nat Commun 4:1983
Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, Walden H (2011) Autoregulation of parkin activity through its ubiquitin-like domain. Embo J 30:2853–2867
Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ (2013) PINK1 drives parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol 200:163–172
Ko HS, Lee Y, Shin JH, Karuppagounder SS, Gadad BS, Koleske AJ, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM (2010) Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function. Proc Natl Acad Sci U S A 107:16691–16696
Imam SZ, Zhou Q, Yamamoto A, Valente AJ, Ali SF, Bains M, Roberts JL, Kahle PJ, Clark RA, Li S (2011) Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson's disease. J Neurosci 31:157–163
Avraham E, Rott R, Liani E, Szargel R, Engelender S (2007) Phosphorylation of parkin by the cyclin-dependent kinase 5 at the linker region modulates its ubiquitin-ligase activity and aggregation. J Biol Chem 282:12842–12850
Yamamoto A, Friedlein A, Imai Y, Takahashi R, Kahle PJ, Haass C (2005) Parkin phosphorylation and modulation of its E3 ubiquitin ligase activity. J Biol Chem 280:3390–3399
Rubio de la Torre E, Luzon-Toro B, Forte-Lago I, Minguez-Castellanos A, Ferrer I, Hilfiker S (2009) Combined kinase inhibition modulates parkin inactivation. Hum Mol Genet 18:809–823
Trempe JF, Chen CX, Grenier K, Camacho EM, Kozlov G, McPherson PS, Gehring K, Fon EA (2009) SH3 domains from a subset of BAR proteins define a Ubl-binding domain and implicate parkin in synaptic ubiquitination. Mol Cell 36:1034–1047
Walden H, Martinez-Torres RJ (2012) Regulation of parkin E3 ubiquitin ligase activity. Cell Mol Life Sci 69:3053–3067
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1
Toma MI, Grosser M, Herr A, Aust DE, Meye A, Hoefling C, Fuessel S, Wuttig D, Wirth MP, Baretton GB (2008) Loss of heterozygosity and copy number abnormality in clear cell renal cell carcinoma discovered by high-density affymetrix 10K single nucleotide polymorphism mapping array. Neoplasia 10:634–642
Gu JA, Ajani JA, Hawk ET, Ye YQ, Lee JH, Bhutani MS, Hofstetter WL, Swisher SG, Wang KK, Wu XF (2010) Genome-wide catalogue of chromosomal aberrations in Barrett's esophagus and esophageal adenocarcinoma: a high-density single nucleotide polymorphism array analysis. Cancer Prev Res 3:1176–1186
Yin D, Ogawa S, Kawamata N, Tunici P, Finocchiaro G, Eoli M, Ruckert C, Huynh T, Liu GT, Kato M et al (2009) High-resolution genomic copy number profiling of glioblastoma multiforme by single nucleotide polymorphism DNA microarray. Mol Cancer Res 7:665–677
Iwakawa R, Okayama H, Kohno T, Sato-Otsubo A, Ogawa S, Yokota J (2012) Contribution of germline mutations to PARK2 gene inactivation in lung adenocarcinoma. Genes Chromosomes Cancer 51:462–472
Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, Zhang S, Lee M, Wu J, Lim KH et al (2012) A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 61:673–684
Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao YJ, Turashvili G, Ding JR, Tse K, Haffari G et al (2012) The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486:395–399
Agirre X, Roman-Gomez J, Vazquez I, Jimenez-Velasco A, Garate L, Montiel-Duarte C, Artieda P, Cordeu L, Lahortiga I, Calasanz MJ et al (2006) Abnormal methylation of the common PARK2 and PACRG promoter is associated with downregulation of gene expression in acute lymphoblastic leukemia and chronic myeloid leukemia. Int J Cancer 118:1945–1953
Toma MI, Wuttig D, Kaiser S, Herr A, Weber T, Zastrow S, Koch R, Meinhardt M, Baretton GB, Wirth MP et al (2013) PARK2 and PACRG are commonly downregulated in clear-cell renal cell carcinoma and are associated with aggressive disease and poor clinical outcome. Genes Chromosomes Cancer 52:265–273
Denison SR, Wang F, Becker NA, Schule B, Kock N, Phillips LA, Klein C, Smith DI (2003) Alterations in the common fragile site gene parkin in ovarian and other cancers. Oncogene 22:8370–8378
Letessier A, Garrido-Urbani S, Ginestier C, Fournier G, Esterni B, Monville F, Adelaide J, Geneix J, Xerri L, Dubreuil P et al (2007) Correlated break at PARK2/FRA6E and loss of AF-6/Afadin protein expression are associated with poor outcome in breast cancer. Oncogene 26:298–307
Wang HX, Liu BB, Zhang C, Peng GY, Liu M, Li DW, Gu F, Chen Q, Dong JT, Fu L et al (2009) Parkin regulates paclitaxel sensitivity in breast cancer via a microtubule-dependent mechanism. Journal of Pathology 218:76–85
