
Abstract
Right ventricular (RV) failure is an important clinical problem with no available therapies, largely because its molecular mechanisms are unknown. Mitochondrial remodeling resulting to a metabolic shift toward glycolysis has been described in RV hypertrophy (RVH), but it is unknown whether this is beneficial or detrimental. While clinically RV failure follows a period of compensation, the transition from a compensated (cRVH) to a decompensated hypertrophied RV (dRVH) is not studied in animal models. We modeled the natural history of RVH and failure in the monocrotaline rat model of pulmonary hypertension by serially assessing clinically relevant parameters in the same animal. We defined dRVH as the stage in which RV systolic pressure started decreasing, along with the cardiac output, while the RV continued to remodel. dRVH was characterized by ascites, weight loss, and high mortality, compared to cRVH. A cRVH myocardium had hyperpolarized mitochondria and low production of mitochondria-derived reactive oxygen species (mROS), activated hypoxia-inducible factor 1α (HIF1α), and increased levels of glucose transporter 1, vascular endothelial growth factor, and stromal-derived factor 1, promoting increased glucose uptake (measured by positron emission tomography–computed tomography) and angiogenesis measured by lectin imaging in vivo. The transition to dRVH was marked by a sharp rise in mROS, inhibition of HIF1α, and activation of p53, both of which contributed to down-regulation of pyruvate dehydrogenase kinase and decreased glucose uptake. This transition was also associated with a sharp decrease in angiogenic factors and angiogenesis. We show that the previously described metabolic shift, promoting HIF1α activation and angiogenesis, is not sustained during the progression of RV failure. The loss of this beneficial remodeling may be triggered by a rise in mROS resulting in HIF1α inhibition and suppressed angiogenesis. The resultant ischemia may contribute to the rapid deterioration of RV function upon entrance to a decompensation phase. The use of clinical criteria and techniques to define and study dRVH facilitates clinical translation of our findings with direct implications for RV therapeutic and biomarker discovery programs.
Key message
-
Decreased RV angiogenesis marks the transition from a cRVH to a dRVH.
-
The RVs in cRVH animals are associated with decreased mROS and increased HIF1α activity compared to dRVH.
-
The RVs in cRVH animals have increased GLUT1 levels and increased glucose uptake compared to the dRVH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In contrast to left ventricular (LV) failure, right ventricular (RV) failure has remained understudied both at the preclinical and clinical level. As the two ventricles are in series, failure of either has the same impact, decreasing cardiac output and resulting in premature death. RV failure is the most important determinant of both morbidity and mortality in patients with pulmonary arterial hypertension (PAH) [1–3]. RV failure is also a strong and independent negative prognostic factor in patients with LV failure that develop secondary pulmonary hypertension [4]. In other words, this is a common and important clinical problem, and the lack of our understanding of what drives RV failure prevents the development of much needed RV-directed therapies.
Although there is a compensatory phase while the RV hypertrophies in PAH, during which the patients show minimal or no symptoms as cardiac output is maintained, there is a clear transition to a decompensatory phase, during which the patients enter a cataclysmic clinical deterioration which may result in death in less than 1 year. Overall, the compensatory phase in RV hypertrophy (RVH) is much shorter than that of the LV, where patients with systemic hypertension and LVH may survive for many decades. Nevertheless, in certain PAH subgroups (like in PAH associated with Eisenmenger's syndrome in congenital heart disease), this compensation phase lasts longer than in others (for example, in idiopathic or scleroderma-associated PAH) [1–3, 5]. The explanation for the longer compensatory phase in patients with Eisenmenger's syndrome is lacking. It has been proposed that the persistence of the “fetal heart gene program” in these patients is the underlying mechanism [2, 4]. This is unlikely to be the only or the most important factor. This is because recent evidence shows that in a model of acquired adult RV hypertrophy, i.e., the pulmonary artery (PA) banding model, the RV is also much more resistant to failure compared to the RVs from other animal models, in which a similar increase in the afterload comes from pulmonary vascular disease [6]. In addition, some PAH patients continue to show a progressive decrease in RV function and have worse outcomes, despite showing a decrease in pulmonary vascular resistance in response to PAH therapies [7]. These data suggest that there may be important mechanisms intrinsic to the myocardium, driving the relentless clinical deterioration of PAH patients, independent of the RV afterload. As the two ventricles have many differences (including different embryology), the knowledge from the mechanisms of LV failure cannot be extrapolated to the RV [2, 8]. There is a critical need to identify the factors that regulate the transition from a compensated to a decompensated RV failure, which remain completely unknown, in order to develop therapies and biomarkers for RV failure. The current preclinical literature suffers from the fact that typically RVH in animals is considered a pathologic condition regardless of its stage. There is typically no attempt to define the decompensation stage in RVH and focus on the mechanisms that drive the transition from a compensated to a decompensated RV.
A metabolic shift in the RVH myocardium toward a glycolytic phenotype (away from the mitochondria-driven glucose oxidation) has been described [9, 10]. However, whether this is beneficial and whether this persists throughout the progression of RVH remain unknown. If this were the case, it would suggest that the metabolic switch is not responsible for the transition toward decompensation and thus may not be an attractive therapeutic target. The lack of definition and modeling of the compensated vs. decompensated RVH prevents the answers to these questions.
