
Abstract
Right ventricular (RV) function is a strong independent determinant of outcomes in a broad range of cardiopulmonary diseases. Despite this recognition, the underlying pathobiology of RV failure remains poorly understood and no RV-specific therapies exist for RV dysfunction. The variable response of RV function to different medical therapies and among etiologies of pulmonary hypertension suggests that elevated afterload is not the sole determinant of RV function. Various molecular mechanisms have been identified that contribute to RV failure. RV ischemia, neurohormonal activation, maladaptive myocardial hypertrophy, metabolic remodeling, and mitochondrial dysfunction are key pathogenic mechanisms that have been demonstrated in both experimental models and humans with RV dysfunction. Genetics may also contribute to RV dysfunction as in heritable pulmonary arterial hypertension and arrhythmogenic RV dysplasia. Metabolic dysregulation and neurohormonal antagonism are currently being tested as RV-specific therapeutic targets in PAH. More detailed understanding of the molecular underpinnings of RV failure will lead to additional therapeutic avenues. Molecular imaging tools such as positron emission tomography may provide a more mechanistic understanding of RV pathophysiology in vivo and allow translation of basic science findings to humans.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Right Ventricular
- Pulmonary Arterial Hypertension
- Right Ventricular Function
- Right Ventricular Dysfunction
- Pulmonary Artery Banding
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Right ventricular (RV) function is a strong, independent prognostic indicator of outcomes in a number of disease states including valvular heart disease, ischemic and non-ischemic cardiomyopathy, pulmonary embolism, and pulmonary arterial hypertension (PAH), among others [1–4]. Despite the recognized importance of RV function in these diseases, the underlying pathobiology of RV failure is poorly understood [5]. Global RV function is primarily determined by three dynamic factors: preload, afterload, and myocardial contractility. The primary inputs of RV afterload are pulsatile reflections from the main pulmonary arteries (PA) and early bifurcations, impedance of the proximal PAs, and arteriolar resistance (pulmonary vascular resistance; PVR). RV contractility is a reflection of loading conditions, adrenergic state, heart rate, medications, metabolic status, and ventricular interdependence. How these three facets of RV function alter or are altered by molecular changes in the RV myocardium are little studied, but undoubtedly powerfully affect outcomes in situations of RV stress and may be independent targets of therapy. This chapter will focus on the pathobiology of right heart failure in chronic pulmonary hypertension and highlight areas of recent advances in our molecular understanding of RV function and dysfunction.
RV Functional Decline and Recovery are Highly Variable
RV failure is a heterogeneous clinical problem. Some patients develop severe RV failure at a given elevation in PA pressure and PVR whereas others maintain long-term preservation of RV function given the same hemodynamic profile. For example, many patients with congenital heart defects who develop Eisenmenger physiology maintain normal RV function for decades despite systemic PA pressures [6]. Relatively good outcomes in Eisenmenger patients are postulated to be related to the development of compensatory RV hypertrophy or persistence of the fetal gene program, but ultimately these mechanisms are not well understood. In many disease, the RV exhibits a remarkable capacity for functional recovery after insult, for example after RV myocardial infarction or pulmonary thromboendarterectomy [7]. A molecular understanding of the mechanisms mediating RV decline and recovery will improve our understanding of RV failure and aid in development of RV-targeted therapy.
RV Failure is in Part Independent of Pulmonary Hemodynamics
In diseases primarily affecting the pulmonary vasculature such as chronic thromboembolic pulmonary hypertension (CTEPH) and PAH, outcomes more closely mirror RV function and reverse remodeling than improvement in pulmonary hemodynamics [5]. There is increasing recognition that elevated RV afterload is not the sole determinant of RV failure and that RV function often declines despite significant improvement in pulmonary hemodynamics in response to medical therapy. Van de Veerdonk et al. showed that decline in RV function despite a clinically significant decline in PVR was associated with significantly worse survival in patients with PAH [8]. Additional evidence includes the good outcomes patients with pulmonic valve stenosis and Eisenmenger’s syndrome who develop adaptive RVH and the lack of RV failure in experimental models of RV pressure overload using pulmonary artery banding [6, 9]. These findings suggest that the development of RV failure depends not just on elevated afterload from pulmonary vascular resistance and large vessel stiffness but additional pathogenic mechanisms. Because RV functional decline is in part independent of pulmonary vascular disease, therapies directed at RV function may lead to improved outcomes. The lack of currently available RV-specific therapies stems from an incomplete understanding of the molecular mechanisms of RV failure.