Denison SR, Callahan G, Becker NA, Phillips LA, Smith DI (2003) Characterization of FRA6E and its potential role in autosomal recessive juvenile parkinsonism and ovarian cancer. Gene Chromosome Canc 38:40–52
Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L et al (2003) Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet 12:2277–2291
Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ et al (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278:43628–43635
von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM (2004) Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci U S A 101:10744–10749
Perez FA, Palmiter RD (2005) Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A 102:2174–2179
Sato S, Chiba T, Nishiyama S, Kakiuchi T, Tsukada H, Hatano T, Fukuda T, Yasoshima Y, Kai N, Kobayashi K et al (2006) Decline of striatal dopamine release in parkin-deficient mice shown by ex vivo autoradiography. J Neurosci Res 84:1350–1357
Kitao Y, Imai Y, Ozawa K, Kataoka A, Ikeda T, Soda M, Nakimawa K, Kiyama H, Stern DM, Hori O et al (2007) Pael receptor induces death of dopaminergic neurons in the substantia nigra via endoplasmic reticulum stress and dopamine toxicity, which is enhanced under condition of parkin inactivation. Hum Mol Genet 16:50–60
Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, Lubbert H (2007) Mono- and double-mutant mouse models of Parkinson's disease display severe mitochondrial damage. Hum Mol Genet 16:2377–2393
Stephenson SEM, Taylor JM, Lockhart PJ (2012) Parkinson's disease and parkin: insights from Park2 knockout mice. In: Dushanova J (ed). Mechanisms in Parkinson's disease—models and treatments InTech.
Fujiwara M, Marusawa H, Wang HQ, Iwai A, Ikeuchi K, Imai Y, Kataoka A, Nukina N, Takahashi R, Chiba T (2008) Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 27:6002–6011
Cookson MR (2012) Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harb Perspect Med 2:a009415
Narendra D, Walker JE, Youle R (2012) Mitochondrial quality control mediated by PINK1 and parkin: links to parkinsonism. Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a011338
Ashrafi G, Schwarz TL (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20:31–42
Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 100:15077–15082
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y et al (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. The Journal of clinical investigation 112:1809–1820
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N (2011) Autophagy-deficient mice develop multiple liver tumors. Gene Dev 25:795–800
Yang F, Jiang Q, Zhao J, Ren Y, Sutton MD, Feng J (2005) Parkin stabilizes microtubules through strong binding mediated by three independent domains. J Biol Chem 280:17154–17162
Ren Y, Jiang H, Yang F, Nakaso K, Feng J (2009) Parkin protects dopaminergic neurons against microtubule-depolymerizing toxins by attenuating microtubule-associated protein kinase activation. J Biol Chem 284:4009–4017
Ren Y, Zhao J, Feng J (2003) Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J Neurosci 23:3316–3324
Moore DJ, Zhang L, Troncoso J, Lee MK, Hattori N, Mizuno Y, Dawson TM, Dawson VL (2005) Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum Mol Genet 14:71–84
Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD, Chin LS (2007) Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J Cell Biol 178:1025–1038
Kaverina I, Straube A (2011) Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol 22:968–974
Watanabe T, Noritake J, Kaibuchi K (2005) Regulation of microtubules in cell migration. Trends Cell Biol 15:76–83
Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A (2003) Parkin is a component of an SCF-like ubipuitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron 37:735–749
Jiang Q, Ren Y, Feng J (2008) Direct binding with histone deacetylase 6 mediates the reversible recruitment of parkin to the centrosome. J Neurosci 28:12993–13002
Zhao J, Ren Y, Jiang Q, Feng J (2003) Parkin is recruited to the centrosome in response to inhibition of proteasomes. J Cell Sci 116:4011–4019
Chen Y, Fang ST, Yeh PC, Yang HH, Chen SY, Chang CJ, Zhai WJ, Chen YC, Juang YL (2012) The C-terminus of PARK2 is required for its self-interaction, solubility and role in the spindle assembly checkpoint. Biochim Biophys Acta 1822:573–580