There is some clinical evidence in PAH patients [11] and preclinical models [6] that RVH is relatively ischemic, potentially because of suppressed angiogenesis. This could explain why the hypertrophied RV myocardium and its increased O2 requirements will fail at a point where an imbalance between O2 demand and delivery occurs. There is also evidence that metabolic remodeling regulates hypoxia-inducible factor 1α (HIF1α) function and thus angiogenesis in cancer [12]. Could this be operative in the myocardium? If so, a dynamic metabolic remodeling during RVH may drive the regulation of HIF1α and thus angiogenesis. Our current work had two objectives: (a) to model the transition of compensated to decompensated RVH and (b) to study the hypothesis that a primary and dynamic metabolic remodeling in RVH myocardium could regulate the HIF–angiogenesis axis (in a paracrine manner) and inhibit angiogenesis signaling, marking the transition from compensated to decompensated RVH.
Methods
Animal model for progressive right ventricular failure
The monocrotaline (MCT)-induced PAH model was used to model the natural history of progressive right ventricular hypertrophy and failure. Male Sprague Dawley rats were randomized to control (sham saline injection) and 60 mg/kg MCT injection (Sigma-Aldrich, St. Louis, MO). All age-matched animals were injected intraperitoneally with 1 mL of the same lot of MCT, made in water and pH set to 7.2, on the same day. Animals were then randomized and sacrificed every week between 2 and 6 weeks post-MCT. Therefore, there were no differences in MCT preparation, injection, or cohort of animals.
Echocardiography
Pulmonary artery acceleration time (using pulsed wave Doppler) was measured in the parasternal short axis view using the VeVo 770 imaging system with a 716 probe (11–24 MHz). Cardiac output (CO) was measured using echocardiography using the formula: CO = stroke volume × heart rate, where stroke volume = 7.85 × LVOT2 × AVOT1. The heart rates of the rats were always between 350 and 400 beats per minute, and LVOT and AVOT1 were measured in parasternal long axis view. Left ventricular function was assessed by fractional shortening measured in the parasternal short axis.
Right heart catheterization
Rats were anesthetized (isoflurane) and placed in a supine position on a heated table. The right jugular vein was cannulated, and right heart catheterization was performed. A sheath was used through which a high-fidelity Millar catheter was advanced (microtip, 1.4 F, Millar Instruments Inc., Houston, TX). The sheath was 0.2 mm in inner diameter and 0.58 mm in outer diameter with a specially curved tip to facilitate passage through the right heart and the pulmonary artery. Pressures in the right atrium, right ventricle, and pulmonary artery were recorded sequentially (Power Lab, using Chart software 5.4, ADInstruments).
Lectin perfusion assay
Rats were anesthetized, and the jugular vein was cannulated. The rats were heparinized using 1,000 IU/kg of heparin (American Pharmaceuticals Partners, Schaumbert, IL) to minimize the formation of thrombi and injected with 5 mg of FITC-conjugated Ricinus Communis Agglutinin I (Vector Laboratories, Burlinghame, CA). After circulation of lectin for 20 min, the rats were euthanized, and the right ventricle was harvested. Tissue processing: Tissue was sliced 50 μm thick from the endocardial side of the RV using a microtome, fixed in 4 % paraformaldehyde, and stained with the nuclear stain DAPI. Tissue imaging: To create a 3D image from the 50-μm-thick slice of endocardial tissue, 20 separate sequential images were taken (230 μm in length × 230 μm in width) systematically 2 μm at a time for a total depth of 40 μm using confocal microscopy. These 20 images were then volumetrically stacked into one 3D image. Therefore, each 3D volumetric image was 230 μm (length) × 230 μm (width) × 40 μm (depth). Tissue analysis: Two different methods were used to assess the number of capillaries in each 3D volumetric stack.
-
1.
We measured total capillary fluorescence intensity (green signal from lectin) from our reconstituted 3D volumetric stacks using the Zeiss software.
-
2.
We also used a nonfluorescence intensity-based quantification method by placing a grid consisting of 2,070 random and evenly distributed points over each 3D lectin volumetric stack and measured the number of points that hit a capillary, which was then divided by the total number of points that hit the RV tissue, as described in [13].
Confocal microscopy
Imaging was performed using a Zeiss LSM 510 confocal microscope (Carl Zeiss). Tetramethyl rhodamine methylester (TMRM) (10 nM, Molecular Probes) to measure mitochondrial membrane potential and MitoSOX (5 μM, Molecular Probes) to measure mitochondria-derived reactive oxygen species like superoxide (mROS) were used as previously described [12, 14, 15]. TMRM and MitoSOX intensity was measured in five random fields per RV per animal. For all TMRM and MitoSOX imaging, the same vials and preparations of these positively charged dyes were used for the staining of the excised endocardium tissues of the free-RV wall (which were similar in size and weight among all animals and experiments). In addition, the excitation and emission remained identical for all animals. For immunofluorescence, to limit any variability between the animals, all RV tissues were sliced from the endocardial side of the RV at 5-μm-thick slices and fixed in 4 % paraformaldehyde. Five-micrometer-thick slices of tissue, in addition to the functional mitochondria-specific dyes, were also stained as needed with appropriate primary and secondary antibodies and imaged using confocal microscopy. All experiments included secondary antibody-only staining, which in all presented experiments showed no signal, supporting the specificity of the antibodies used. For all immunofluorescence data, a minimum of five animals was used per group on at least five separate 5-μm-thick slices of tissue for each animal and at least five random images for each slice.
Immunoblotting
RV tissues were collected, and immunoblotting was performed on samples from four to five rats/group (25 μg protein in sample/lane). The films were digitized and quantified using 1D Image Analysis Software (Kodak, Rochester, NY). HIF1α, vascular endothelial growth factor (VEGF), stromal-derived factor 1 (SDF1), glucose transporter 1 (Glut1), heme-oxygenase-1 (HO-1), von Willebrand factor (vWF), p53, p21, pyruvate dehydrogenase kinase (PDK) I, PDKII, PDKIV, and actin were purchased from Santa Cruz Biotechnology and used at the recommended dilution. Expression was normalized to actin to correct for loading differences.