Pathology of RV Failure
Despite well-characterized pulmonary vascular pathology, the pathology of RV failure has not been well studied. In addition to gross increase in mass, RV myocyte hypertrophy is well described in the context of PAH [5]. Changes in capillary density or size, the role of fibrosis and differences across the clinically variable causes of RV failure are little described in humans. Several causes of acute RV failure, such as pulmonary embolism and RV infarction are associated with RV myocardial necrosis [10], but this is not described in chronic causes of RV failure such as PAH. Little comparative information is available about RV pathology in the WHO Groups of pulmonary hypertension, but data from humans suggesting diseases such as scleroderma-associated PAH has disproportionate RV failure [11, 12] may point to different patterns of RV pathology in this disease.
Molecular Mechanisms of RV Failure
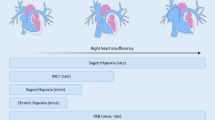
Several molecular mechanisms have been identified to contribute to RV failure in animal models and humans including myocardial ischemia, neurohormonal activation, metabolic dysregulation and mitochondrial dysfunction, and maladaptive myocyte hypertrophy. Ultimately, many of these processes potentiate one another leading to a cycle of worsening myocardial failure (Fig. 4.1). Chronic myocardial ischemia leads to mitochondrial dysfunction and abnormal energy substrate utilization that then fails to provide adequate ATP for efficient myocardial contraction. Ischemia is worsened by the development of maladaptive RVH and a compensatory increase in contractility is compromised by β(beta)-receptor down-regulation from chronic neurohormonal stimulation. Much progress has been made in our understanding of these processes in recent years with most available data coming from experimental models of PAH and human cardiac imaging.

Molecular mechanisms of RV failure. Alone or in combination, the various mechanisms pictured have been implicated in the development of RV failure. Several molecular mechanisms of RV failure such as RV ischemia and hypertrophy occur primarily as a result of elevated RV afterload. However, others such as metabolic dysregulation and mitochondrial dysfunction may by inherent features of PAH
Ischemia
Patients with PH develop increased myocardial wall stress due to increased RV pressure and dilation resulting in an increased myocardial oxygen demand [13]. A decrease in systemic blood pressure resulting from poor cardiac output combined with an increase in RV pressure augments a decrease in coronary perfusion pressure resulting in ischemia, often manifesting as chest pain in patients with PAH both at rest and with exercise [14]. Right ventricular myocardial ischemia has been documented in PAH using myocardial scintigraphy and correlates directly with increases in RV diastolic pressure [15]. Detailed study of coronary flow patterns in PAH demonstrates a decrease in systolic flow in the right coronary artery compared to healthy controls and a decrease in total flow with increasing RV mass, indicating an imbalance between myocardial supply and demand [16].
Additional factors may contribute to supply/demand mismatch in PAH such as coronary compression from increased wall tension, hypoxemia due to impaired gas exchange, microvascular dysfunction, and impaired NO signaling [17]. Capillary loss and failure of new capillary growth in proportion to myocyte hypertrophy have recently been shown to differentiate angio-proliferative models of experimental PAH from RV pressure-overload models. In an experimental model of PAH using the vascular endothelial growth factor (VEGF) receptor blocker SU5146 and hypoxia (SuHx model), RV failure was associated with decreased RV capillary density accompanied by a reduction in VEGF mRNA and protein transcription [9]. Capillary density and VEGF expression were unchanged in a model of early, adaptive hypertrophy using pulmonary artery banding, providing further evidence that RV failure is not governed solely by elevated afterload and highlighting the potentially critical role of decreased oxygen delivery in the failing right heart. Reductions in both VEGF and capillary density were shown in the monocrotaline model of PAH and genetically engineered reductions in VEGF have been shown to reduce capillary volume in the mouse myocardium. Capillary density is also decreased in RV myocardium from humans with PAH who died of RV failure [18]. These data suggest that VEGF production in PAH may be insufficient to induce adequate angiogenesis relative to cardiac hypertrophy, resulting in RV ischemia.