Liu M, Aneja R, Sun X, Xie S, Wang H, Wu X, Dong JT, Li M, Joshi HC, Zhou J (2008) Parkin regulates Eg5 expression by Hsp70 ubiquitination-dependent inactivation of c-Jun NH2-terminal kinase. J Biol Chem 283:35783–35788
Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, Matsumoto T (2006) Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet 15:883–895
Rothfuss O, Fischer H, Hasegawa T, Maisel M, Leitner P, Miesel F, Sharma M, Bornemann A, Berg D, Gasser T et al (2009) Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet 18:3832–3850
Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM (2011) PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell 144:689–702
Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ (2010) Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A 107:11835–11840
Brandon M, Baldi P, Wallace DC (2006) Mitochondrial mutations in cancer. Oncogene 25:4647–4662
Chatterjee A, Mambo E, Sidransky D (2006) Mitochondrial DNA mutations in human cancer. Oncogene 25:4663–4674
Zanssen S, Schon EA (2005) Mitochondrial DNA mutations in cancer. PLoS Med 2:e401
Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M et al (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for parkin recruitment to damaged mitochondria. Nat Commun 3:1016
Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and parkin recruitment. EMBO Rep 13:378–385
Lazarou M, Jin SM, Kane LA, Youle RJ (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase parkin. Dev Cell 22:320–333
Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M et al (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2:120080
Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N (2012) PINK1-mediated phosphorylation of the parkin ubiquitin-like domain primes mitochondrial translocation of parkin and regulates mitophagy. Sci Rep 2:1002
Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496:372–376
Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC (2011) Broad activation of the ubiquitin-proteasome system by parkin is critical for mitophagy. Hum Mol Genet 20:1726–1737
Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147:893–906
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. Journal of Cell Biology 183:795–803
Van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, De Strooper B, Vandenberghe W (2011) Parkin interacts with Ambra1 to induce mitophagy. J Neurosci 31:10249–10261
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12:119–131
Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ (2010) p62/SQSTM1 is required for parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6:1090–1106
Gogvadze V, Orrenius S, Zhivotovsky B (2008) Mitochondria in cancer cells: what is so special about them? Trends Cell Biol 18:165–173
Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 279:18614–18622
Berger AK, Cortese GP, Amodeo KD, Weihofen A, Letai A, LaVoie MJ (2009) Parkin selectively alters the intrinsic threshold for mitochondrial cytochrome c release. Hum Mol Genet 18:4317–4328
Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M et al (2003) Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet 12:517–526
Johnson BN, Berger AK, Cortese GP, LaVoie MJ (2012) The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc Natl Acad Sci U S A 109:6283–6288
Ekholm-Reed S, Goldberg MS, Schlossmacher MG, Reed SI (2013) Parkin-dependent degradation of the f-box protein fbw7beta promotes neuronal survival in response to oxidative stress by stabilizing mcl-1. Mol Cell Biol 33:3627–3643
Chen D, Gao F, Li B, Wang HF, Xu YX, Zhu CQ, Wang GH (2010) Parkin mono-ubiquitinates Bcl-2 and regulates autophagy. J Biol Chem 285:38214–38223
Kahns S, Lykkebo S, Jakobsen LD, Nielsen MS, Jensen PH (2002) Caspase-mediated parkin cleavage in apoptotic cell death. J Biol Chem 277:15303–15308
Kahns S, Kalai M, Jakobsen LD, Clark BF, Vandenabeele P, Jensen PH (2003) Caspase-1 and caspase-8 cleave and inactivate cellular parkin. J Biol Chem 278:23376–23380
Lee K, Lee MH, Kang YW, Rhee KJ, Kim TU, Kim YS (2012) Parkin induces apoptotic cell death in TNF-alpha-treated cervical cancer cells. BMB Rep 45:526–531
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Periquet M, Corti O, Jacquier S, Brice A (2005) Proteomic analysis of parkin knockout mice: alterations in energy metabolism, protein handling and synaptic function. J Neurochem 95:1259–1276