Positron emission tomography imaging
Positron emission tomography (PET) imaging was performed using a Triumph PET/SPECT/CT system (GE Healthcare, Northridge, CA, USA). Rats were injected with 500 μCi of fludeoxyglucose (FDG) and imaged 60 min after FDG injection. PET data acquisition images are shown using a pseudo-color map (red indicates high glucose uptake). Data were quantified as mean PET signal by manual segmentation of the RV using computed tomography (CT). Mean data are displayed as RV signal/LV signal to control for potential differences in FDG injection and imaging, as the left ventricle is typically considered unaltered in PAH animals, at least in terms of structural and functional remodeling.
Quantitative RT-PCR
Samples were added to a microwell plate with TaqMan probes and RT-PCR reagents (Applied Biosystems, Foster City, CA). Quantitative RT-PCR (qRT-PCR) was performed with an ABI PRISM 7700 Sequence Detector (Applied Biosystems, Foster City, CA), and primers for rat SDF1, VEGF, and 18s (housekeeping gene) were used.
Statistics
Values are expressed as mean ± SEM. Inter-group differences were appropriately assessed by one-way ANOVA with a Fisher's least significance test post hoc analysis (SPSS 19). p < 0.05 was considered significant.
Results
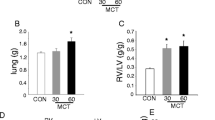
We used the MCT-induced PAH model of RV failure [16, 17]. Although this model has been criticized that it does not exactly recapitulate the human PAH pathology (for example, lack of plexogenic arteriopathy) and that myocarditis is evident in both the RV and LV [18], it does mimic other important aspects of human PAH, particularly in regard to RV failure. For example, it is characterized by a relatively rapid clinical deterioration (6 weeks) and almost 100 % mortality due to RV failure, following a phase of relative stability; it is also characterized by a strong inflammatory environment, similar to human PAH. We defined the decompensation stage based on (a) a decrease in RV systolic pressure (i.e., pump failure) and cardiac output while mean PA pressure remains elevated and RV remodeling continues to progress and (b) development of clinical signs of heart failure like ascites and an overall weight loss despite the fluid retention. Based on preliminary longitudinal weekly hemodynamic and clinical evaluation of rats following an injection of MCT, we found that the transition to decompensation takes place after the fourth week of MCT-PAH. Thus, we studied three groups of rats: (1) baseline (2) compensated RVH (cRVH) during the third and fourth week of MCT-PAH, and (3) decompensated RVH (dRVH) during the fifth and sixth week of MCT-PAH. During these stages, we assessed right-sided pressures both invasively, with measurement of the RV systolic pressure (RVSP) and mean PA pressure (PAP), and noninvasively with Doppler echo measurement of the pulmonary artery acceleration time (PAAT, a clinically used parameter that is inversely related to the mean PA pressure), and cardiac output, RV/LV + septum weight, body weight, ascites, and mortality.
In cRVH, the RVSP, mean PAP, PAAT, and RVH worsened significantly compared to baseline while the cardiac output and weight showed a trend to decrease, but were not statistically significant (Fig. 1a–c). There was no ascites, and none of the animals died. In dRVH, the RVSP and cardiac output decreased significantly whereas the mean PAP remained elevated (Fig. 1a–c). Ascites was present in all rats while the body weight decreased significantly (Fig. 1c). There was a rapid increase in mortality in this stage, reaching 90 % by the end of the sixth week. This clinical evolution in rat MCT-PAH mimics that of human PAH, with the cRVH stage mimicking WHO functional class I and II (minimal symptoms with exertion) and the dRVH mimicking WHO class III and IV (severe symptoms even at rest with overt signs of heart failure and high mortality). In addition, LV function as assessed by LV ejection fraction, fractional shortening, and mass remained similar between MCT animals and baseline control animals (Supplementary Fig. 1A–B). This model of the natural history of RV failure allowed us to obtain RV tissues from the three groups and perform a mechanistic comparison.

A rat model of the natural history or RV hypertrophy and failure. a The relationship between RV function (RVSP) and RV weight is plotted. cRVHs (compensated) have increased RV size associated with increased RVSP, while dRVHs (decompensated) have increased RV size but decreased RVSP (n = 6 animals for baseline, 10 for cRVH, and 9 for dRVH). b Representative hemodynamic traces for the pulmonary artery, right ventricle, and right atrium of a baseline control animal. By slowly pulling back the catheter from the PA position, we are able to record mean PA pressure, RVSP, and RV diastolic pressure (RVDP) for the same animal. c Animals with a cRVH have increased RVSP, mean RV pressure, RV mass, and RV/LV + septum ratio compared to baseline. Animals with a dRVH have decreased RVSP and increased RV mass and RV/LV + septum compared to cRVH. Animals with a dRVH also have a trend for increased RVDP compared to baseline animals. While cardiac output and body weight remain unchanged in cRVH, dRVH animals have decreased cardiac output and body weight, in the presence of ascites and malaise. Both cRVH and dRVH animals had decreased levels of PAAT and increased mean pulmonary artery pressure compared to baseline, indicating the development of pulmonary arterial hypertension; n = 6–10 animals per group for all parameters studied, *p < 0.05 vs. baseline, # p < 0.05 vs. cRVH
We have previously shown that a mitochondrial hyperpolarization and a decrease in mROS, both indicators of mitochondrial function and compatible with a mitochondrial suppression of glucose oxidation and a switch to a glycolytic phenotype, characterize RVH tissues (studied around the third week of MCT-PAH) [19]. We performed TMRM and MitoSOX imaging of intact RVs immediately following euthanasia. These positively charged dyes are preferentially taken up by the negatively charged mitochondria and detect dynamic changes in mitochondrial membrane potential (ΔΨm) and mROS levels. ΔΨm was increased in cRVH and remained elevated in dRVH, compared to baseline (Fig. 2a). mROS on the other hand were decreased in cRVH compared to baseline, but increased significantly in dRVH (Fig. 2b). To evaluate if the increase in mROS was also due to a decrease in anti-oxidants, we measured levels of HO-1. There were no major differences in HO-1 levels between cRVH and dRVH animals (Fig. 2c and Supplementary Fig. 2), suggesting a primary increase in mROS.