Neurohormonal Activation in RV Failure
Neurohormonal activation is likely both a cause and a consequence of RV failure. Elevated RV afterload results in an increase in norepinephrine to increase inotropy and renal vein hypertension decreases renal perfusion resulting in RAAS activation. As in left heart failure, the initially compensatory mechanisms of sympathetic system and RAAS activation ultimately become detrimental in patients with right heart failure. After prolonged stimulation, this results in down-regulation of β(beta)-receptors impairing RV inotropic reserve and worsening RV failure. There is abundant evidence of neurohormonal activation in patients with RV failure: increased heart rate with reduced heart rate variability [19], increased plasma norepinephrine levels [20], decreased β1-receptor density in the RV in PAH, and increased muscle sympathetic nerve activity [21]. In addition, hyponatremia, an indirect marker of renin-angiotensin-aldosterone systemic activation is associated with reduced survival and RV failure in PAH [22].
There are limited data on therapeutic interventions to blunt neurohormonal activation in RV failure. In the case of β-blockers, clinical dogma has held that patients with RV failure are heart rate dependent and beta-blockers would impair both chronotropic and inotropic reserve. However, recent evidence from preclinical models of RV failure suggests a potential benefit from beta-blockers. In the SuHx and monocrotaline PAH models, carvedilol, a β1,2- and α(alpha)1-blocker with potentially beneficial pleiotropic effects was found to improve RV function and increase exercise capacity compared to vehicle treated animals. These effects were associated with an increase in protein kinase G, decreased myocardial fibrosis and increased RV capillary density. Similarly, the β1-receptor blocker bisoprolol improved RV function in experimental PAH [23]. Early-phase clinical trials are now underway testing the effect of beta-blockade on RV function in patients with PAH. Elevated pulmonary aldosterone expression is present in PAH and correlates directly with endothelin-1 secretion [24]. Treatment with aldosterone antagonists in the SuHx and monocrotaline PAH models reduces pulmonary pressure and PVR without significant systemic side effects [25].
A common clinical dilemma in the treatment of RV failure is the choice of inotrope or vasopressor during acute decompensation. There are no specific recommendations in published guidelines and no clinical trial data to support evidence-based use of a specific inotrope. Recent findings regarding adrenergic remodeling in animal models of RVH may provide some guidance. PAH-associated (maladaptive) RVH as compared to RVH secondary to pulmonary artery banding is associated with downregulation of β1, α1, and dopamine-1 receptors resulting in reduced inotropic reserve. As a result of superior coupling to adenylyl cyclase, dobutamine outperformed dopamine as an RV inotrope in both in vivo and ex vivo models [26]. Larger clinical trials are needed to translate this data to humans, but this study demonstrates the influence of neurohormonal activation on myocardial response to inotropic therapy.
Right Ventricular Metabolism and Mitochondrial Function
In patients with PH, chronically increased pulmonary pressure and PVR results in a stimulus for compensatory RV hypertrophy (RVH), thereby increasing myocardial metabolic demand. Decreased coronary perfusion pressure and capillary rarefaction limit oxygen supply, leading to RV ischemia and oxygen supply/demand mismatch. Emerging evidence from both experimental models and human PAH suggests that the ability of the myocardium to maintain energy substrate flexibility in the setting of RVH and ischemia is an important determinant of RV failure. In the normal adult heart, fatty acid oxidation accounts for the majority of energy supply and metabolic flexibility exists to use glucose as an additional fuel source. Recent evidence suggests that RVH and RV failure are associated with increased utilization of glycolysis for ATP production, even in the setting of abundant oxygen when oxidative metabolism would otherwise be used [27, 28]. This process, well-described in cancer cells, is hypothesized to be advantageous because these cells are less reliant on oxygen for energy production and can therefore proliferate in regions of relative hypoxia [29]. Direct measurement of increased RV glycolysis has been demonstrated in the monocrotaline PAH model [30]. Increased glycolysis (and decreased glucose oxidation) in this model is shown to be due to increased pyruvate dehydrogenase kinase (PDK) activty, which inhibits conversion of pyruvate (the product of glycolysis) to acetyl CoA, the substrate for Krebs’ cycle initiation. Failure to produce additional ATP from glucose oxidation results in decreased oxygen consumption and impaired RV function. Increased RV glucose uptake in human PAH has been shown in several studies using 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET) [31–33]. Although this suggests an increase in glycolysis given findings in experimental PAH, it is difficult to draw definitive conclusions because FDG uptake does not directly measure glycolysis but simply glucose uptake. Combination of FDG with other PET tracers measuring oxidative metabolism may help determine the relative activity of glycolysis and fatty acid oxidation in the human RV.