Davison EJ, Pennington K, Hung CC, Peng JH, Rafiq R, Ostareck-Lederer A, Ostareck DH, Ardley HC, Banks RE, Robinson PA (2009) Proteomic analysis of increased parkin expression and its interactants provides evidence for a role in modulation of mitochondrial function. Proteomics 9:4284–4297
Xun Z, Kaufman TC, Clemmer DE (2009) Stable isotope labeling and label-free proteomics of Drosophila parkin null mutants. J Proteome Res 8:4500–4510
Greene JC, Whitworth AJ, Andrews LA, Parker TJ, Pallanck LJ (2005) Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum Mol Genet 14:799–811
Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G (2004) Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 131:2183–2194
Saini N, Oelhafen S, Hua H, Georgiev O, Schaffner W, Bueler H (2010) Extended lifespan of Drosophila parkin mutants through sequestration of redox-active metals and enhancement of anti-oxidative pathways. Neurobiol Dis 40:82–92
Yu F, Zhou J (2008) Parkin is ubiquitinated by Nrdp1 and abrogates Nrdp1-induced oxidative stress. Neurosci Lett 440:4–8
Hyun DH, Lee M, Hattori N, Kubo S, Mizuno Y, Halliwell B, Jenner P (2002) Effect of wild-type or mutant parkin on oxidative damage, nitric oxide, antioxidant defenses, and the proteasome. J Biol Chem 277:28572–28577
Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V et al (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Gene Dev 25:460–470
Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107:8788–8793
LaVoie MJ, Cortese GP, Ostaszewski BL, Schlossmacher MG (2007) The effects of oxidative stress on parkin and other E3 ligases. J Neurochem 103:2354–2368
Winklhofer KF, Henn IH, Kay-Jackson PC, Heller U, Tatzelt J (2003) Inactivation of parkin by oxidative stress and C-terminal truncations: a protective role of molecular chaperones. J Biol Chem 278:47199–47208
Meng F, Yao D, Shi Y, Kabakoff J, Wu W, Reicher J, Ma Y, Moosmann B, Masliah E, Lipton SA et al (2011) Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol Neurodegener 6:34
Wong ES, Tan JM, Wang C, Zhang Z, Tay SP, Zaiden N, Ko HS, Dawson VL, Dawson TM, Lim KL (2007) Relative sensitivity of parkin and other cysteine-containing enzymes to stress-induced solubility alterations. J Biol Chem 282:12310–12318
Fallon L, Belanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, Voortman J, Haber M, Rouleau G, Thorarinsdottir T et al (2006) A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol 8:834–842
Cha GH, Kim S, Park J, Lee E, Kim M, Lee SB, Kim JM, Chung J, Cho KS (2005) Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci U S A 102:10345–10350
Hwang S, Kim D, Choi G, An SW, Hong YK, Suh YS, Lee MJ, Cho KS (2010) Parkin suppresses c-Jun N-terminal kinase-induced cell death via transcriptional regulation in Drosophila. Mol Cells 29:575–580
Henn IH, Bouman L, Schlehe JS, Schlierf A, Schramm JE, Wegener E, Nakaso K, Culmsee C, Berninger B, Krappmann D et al (2007) Parkin mediates neuroprotection through activation of IkappaB kinase/nuclear factor-kappaB signaling. J Neurosci 27:1868–1878
Rawal N, Corti O, Sacchetti P, Ardilla-Osorio H, Sehat B, Brice A, Arenas E (2009) Parkin protects dopaminergic neurons from excessive Wnt/beta-catenin signaling. Biochem Bioph Res Co 388:473–478
Ren Y, Jiang H, Ma D, Nakaso K, Feng J (2011) Parkin degrades estrogen-related receptors to limit the expression of monoamine oxidases. Hum Mol Genet 20:1074–1083
Vos M, Esposito G, Edirisinghe JN, Vilain S, Haddad DM, Slabbaert JR, Van Meensel S, Schaap O, De Strooper B, Meganathan R et al (2012) Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science 336:1306–1310
Acknowledgments
We thank Chen Ye for the kind help in generation figures. This work was funded by the Singapore Ministry of Health's National Medical Research Council (NMRC) under its Singapore Translational Research (STaR) Investigator Award to H. Phillip Koeffler, NMRC Individual Research Grant (NMRC/1311/2011), and NIH grant R01CA026038-23. This work was also partially supported by grants from the Natural Science Foundation of China (81071788, 81272956).
Conflict of interest
The authors declare no conflict of interest related to this study.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 268 kb)
Rights and permissions
About this article
Cite this article
Xu, L., Lin, Dc., Yin, D. et al. An emerging role of PARK2 in cancer. J Mol Med 92, 31–42 (2014). https://doi.org/10.1007/s00109-013-1107-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-013-1107-0