Differences in mitochondrial function between the cRVH and dRVH myocardium. a Freshly excised RVs from cRVH and dRVH animals show hyperpolarized mitochondrial membrane potential compared to baseline RVs (TMRM: red, nuclear stain DAPI: blue; n = 5 animals per group, *p < 0.05 vs. baseline). b Freshly excised RVs from cRVH animals show decreased levels of mitochondrial-derived reactive oxygen species (i.e., superoxide) measured by MitoSOX (red, nuclear stain DAPI: blue) compared to baseline RVs. dRVH tissues show increased levels of mROS compared to cRVH (n = 5 animals per group, *p < 0.05 vs. baseline, # p < 0.05 vs. cRVH). c Protein levels of the anti-oxidant HO-1 are increased in the RVs of cRVH and dRVH animals compared to baseline (n = 4 animals for baseline, 5 for cRVH, and 5 for dRVH). Immunoblots (top) and quantified mean data normalized to actin (bottom) are shown. On top of each band representing each rat, we indicate the week post-MCT at which its RV was harvested. Also, see Ponseau-S for additional loading control in Supplementary Fig. 2, which shows a similar pattern to actin
We have recently shown that suppressed mitochondrial function and decreased mROS increase HIF1α activity and glucose uptake in cancer (due to the well-described HIF1α-driven up-regulation of glucose transporters) [12]. To assess HIF1α activity in vivo, we measured nuclear localization of HIF1α in baseline, cRVH, and dRVH tissues. Nuclear levels of HIF1α (immunofluorescence) and overall protein levels (immunoblots) along with mRNA levels (qRT-PCR) were increased in cRVH compared to baseline, but were decreased again in the dRVH myocardium (Fig. 3a–b and Supplementary Fig. 3A). In addition, the HIF1α-regulated protein Glut1 followed a similar pattern to HIF1α (immunoblot) and in the same cells with HIF1α induction; there was an increase in Glut1 levels and the clear presence in myosin heavy chain (MHC)-positive cardiomyocytes (immunofluorescence) (Fig. 3b and Supplementary Fig. 3B). These all suggest that HIF1α activity was effectively decreased in dRVH compared to cRVH animals.

HIF1α protein levels and activity are decreased in dRVH compared to cRVH. a Nuclear levels of HIF1α (HIF1α: red, nuclear stain DAPI: blue) are increased in the RVs of cRVH animals compared to baseline while dRVH animals show decreased nuclear levels of HIF1α compared to cRVH animals, as measured by confocal microscopy and immunofluorescence. Representative confocal images are shown with lower magnification (above) and a higher magnification of the white box (below) (n = 5 animals per group, *p < 0.05 vs. baseline, # p < 0.05 vs. cRVH). b Overall protein levels of HIF1α and its downstream target Glut1 are increased in the RV of cRVH animals compared to baseline, while dRVH animals show decreased protein levels of HIF1α and Glut1 compared to cRVH, as measured by immunoblots. Each animal represents one band (n = 4 animals for baseline, 5 animals for cRVH, and 5 animals for dRVH). On top of each band, we indicate the week post-MCT at which its RV was harvested. Nuclear levels of HIF1α (HIF1α: red, nuclear stain DAPI: blue) are increased in the same cells which show increased Glut 1 (green) (see white arrows) as indicated by confocal microscopy and immunofluorescence (top images). In addition, nuclear levels of HIF1α (HIF1α: red, nuclear stain DAPI: blue) are present in cardiomyocytes as it colocalizes with myosin heavy chain (MHC, green) positive cells (bottom images). For similar photomicrographs in the baseline and dRVH groups (which do not have significant Glut1 expression and nuclear HIF1α) see Supplement Fig. 3B
Activation of HIF1α is prevalent in cancer or vascular cells that undergo a switch from a mitochondria-based glucose oxidation to a glycolytic phenotype (Warburg effect) [20–22], a metabolic switch that has also been described in RVH [9, 10]. This switch to glycolysis is associated with an up-regulation of glucose uptake, shown by the increased uptake of glucose measured by 18fludeoxyglucose–PET imaging in animal and human RVH [10, 23]. We performed in vivo 18fludeoxyglucose–PET–CT imaging, and similar to the Glut 1 levels in Fig. 3b, we observed a significant increase in glucose uptake in cRVH compared to baseline, which was reversed in dRVH (Fig. 4).

The dRVH has decreased RV glucose uptake and HIF1α activation compared to cRVH. Glucose uptake in the RVs of cRVH animals is increased compared to baseline; dRVH, on the other hand, has decreased glucose uptake compared to cRVH, as measured by micro-PET–CT imaging with [18F]-deoxyglucose. Pseudo-color map indicates FDG levels (red is high and purple is low levels of FDG uptake, n = 5 animals per group, *p < 0.05 vs. baseline, # p < 0.05 vs. cRVH)
We then measured mRNA and protein levels of two HIF1α-regulated products important in angiogenic signaling: VEGF and SDF1. Both VEGF and SDF1 mRNA and protein were increased in cRVH compared to baseline, but were decreased in the dRVH tissues (Fig. 5a–b).