Glucose oxidation and fatty acid oxidation are reciprocating processes in the mitochondria – as one increases the other decreases and vice versa through Randle’s cycle [34]. This feedback facilitates metabolic flexibility, which is particularly critical for myocardial function in times of nutritional restriction. This cycle has been exploited for therapeutic purposes in experimental PAH in which the PDK inhibitor dichloroacetate and fatty acid oxidation inhibitors trimetazidine and ranolazine have improved RV function [35, 36]. It is unclear whether therapeutic strategies that impair fatty acid oxidation either directly or by increasing glucose oxidation will benefit patients with PAH.
Recent evidence suggests that mitochondrial dysfunction is present in both adaptive and maladaptive RVH and forms the basis for the abnormal energy metabolism observed in RV failure described above. RV failure in the monocrotaline model of PAH is associated with decreased expression of genes required for fatty acid oxidation and mitochondrial biogenesis as well as reduction in mitochondria number per gram of tissue and oxidative capacity [37]. Similar mitochondrial dysfunction was not observed in a pulmonary artery banding model suggesting that mitochondrial metabolic remodeling may be an inherent feature of RV failure in PAH and not simply a consequence of elevated RV afterload. The observation of mitochondrial hyperpolarization in RV tissue from humans with PH further indicates the presence of mitochondrial dysfunction in RVH [38].
While changes in mitochondrial metabolism shifting from fatty acid oxidation to glycolysis in RV failure are clearly present, the underlying triggers for this shift are unknown. Understanding the influence of RVH on metabolic adaptation, and the potential for metabolic modulation as an RV-specific therapeutic strategy for RV failure are areas of intense investigation and hold promise for future therapies in RV failure from many causes, not just PAH.
Myocyte Hypertrophy
At the time of birth and switch from fetal to adult circulation patterns, the RV undergoes a profound shift from being a high pressure, high resistance pump to a low pressure, high flow conduit for blood to enter the pulmonary circulation. Mechanical changes including high oxygen tension in the lungs and closure of the patent ductus arteriosus facilitate this switch, but the molecular changes of the RV at this time are unknown. What is clear is that the RV in the adult is a thin-walled structure with a morphology that is described elsewhere in this edition. In situations of acquired increased PVR, the RV increases in size, i.e. hypertrophies, to transition from a flow conduit to a pressure pump. This switch is thought to be required to maintain cardiac output in the face of increased load stress and thus adaptive. However, over time, the RV often fails and thus transitions to maladaptive hypertrophy. In congenital heart disease lesions that include persistent elevations in pulmonary pressure after birth, the RV often retains the capacity to generate high pressures through persistent adaptive hypertrophy. This may underlie the well-described improved survival in congenital heart disease-associated PAH compared with idiopathic PAH in which the elevated pulmonary vascular resistance occurs in the adult circulation [39]. The molecular mediators of this shift from adaptive to maladaptive hypertrophy and RV failure are presently unknown, but animal models are beginning to provide some insight by comparing models of adaptive and maladaptive hypertrophy. Bogaard et al. has used pulmonary artery banding as a tool to study chronic pressure overload of the RV and compared the effects with the sugen + hypoxia model [9]. They found that in the pulmonary artery banding model, there is little mortality compared with the SuHx model, RV hypertrophy is present but to a lesser degree and there is less fibrosis in pulmonary artery banding. In the pulmonary artery banding model, cardiac output and tricuspid annular plane systolic excursion are preserved for up to 22 weeks. These studies have allowed better interpretation of animal models, suggesting that pulmonary artery banding is most informative as a model of early, adaptive hypertrophy. This paper suggests that angioproliferative pulmonary hypertension in SuHx is associated with capillary rarefaction in the RV, which is not present in pulmonary artery banding. The RV consequences of ischemia have been described and include normoxic HIF signaling [40] and mitochondrial metabolic changes discussed in the section on metabolism above. With inefficient production of ATP through glycolysis, there is postulated to be less substrate availability for myocardial cell growth required for hypertrophy. There is some animal data that dichloroacetate may reverse maladaptive hypertrophy associated with mitochondrial dysfunction in RV failure and ongoing human trials will provide much anticipated information about the utility of this other RV-directed metabolic interventions in the future [27].