HIF1α products VEGF and SDF1 are decreased in dRVH compared to cRVH. a mRNA levels of VEGF and SDF1 are increased in the RVs of cRVH animals compared to baseline while they are decreased in dRVH animals compared to cRVH animals, measured by qRT-PCR (n = 5 animals per group, *p < 0.05 vs. baseline, # p < 0.05 vs. cRVHs). b Protein levels of VEGF and SDF1 are increased in RVs of cRVH animals compared to baseline but decreased in dRVH animals compared to cRVH animals, measured by immunoblots. Each animal represents one band (n = 4 animals for baseline, 9 animals for cRVH, and 6 animals for dRVH, *p < 0.05 vs. baseline, # p < 0.05 vs. cRVHs). On top of each band, we indicate the week post-MCT at which its RV was harvested. Note that we added three rats out of our protocol from week 2 post-MCT in order to show a potential gradient in the increase of these HIF1α products earlier than week 3. The data confirm that the increase in the angiogenesis signaling occurs indeed within week 3 and 4. Immunoblots of VEGF and SDF1 are shown above, and normalized data to actin are shown below
To assess RV perfusion and angiogenesis, we injected lectin (a natural ligand to endothelial cells that when injected in vivo binds to endothelial cells of perfused vessels and can be imaged by fluorescence postmortem) into the jugular vein of rats prior to euthanasia. To measure capillaries (i.e., angiogenesis) and not arterioles or larger vessels, we used a magnification on our confocal microscope that allows the capture of a one-cell-layer thickness plane imaging as shown in Fig. 6a. In the smaller magnifications in the figure, one can see the larger vessels and arterioles (arrowheads) in addition to the capillary network (shown by white arrows). In the larger magnifications (one-cell-layer thickness plane), the vast majority of the signal comes from the capillaries, and by using “stacked imaging,” a volumetric assessment of several planes of images can be performed as can be observed in Supplemental Videos 1–3. By comparing similar volumes of tissues (230 μm × 230 μm × 40 μm), we measured the amount of capillaries in the different stages of RVH. We used two different methods to assess capillaries. We measured the overall fluorescence intensity of our stacked images, and we also measured the number of randomly distributed points that directly overlapped with a capillary (see “Methods”). The second method was used to address any potential confounding factor due to differences in lectin binding or uptake between animals. For example, a decrease in the fluorescence signal may not be due to decreased endothelial cells but due to decreased fluorescence signal despite the same number of capillaries. With the second method, we addressed this since we did not quantify the fluorescence intensity (like in the first method), but we directly measured the actual number of capillary structures. Capillary density was increased in the RVs of cRVH animals compared to baseline animals, but this was decreased in dRVH animals, as shown by both methods that showed similar results (Fig. 6b and Supplementary Fig. 4).

Capillary perfusion is decreased in the dRVH compared to cRVH. a Lectin fluorescence (injected intravenously prior to euthanasia, thus marking the endothelium of perfused vessels) at a lower magnification shows branch points from medium and small arteries (arrowheads) to capillaries at the last visible branch point (left). Capillaries are shown by white arrows (middle). High magnification (right) shows that the capillaries measured at this magnification are one-cell-layer thick as assessed by individual endothelial cells as seen by the blue nuclear stain DAPI. b Capillary networks at high magnification marked by lectin fluorescence were increased in the RVs of cRVH animals compared to baseline. dRVH animals had decreased lectin fluorescence at high magnification at the one-cell-layer thickness level, in the RV compared to cRVH animals (n = 5–10 animals/group, *p < 0.05 vs. baseline, # p < 0.05 vs. cRVH; bottom left). In order to show the trend of decreased lectin perfusion in the RV, we also analyzed animals within each group according to the week post-MCT injection, shown in bottom right [n = 5 animals for baseline, 5 animals at 3 weeks, and 5 animals at 4 weeks post-MCT (i.e., cRVH); and 5 animals at 5 weeks and 5 animals at 6 weeks post-MCT (i.e., dRVH)]. Note that a trend for decreased angiogenesis begins within week 4 but continues to decrease entering week 5, which marks the “clinical” definition of dRVH in our model. Representative photomicrographs and quantified mean data are shown. c Protein levels of the endothelial marker vWF are increased in the RVs of cRVH animals compared to baseline and decreased in dRVH animals compared to cRVH animals, measured by immunoblots (n = 5 animals per group; *p < 0.05 vs. baseline, # p < 0.05 vs. cRVH). On top of each band representing each rat, we indicate the week post-MCT at which its RV was harvested. Immunoblots and quantified mean data normalized to actin are shown below
Using immunoblots, we also measured the endothelial marker vWF and observed a similar pattern (Fig. 6c). However, the differences in vWF may also be due to altered endothelial cell proliferation rates or induction of apoptosis. To exclude the possibility that the decrease in capillaries or the decrease in RV function was also due to an increase in capillary or myocyte death, respectively, we assessed apoptosis in our samples. We observed a minimal increase (∼1 %) in apoptosis in the RV tissues of dRVH compared to cRVH animals (Supplementary Fig. 5). This suggests that apoptosis is not a major determinant for the decrease in the observed capillary density or functional decline at least at the stage of the disease that we studied.
What is the cause of the dynamic changes in HIF1α? We have recently described a number of mechanisms by which mitochondrial signals can directly regulate HIF1α, including (a) direct effects of mROS on specific sulfhydryl groups on HIF1α, inhibiting its ability to bind DNA [24–26]; (b) direct effects of mitochondria-derived H2O2 (which is more stable and diffusible compared to superoxide) and α-ketoglutarate on prolyl-hydroxylases, which hydroxylate HIF1α and signal it for ubiquination [27–30]; and (c) indirect effects driven by a mROS-mediated activation of the redox-sensitive p53 [31–34], an inhibitor of HIF1α signaling [35–38]. Similar to the mROS levels observed in Fig. 2b, nuclear levels of p53 (i.e., an index of its activation) are increased in dRVH compared to cRVH (Fig. 7). To further assess p53 activity, we measured the levels of the p53-target gene, p21, and observed a slight increase in the RVs of dRVH animals compared to cRVH animals (Fig. 7). Recently, p53 was shown to decrease the expression of PDK, a mitochondrial protein that phosphorylates and inhibits pyruvate dehydrogenase (PDH), suppressing glucose oxidation and mitochondrial function. In addition, HIF1α can directly increase the expression of PDK. Of the four known PDK isoenzymes, PDKI–II are ubiquitously expressed, PDKIV is primarily expressed in the heart, and PDKIII is primarily expressed in the testis [39]. In keeping with the changes in p53 and HIF1α, the expression of PDK I and II first increased in cRVH and then decreased in dRVH, while PDKIV showed a continuous decline (Fig. 8).

Nuclear levels of p53 are increased in the dRVHs compared to cRVHs. dRVH myocardium has increased nuclear levels of p53 (p53: green, nuclear stain DAPI: blue) compared to cRVH, measured by confocal microscopy and immunofluorescence. Representative confocal images at a low (top) and high (bottom) magnification are shown. White arrows denote nuclear localization of p53. Quantified data are shown to the right (n = 5 animals/group, *p < 0.05 vs. baseline). Protein levels of p21 (a p53 gene product) are increased in the RVs of dRVH animals compared to cRVH animals, measured by immunoblots. Immunoblots for each individual animal and the week post-MCT in which the RV was harvested are shown above (n = 4 animals for baseline, 5 animals for cRVH, and 5 animals for dRVH

PDK are decreased in the dRVHs compared to cRVHs. dRVH myocardium has decreased levels of PDKI, PDKII, and PDKIV compared to cRVH, measured by immunoblots (top). Quantified mean data normalized to actin are shown in the bottom (n = 4 animals for baseline, 5 animals for cRVH, 5 animals for dRVH; *p < 0.05 vs. cRVH). On top of each band representing each rat, we indicate the week post-MCT at which its RV was harvested. The actin for PDKII is identical to the one presented in Fig. 7 for p21 as the same membrane was used to probe for both proteins, which are of completely different molecular weights
Discussion
Here, we established a model of the natural history of RV hypertrophy and failure in PAH. We showed that while the glycolytic shift in cRVH is associated with an appropriate activation of the HIF1α axis promoting angiogenesis, the loss of this mitochondrial-metabolic remodeling marks the transition toward dRVH, where the suppression of angiogenesis may drive the accelerated decline of function in an RV that is still hypertrophied but ischemic (Fig. 9). We believe our work may offer clarity to the role of the previously described metabolic shift in animal and human RVH. While this cancer-like shift to glycolysis, due to suppressed mitochondrial function, may be beneficial early in the response of the RV to increased afterload, this is not sustained as the RVH progresses. This knowledge will be important in the application of metabolic modulators as potential treatments of RV failure. In addition, our work may clarify the interpretation of important imaging approaches (like PET) or metabolism-based biomarkers in RV failure.

A mitochondria–HIF1α axis in cRVH and dRVH. In cRVH, low levels of mROS result in suppression of p53 and induction of HIF1α, increasing PDK expression and decreasing the glucose oxidation (GO) to glycolysis (GLY) ratio. The activation of HIF1α increases the levels of proangiogenic factors and chemokines like vascular endothelial growth factor (VEGF) and stromal-derived factor 1 (SDF1), increasing angiogenesis. In dRVH, the increase in mROS results in activation of p53 and suppression of HIF1α, decreasing PDK expression, reversing the glycolytic phenotype, and suppressing angiogenesis in the hypertrophied myocardium, contributing to ischemia and RV failure. Note a potential positively reenforced feedback loop between PDK and p53-HIF1α, “trapping” the RV in sustained decompensation
Our study is in keeping with recent work by Boggard et al., who suggested that capillary density in RV failure is decreased [6]. In that study, the investigators did not provide a precise mechanism for this, and also, instead of studying the role of angiogenesis longitudinally in the same model (as in our study), they compared RVs from the PA banding model (which shows more prolonged RV compensation in response to increased afterload) to a sugen-hypoxia PAH model of a failing RV [6]. The results from that study were also difficult to interpret given the fact that the sugen compound (which was given systemically) is a VEGF receptor antagonist and thus was directly interfering with the regulation of angiogenesis in these RVs.
Our study suffers from a potential mechanistic weakness in that we do not establish a direct causal relationship between the mitochondria signals, the HIF1α axis, and RV failure, but rather provide “associative” findings. On the other hand, all the parameters that we studied follow an impressively similar temporal pattern, suggesting a direct and strong association. More importantly, our recent publications have characterized in detail a direct causal relationship between mitochondrial suppression (with a resulting glycolytic shift) and HIF1α-driven angiogenesis [12]. Therefore, we believe that it is the pseudo-hypoxic signaling due to mitochondrial suppression and not a direct hypoxic signaling that results in the observed HIF1α activation in our cRVH animals.