Ischemia associated with reduced capillary volume may underlie the transition from adaptive to maladaptive hypertrophy, but alternative or contributory mechanisms have been implicated including altered electrophysiology [30] and perhaps geometry. Other hypertrophy triggers that are thought to be unrelated to ischemia have also been reported. Recent work by Nagendran et al. has demonstrated upregulation of endothelin-1 protein expression and endothelin receptor A in the hypertrophied human RV and in animal models of pulmonary hypertension [41]. Similarly, the phosphodiesterase 5 inhibitor sildenafil has been shown to increase cardiac index in its pivotal PAH trial [42]. Animal data suggests that increased NO signaling though PDE5 inhibition attenuates maladaptive RV hypertrophy in rodent models of right ventricular hypertrophy [43, 44]. The clinically evident RV effects of these disparate pharmaceutical classes has led to interesting insight into molecular basis of RV failure and may point to future therapeutics or new uses of older drug classes.
Other Causes of RV Failure
Additional contributing mechanisms for RV failure may include inflammation, oxidant stress, and fibrosis though the relative importance of these processes is not well understood. Myocardial fibrosis is present on cardiac MRI (CMR) in patients with RV failure as well as histology in experimental models of PAH. Fibrosis on CMR correlates with pulmonary hemodynamics and independently predicts clinical worsening in PAH [45]. Whether fibrosis is a consequence of chronic myocardial mechanical strain or myocardial ischemia [46] or an independent pathologic process as a result of endothelial cell dysfunction is not known [47, 48]. The role of sex hormones the development of RV failure may also be important given the observation that PAH is more prevalent in females but associated with worse outcomes in males. Recent data from our group has shown that testosterone negatively regulates right ventricular function in the pulmonary artery banding model of RV hypertrophy and that testosterone deprivation via castration is associated with prolonged survival in this model [49]. Castration was associated with reduced myocyte hypertrophy and reduced expression of natriuretic peptides associated with hypertrophy in this model. It is possible that the worse survival in male PAH patients is mediated by this negative effect of testosterone on RV hypertrophic responses.
Genetics of RV Failure
BMPR2
A major hindrance to the study of right heart failure has been the limitations of currently available animal models. Monocrotaline, SuHx and pulmonary artery banding all have heterogeneous effects on RV size and function and are of questionable relevance to human disease. Transgenic models would facilitate a more detailed molecular dissection of the signaling pathways key to development of RV failure. Archer et al. have used the fawn-hooded rat to identify the key metabolic derangements in RV failure [50]. We have recently described worse survival in patients with heritable PAH compared with idiopathic PAH that may be due to impaired RV compensation in heritable PAH [51, 52]. Heritable PAH is most commonly associated with mutations in the bone morphogenic protein receptor type 2 (BMPR2), for which there are transgenic rodent models [53, 54]. In unpublished data we have found evidence of reduced fatty acid oxidation intermediaries associated with lipid deposition in humans with heritable PAH and in transgenic mice with similar mutation that is universally expressed [55, 56]. Moreover, hypertrophic responses in this strain appear to be impaired. The use of transgenic models to study RV failure will facilitate a deeper molecular understanding of the mechanisms that drive development of this syndrome and potentially point to new, effective therapeutic targets.
ARVC/D
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ACRC/D) is an inherited cardiomyopathy characterized by replacement of myocardium with fibrofatty tissue [57]. The genetic underpinnings of ARVC/D primarily involve mutation in desmosomal proteins resulting in disruptions in cell-cell adhesion and intracellular signaling; over a dozen unique mutations have been identified to cause disease [58]. Clinical manifestations typically occur in early adulthood and often the first manifestation is life-threatening ventricular arrhythmia or sudden cardiac death. Although ARVD is not known to be common in PAH or other WHO pulmonary hypertension groups, it is a genetic cause of RV-related disease. Its study may aid in understanding how the RV fails in the absence of load stress, as patients with this condition do not have pulmonary hypertension.
Future Directions
Preclinical studies of metabolic modulation have shown promise for the treatment of RV failure and pilot clinical trials of metabolic therapy for RV failure in human PAH are underway (trials of carvedilol on clinicaltrials.gov: NCT01586156, NCT01723371; trial of dichloroacetate: NCT01083524). If successful, metabolic modulators may represent the first RV-specific heart failure therapies. Whether these therapies will be efficacious in RV failure of other etiologies is unknown. A parallel focus on RV function in addition to pulmonary vascular disease is critical given the strong prognostic power of RV function in PAH. Future clinical trials of pulmonary vasodilator therapies should also include RV-specific functional outcomes [59]. Additional clinical studies are needed to determine the optimal medical management for acute RV failure, including a direct comparison of different inotropic agents.
The current non-invasive evaluation of RV function with echocardiography and CMR is largely descriptive and does not allow for early detection of impending RV failure. Molecular imaging tools that provide a more mechanistic understanding of RV pathophysiology should be expanded and potentially extended into clinical practice. PET imaging with metabolic tracers such as FDG, 11C acetate, and others can provide detailed information about mitochondrial function and metabolic remodeling in the RV. PET may ultimately provide superior biomarkers and clinical trial endpoints of therapeutic efficacy compared to conventional metrics. Finally, given the impact of metabolic dysregulation on RV function, broad metabolite profiling may provide novel insights into the metabolic etiology of both acute and chronic RV failure. Because they are downstream of transcription, translation and posttranslational modifications, metabolites reflect early alteration in the body’s response to disease. A metabolomics approach has previously been used to detect early metabolic changes after myocardial infarction and identify markers cardiopulmonary fitness [60, 61] and may bear fruit in the study of RV failure.
References
Borer JS, Bonow RO. Contemporary approach to aortic and mitral regurgitation. Circulation. 2003;108(20):2432–8. doi:10.1161/01.CIR.0000096400.00562.A3.
Ghio S, Gavazzi A, Campana C, et al. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol. 2001;37(1):183–8.
Goldhaber SZ, Visani L, De Rosa M. Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER). Lancet. 1999;353(9162):1386–9.
Forfia PR, Fisher MR, Mathai SC, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med. 2006;174(9):1034–41. doi:10.1164/rccm.200604-547OC.
Voelkel NF, Quaife RA, Leinwand LA, et al. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation. 2006;114(17):1883–91. doi:10.1161/CIRCULATIONAHA.106.632208.
Hopkins WE, Ochoa LL, Richardson GW, Trulock EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. J Heart Lung Transplant. 1996;15(1 Pt 1):100–5.
Brittain EL, Hemnes AR, Keebler M, Lawson M, Byrd BF, Disalvo T. Right ventricular plasticity and functional imaging. Pulm Circ. 2012;2(3):309–26. doi:10.4103/2045-8932.101407.
van de Veerdonk MC, Kind T, Marcus JT, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol. 2011;58(24):2511–9. doi:10.1016/j.jacc.2011.06.068.
Bogaard HJ, Natarajan R, Henderson SC, et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120(20):1951–60. doi:10.1161/CIRCULATIONAHA.109.883843.
Watts JA, Marchick MR, Kline JA. Right ventricular heart failure from pulmonary embolism: key distinctions from chronic pulmonary hypertension. J Card Fail. 2010;16(3):250–9. doi:10.1016/j.cardfail.2009.11.008.
Tedford RJ, Mudd JO, Girgis RE, et al. Right ventricular dysfunction in systemic sclerosis associated pulmonary arterial hypertension. Circ Heart Fail. 2013;6(5):953–63. doi:10.1161/CIRCHEARTFAILURE.112.000008.
Mathai SC, Bueso M, Hummers LK, et al. Disproportionate elevation of N-terminal pro-brain natriuretic peptide in scleroderma-related pulmonary hypertension. Eur Respir J. 2010;35(1):95–104. doi:10.1183/09031936.00074309.
Hein S, Arnon E, Kostin S, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107(7):984–91.
Ross RS. Right ventricular hypertension as a cause of precordial pain. Am Heart J. 1961;61:134–5.
Gomez A, Bialostozky D, Zajarias A, et al. Right ventricular ischemia in patients with primary pulmonary hypertension. J Am Coll Cardiol. 2001;38(4):1137–42.
van Wolferen SA, Marcus JT, Westerhof N, et al. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J. 2008;29(1):120–7. doi:10.1093/eurheartj/ehm567.
Kajiya M, Hirota M, Inai Y, et al. Impaired NO-mediated vasodilation with increased superoxide but robust EDHF function in right ventricular arterial microvessels of pulmonary hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;292(6):H2737–44. doi:10.1152/ajpheart.00548.2006.
Ruiter G, Ying Wong Y, de Man FS, et al. Right ventricular oxygen supply parameters are decreased in human and experimental pulmonary hypertension. J Heart Lung Transplant. 2013;32(2):231–40. doi:10.1016/j.healun.2012.09.025.
Wensel R, Jilek C, Dörr M, et al. Impaired cardiac autonomic control relates to disease severity in pulmonary hypertension. Eur Respir J. 2009;34(4):895–901. doi:10.1183/09031936.00145708.