A recent study showed that in the dRVH, there were decreased mRNA levels of mitochondrial proteins involved in glucose oxidation and the Krebs cycle [40], while we suggest an increase in mitochondrial function in dRVH. A potential explanation may be differences in the models studied as the authors had compared a PA banding model of cRVH to a sugen-hypoxia model of dRVH [40], while we used a single model and serial measurements for both cRVH and dRVH, mimicking perhaps the natural history of clinical RV failure. Our data suggest that perhaps the dRVH myocardium attempts to defend the decreased contractility by improving energetic efficiency and activating mitochondria, but this comes at the expense of suppressed angiogenesis.
Our work points to a potential critical role for mROS generation in dRVH (as opposed to their decrease in cRVH compared to baseline), which appears to drive the inhibition of HIF1α. The trigger for this significant change in the regulation of mROS production that takes place at the transition from cRVH to dRVH is unknown. It remains to be determined whether this increase in mROS is a part of an overall reversal of the mitochondrial/metabolic remodeling or is due to a mechanism specific to the regulation of the mROS in the electron transport chain of RV myocardial mitochondria and its dismutation by the mitochondrial manganese superoxide dismutase. Although our PET data would suggest that the glycolytic shift is overall reversed in dRVH, these data, although intriguing, need to be interpreted with caution, as the decrease in the PET signal could also be due to the decreased delivery of the FDG, as the dRVH myocardium becomes more ischemic. Either way, however, our PET data also show that this change (whatever its exact cause may be) does happen in vivo at precisely the same time and with the same pattern as all the other hemodynamic and tissue parameters that we studied. Intriguingly, our work is relevant to recent clinical work, showing that PAH patients have increased PET signal in the RV and induction of nuclear HIF1α in the myocardium (taken from autopsy) compared to controls [41], although in this study, there was also no attempt to identify the RVs as compensated vs. decompensated.
Our work also supports a recent study in which carvedilol, but not other beta blockers, resulted in prevention of the decline in RV function in animal models [42]. Our work suggests that the beneficial effects of carvedilol may be due to its unique ability (compared to other beta blockers) to also scavenge ROS [43, 44].
Our data agree with similar work in the LV, where the transition from compensated LVH to a decompensated LVH was associated with induction of p53, decreased HIF1α activity, and decreased angiogenesis [45, 46]. Mitochondrial signals can activate p53 through two pathways. First, by producing citrate, the mitochondria provide the cytosol and nucleus with acetyl-CoA in the presence of ATP-citrate lyase [47]. This is important since acetylation of p53 is necessary for its activation [48]. Second, mROS have also been shown to directly activate the redox-sensitive p53 [31–33, 36]. In other words, the mitochondrial suppression that underlies the glycolytic shift in cRVH could, at least in part, explain both the inhibition of p53 and the activation of HIF1α, and loss of this switch may explain the loss of a beneficial HIF1α activation and p53 inhibition in dRVH.
Both the increase in p53 activity and the inhibition of HIF1α in dRVH can promote inhibition of PDK expression, activating PDH, increasing glucose oxidation, and thus reversing the glycolytic shift that appears to be protective during cRVH. While the mitochondrial remodeling can potentially primarily drive these changes in p53 and HIF1α and thus in PDK, the changes in PDK in turn will sustain this mitochondria remodeling, thus “trapping” the dRVH in a positively reinforced vicious feedback loop (Fig. 9), perhaps explaining the rapid deterioration of RV function in animal and human pulmonary hypertension.
References
Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF (2009) The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 135:794–804
Haddad F, Ashley E, Michelakis ED (2010) New insights for the diagnosis and management of right ventricular failure, from molecular imaging to targeted right ventricular therapy. Curr Opin Cardiol 25:131–140
Haddad F, Hunt SA, Rosenthal DN, Murphy DJ (2008) Right ventricular function in cardiovascular disease, part I: anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation 117:1436–1448
Haddad F, Doyle R, Murphy DJ, Hunt SA (2008) Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation 117:1717–1731
McLaughlin VV, Presberg KW, Doyle RL, Abman SH, McCrory DC, Fortin T, Ahearn G (2004) Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 126:78S–92S
Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF (2009) Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 120:1951–1960
van de Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, Boonstra A, Marques KM, Westerhof N, Vonk-Noordegraaf A (2011) Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 58:2511–2519
Srivastava D (2006) Making or breaking the heart: from lineage determination to morphogenesis. Cell 126:1037–1048
Bishop SP, Altschuld RA (1970) Increased glycolytic metabolism in cardiac hypertrophy and congestive failure. Am J Physiol 218:153–159
Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang HJ, Haber I, Rehman J et al (2010) The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med 88:47–60
Gomez A, Bialostozky D, Zajarias A, Santos E, Palomar A, Martinez ML, Sandoval J (2001) Right ventricular ischemia in patients with primary pulmonary hypertension. J Am Coll Cardiol 38:1137–1142
Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, McMurtry MS, Michelakis ED (2013) Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 32:1638–1650
Bruel A, Oxlund H, Nyengaard JR (2005) The total length of myocytes and capillaries, and total number of myocyte nuclei in the rat heart are time-dependently increased by growth hormone. Growth Horm IGF Res 15:256–264
Sutendra G, Dromparis P, Bonnet S, Haromy A, McMurtry MS, Bleackley RC, Michelakis ED (2011) Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFalpha contributes to the pathogenesis of pulmonary arterial hypertension. J Mol Med (Berl) 89:771–783
Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC et al (2011) The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med 3:88ra55
Ceconi C, Condorelli E, Quinzanini M, Rodella A, Ferrari R, Harris P (1989) Noradrenaline, atrial natriuretic peptide, bombesin and neurotensin in myocardium and blood of rats in congestive cardiac failure. Cardiovasc Res 23:674–682
Muders F, Elsner D (2000) Animal models of chronic heart failure. Pharmacol Res 41:605–612
Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, Bogaard HJ (2012) The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol 302:L363–L369
Nagendran J, Gurtu V, Fu DZ, Dyck JR, Haromy A, Ross DB, Rebeyka IM, Michelakis ED (2008) A dynamic and chamber-specific mitochondrial remodeling in right ventricular hypertrophy can be therapeutically targeted. J Thorac Cardiovasc Surg 136:168–178, 178 e161-163
Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G et al (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11:37–51
Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, Dyck JR, Michelakis ED (2010) Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med 2:44ra58
Dromparis P, Sutendra G, Michelakis ED (2010) The role of mitochondria in pulmonary vascular remodeling. J Mol Med (Berl) 88:1003–1010
Oikawa M, Kagaya Y, Otani H, Sakuma M, Demachi J, Suzuki J, Takahashi T, Nawata J, Ido T, Watanabe J et al (2005) Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol 45:1849–1855
Huang LE, Arany Z, Livingston DM, Bunn HF (1996) Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem 271:32253–32259
Salceda S, Caro J (1997) Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 272:22642–22647
Wang GL, Jiang BH, Semenza GL (1995) Effect of altered redox states on expression and DNA-binding activity of hypoxia-inducible factor 1. Biochem Biophys Res Commun 212:550–556
Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS (2005) Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab 1:409–414
Denko NC (2008) Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8:705–713
Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC (2005) Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab 1:393–399
MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG, Gottlieb E (2007) Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol 27:3282–3289
Huang C, Zhang Z, Ding M, Li J, Ye J, Leonard SS, Shen HM, Butterworth L, Lu Y, Costa M et al (2000) Vanadate induces p53 transactivation through hydrogen peroxide and causes apoptosis. J Biol Chem 275:32516–32522
Wang S, Leonard SS, Ye J, Ding M, Shi X (2000) The role of hydroxyl radical as a messenger in Cr(VI)-induced p53 activation. Am J Physiol Cell Physiol 279:C868–C875
Xie S, Wang Q, Wu H, Cogswell J, Lu L, Jhanwar-Uniyal M, Dai W (2001) Reactive oxygen species-induced phosphorylation of p53 on serine 20 is mediated in part by polo-like kinase-3. J Biol Chem 276:36194–36199
Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS (2003) Glycogen synthase kinase-3beta (GSK3beta) binds to and promotes the actions of p53. J Biol Chem 278:48872–48879
Schmid T, Zhou J, Kohl R, Brune B (2004) p300 relieves p53-evoked transcriptional repression of hypoxia-inducible factor-1 (HIF-1). Biochem J 380:289–295
Vousden KH, Ryan KM (2009) p53 and metabolism. Nat Rev Cancer 9:691–700
Kaluzova M, Kaluz S, Lerman MI, Stanbridge EJ (2004) DNA damage is a prerequisite for p53-mediated proteasomal degradation of HIF-1alpha in hypoxic cells and downregulation of the hypoxia marker carbonic anhydrase IX. Mol Cell Biol 24:5757–5766
Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A (2000) Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev 14:34–44
Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM (1995) Diversity of the pyruvate dehydrogenase kinase gene family in humans. J Biol Chem 270:28989–28994
Gomez-Arroyo J, Mizuno S, Szczepanek K, Van Tassell B, Natarajan R, dos Remedios CG, Drake JI, Farkas L, Kraskauskas D, Wijesinghe DS et al (2012) Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ Heart Fail 6:136–144
Lundgrin EL, Park MM, Sharp J, Tang WH, Thomas JD, Asosingh K, Comhair SA, Difilippo FP, Neumann DR, Davis L et al (2013) Fasting 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann Am Thorac Soc 10:1–9
Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN et al (2010) Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med 182:652–660
Yasunari K, Maeda K, Nakamura M, Yoshikawa J (2002) Carvedilol inhibits pressure-induced increase in oxidative stress in coronary smooth muscle cells. Hypertens Res 25:419–425
Nakamura K, Kusano K, Nakamura Y, Kakishita M, Ohta K, Nagase S, Yamamoto M, Miyaji K, Saito H, Morita H et al (2002) Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation 105:2867–2871
Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M et al (2007) p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 446:444–448
Song H, Conte JV Jr, Foster AH, McLaughlin JS, Wei C (1999) Increased p53 protein expression in human failing myocardium. J Heart Lung Transplant 18:744–749
Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324:1076–1080
Tang Y, Zhao W, Chen Y, Zhao Y, Gu W (2008) Acetylation is indispensable for p53 activation. Cell 133:612–626
Acknowledgments
This study was funded by grants from the Canadian Institutes for Health Research (CIHR), the Heart and Stroke Foundation of Canada, and the Mazankowski Heart Institute–Hospital Foundation.
Conflict of interest
The authors have no conflicts to disclose.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 763 kb)
Supplementary Videos 1–3
Stereological representation of capillary networks in the right ventricle at a high magnification marked by lectin fluorescence (green) and the nuclear stain DAPI (blue) are shown. Also please refer to Fig. 6B for quantified mean data. Supplementary Video 1 is a stereological representation of the right ventricle of a baseline control animal, while Supplementary Video 2 is a stereological representation of the right ventricle of a cRVH animal and Supplementary Video 3 is a stereological representation of the right ventricle of a dRVH animal. (MOV 279722 kb)
Supplementary Video 2
(MOV 279722 kb)
Supplementary Video 3
(MOV 280262 kb)
Rights and permissions
About this article
Cite this article
Sutendra, G., Dromparis, P., Paulin, R. et al. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med 91, 1315–1327 (2013). https://doi.org/10.1007/s00109-013-1059-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-013-1059-4