Nootens M, Kaufmann E, Rector T, et al. Neurohormonal activation in patients with right ventricular failure from pulmonary hypertension: relation to hemodynamic variables and endothelin levels. J Am Coll Cardiol. 1995;26(7):1581–5. doi:10.1016/0735-1097(95)00399-1.
Velez-Roa S, Ciarka A, Najem B, Vachiery J-L, Naeije R, van de Borne P. Increased sympathetic nerve activity in pulmonary artery hypertension. Circulation. 2004;110(10):1308–12. doi:10.1161/01.CIR.0000140724.90898.D3.
Forfia PR, Mathai SC, Fisher MR, et al. Hyponatremia predicts right heart failure and poor survival in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;177(12):1364–9. doi:10.1164/rccm.200712-1876OC.
de Man FS, Handoko ML, van Ballegoij JJM, et al. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail. 2012;5(1):97–105. doi:10.1161/CIRCHEARTFAILURE.111.964494.
Maron BA, Opotowsky AR, Landzberg MJ, Loscalzo J, Waxman AB, Leopold JA. Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur J Heart Fail. 2013;15(3):277–83. doi:10.1093/eurjhf/hfs173.
Maron BA, Zhang Y-Y, White K, et al. Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation. 2012;126(8):963–74. doi:10.1161/CIRCULATIONAHA.112.094722.
Piao L, Fang Y-H, Parikh KS, et al. GRK2-mediated inhibition of adrenergic and dopaminergic signaling in right ventricular hypertrophy: therapeutic implications in pulmonary hypertension. Circulation. 2012;126(24):2859–69. doi:10.1161/CIRCULATIONAHA.112.109868.
Piao L, Fang Y-H, Cadete VJJ, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med (Berl). 2010;88(1):47–60. doi:10.1007/s00109-009-0524-6.
Drake JI, Bogaard HJ, Mizuno S, et al. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. Am J Respir Cell Mol Biol. 2011;45(6):1239–47. doi:10.1165/rcmb.2010-0412OC.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi:10.1126/science.1160809.
Piao L, Marsboom G, Archer SL. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J Mol Med (Berl). 2010;88(10):1011–20. doi:10.1007/s00109-010-0679-1.
Oikawa M, Kagaya Y, Otani H, et al. Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol. 2005;45(11):1849–55. doi:10.1016/j.jacc.2005.02.065.
Mielniczuk LM, Birnie D, Ziadi MC, et al. Relation between right ventricular function and increased right ventricular [18F]fluorodeoxyglucose accumulation in patients with heart failure. Circ Cardiovasc Imaging. 2011;4(1):59–66. doi:10.1161/CIRCIMAGING.109.905984.
Bokhari S, Raina A, Berman Rosenweig E, et al. Positron emission tomography imaging may provide a novel biomarker and understanding of right ventricular dysfunction in patients with idiopathic pulmonary arterial hypertension. Circ Cardiovasc Imaging. 2011;4(6):641–7. doi:10.1161/CIRCIMAGING.110.963207.
Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1(7285):785–9.
Fang YH, Piao L, Hong Z, et al. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. J Mol Med (Berl). 2012;90(1):31–43. doi:10.1007/s00109-011-0804-9.
Archer SL, Fang Y-H, Ryan JJ, Piao L. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm Circ. 2013;3(1):144–52. doi:10.4103/2045-8932.109960.
Gomez-Arroyo J, Mizuno S, Szczepanek K, et al. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ Heart Fail. 2013;6(1):136–44. doi:10.1161/CIRCHEARTFAILURE.111.966127.
Nagendran J, Gurtu V, Fu DZ, et al. A dynamic and chamber-specific mitochondrial remodeling in right ventricular hypertrophy can be therapeutically targeted. J Thorac Cardiovasc Surg. 2008;136(1):168–78, 178 e1–3. doi:10.1016/j.jtcvs.2008.01.040.
McLaughlin VV, Presberg KW, Doyle RL, et al. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):78S–92. doi:10.1378/chest.126.1_suppl.78S.
Bonnet S, Michelakis ED, Porter CJ, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113(22):2630–41. doi:10.1161/CIRCULATIONAHA.105.609008.
Nagendran J, Sutendra G, Paterson I, et al. Endothelin axis is upregulated in human and rat right ventricular hypertrophy. Circ Res. 2013;112(2):347–54. doi:10.1161/CIRCRESAHA.111.300448.
Galiè N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–57. doi:10.1056/NEJMoa050010.
Nagendran J, Archer SL, Soliman D, et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007;116(3):238–48. doi:10.1161/CIRCULATIONAHA.106.655266.
Hemnes AR, Zaiman A, Champion HC. PDE5A inhibition attenuates bleomycin-induced pulmonary fibrosis and pulmonary hypertension through inhibition of ROS generation and RhoA/Rho kinase activation. Am J Physiol Lung Cell Mol Physiol. 2008;294(1):L24–33. doi:10.1152/ajplung.00245.2007.
Freed BH, Gomberg-Maitland M, Chandra S, et al. Late gadolinium enhancement cardiovascular magnetic resonance predicts clinical worsening in patients with pulmonary hypertension. J Cardiovasc Magn Reson. 2012;14:11. doi:10.1186/1532-429X-14-11.
Benza R, Biederman R, Murali S, Gupta H. Role of cardiac magnetic resonance imaging in the management of patients with pulmonary arterial hypertension. J Am Coll Cardiol. 2008;52(21):1683–92. doi:10.1016/j.jacc.2008.08.033.
Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13(8):952–61. doi:10.1038/nm1613.
Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard HJ. Mechanisms of right heart failure-A work in progress and a plea for failure prevention. Pulm Circ. 2013;3(1):137–43. doi:10.4103/2045-8932.109957.
Hemnes AR, Maynard KB, Champion HC, et al. Testosterone negatively regulates right ventricular load stress responses in mice. Pulm Circ. 2012;2(3):352–8. doi:10.4103/2045-8932.101647.
Piao L, Fang Y-H, Parikh K, Ryan JJ, Toth PT, Archer SL. Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med (Berl). 2013;91:1185–97. doi:10.1007/s00109-013-1064-7.
Brittain EL, Pugh ME, Wheeler LA, et al. Shorter survival in familial versus idiopathic pulmonary arterial hypertension is associated with hemodynamic markers of impaired right ventricular function. Pulm Circ. 2013;3(3):589–98. doi:10.1086/674326. PMID: 24618543.
Hemnes AR, Brittain EL, Trammell AW, et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;189(3):325–34. doi:10.1164/rccm.201306-1086OC. PMID: 24274756.
Johnson JA, Hemnes AR, Perrien DS, et al. Cytoskeletal defects in Bmpr2-associated pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;302(5):L474–84. doi:10.1152/ajplung.00202.2011.
West J, Fagan K, Steudel W, et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94(8):1109–14. doi:10.1161/01.RES.0000126047.82846.20.
Hemnes AR, Fessel JP, Penner N, Gleaves L, Robinson L, West J. Universal expression of BMPR2 mutation is associated with impairment of right ventricular hypertrophy and steatosis in mice. Am J Respir Crit Care Med (abstr). 2012;185:A3454.
Brittain E, Fessel J, Fox K, et al. Bone morphogenetic protein receptor type II-associated heritable pulmonary arterial hypertension is associated with fatty acid oxidation defects and cardiac steatosis. Circulation (Abstract). 2012;126, A14564.
Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373(9671):1289–300. doi:10.1016/S0140-6736(09)60256-7.
Corrado D, Basso C, Pilichou K, Thiene G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart. 2011;97(7):530–9. doi:10.1136/hrt.2010.193276.
Archer SL. Riociguat for pulmonary hypertension – a glass half full. N Engl J Med. 2013;369(4):386–8. doi:10.1056/NEJMe1306684.
Lewis GD, Farrell L, Wood MJ, et al. Metabolic signatures of exercise in human plasma. Sci Transl Med. 2010;2(33):33ra37. doi:10.1126/scitranslmed.3001006.
Lewis GD, Wei R, Liu E, et al. Metabolite profiling of blood from individuals undergoing planned myocardial infarction reveals early markers of myocardial injury. J Clin Invest. 2008;118(10):3503–12. doi:10.1172/JCI35111.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag London
About this chapter
Cite this chapter
Brittain, E.L., Hemnes, A.R. (2014). Right Ventricular Pathobiology. In: Gaine, S., Naeije, R., Peacock, A. (eds) The Right Heart. Springer, London. https://doi.org/10.1007/978-1-4471-2398-9_4
Download citation
DOI: https://doi.org/10.1007/978-1-4471-2398-9_4
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-2397-2
Online ISBN: 978-1-4471-2398-9
eBook Packages: MedicineMedicine (R0)