
Abstract
Though the existence of hydrogen sulfide (H2S) in biological tissues has been known for over 300 years, it is the most recently appreciated of the gasotransmitters as a physiologic messenger molecule. The enzymes cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS) had long been speculated to generate H2S, and inhibitors of these enzymes had been employed to characterize influences of H2S in various organs. Definitive evidence that H2S is a physiologic regulator came with the development of mice with targeted deletion of CSE and CBS. Best characterized is the role of H2S, formed by CSE, as an endothelial derived relaxing factor that normally regulates blood pressure by acting through ATP-sensitive potassium channels. H2S participates in various phases of the inflammatory process, predominantly exerting anti-inflammatory actions. Currently, the most advanced efforts to develop therapeutic agents involve the combination of H2S donors with non-steroidal anti-inflammatory drugs (NSAIDs). The H2S releasing moiety provides cytoprotection to gastric mucosa normally adversely affected by NSAIDs while the combination of H2S and inhibition of prostaglandin synthesis may afford synergistic anti-inflammatory influences.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hydrogen sulfide (H2S) is the most recently appreciated of the three gasotransmitters, joining nitric oxide (NO), and carbon monoxide (CO). Though only recently recognized as being physiologically formed in mammalian tissues, H2S has been known to exist in animal tissues for many years. Like NO and CO, H2S is toxic, about five times more so than CO [1]. Recently, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) have been established as the major physiologic sources of mammalian H2S based on studies showing that their deletion or inhibition markedly diminishes mammalian H2S levels [2, 3]. As with NO and CO, identification of the biosynthetic enzymes now provides a firm basis for elucidating how H2S is produced, signals to intracellular targets, and affects diverse physiologic processes. In the interest of brevity, the review will be limited to a few areas of H2S disposition: focusing on physiologic roles in the cardiovascular system and inflammation and a brief discussion of regulatory mechanisms and signaling modalities.
Understanding the disposition of H2S can be facilitated by comparisons with NO and CO. NO was identified as endothelial-derived relaxing factor and as regulating macrophage function years before the first NO synthase (NOS) was purified and cloned [4, 5]. NO is formed by a family of NO synthase isoforms. Cloning and characterization of neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS) greatly facilitated research in the field and led to a vast expansion of NO literature [6]. The NOS isoforms are all heme-requiring enzymes, a property they share with CBS. nNOS and eNOS are constitutively expressed but activated in response to Ca2+/calmodulin [7], which also activates CSE [8].
NO relaxes blood vessels by binding to heme in the active site of guanylyl cyclase to facilitate formation of cyclic GMP which, via protein kinase G, relaxes blood vessels [9]. S-nitrosylation is a more prominent and ubiquitous physiological signaling mechanism for NO whereby NO reacts with the SH group of cysteines in target proteins to inhibit or activate them [10]. As described below, H2S appears to signal predominantly by an analogous mechanism—sulfhydration of target proteins, whereas no major action via cyclic nucleotides has been reported for H2S [11].
nNOS is highly localized to discrete neuronal systems in the brain and to autonomic nerves in the periphery [12]. eNOS occurs in the endothelial layer of blood vessels and the respiratory system, while iNOS occurs in all cells of the body, but is notably enriched in macrophages [4]. Localizations of CBS and CSE are less well characterized, though the enzymes are highly expressed in liver and kidney and at lower levels in pancreas, adipose tissue, small intestine and brain [13]. In the brain, CBS is largely glial while CSE occurs in neurons and endothelial cells [14, 15].
CO also displays some analogies to NO. It is generated by two isoforms of heme oxygenase (HO) with HO1 being inducible, similar to iNOS while HO2 is constitutive. Like nNOS and eNOS, HO2 is activated by calcium/calmodulin [16]. HO2 is highly localized to neurons in the brain and the periphery and fulfills many characteristics of a neurotransmitter [17]. In the intestine HO2 and nNOS are co-localized in myenteric neurons where both appear to serve as neurotransmitters of non-adrenergic-non-cholinergic neurotransmission [18, 19]. As related below, there is some evidence for myenteric localization of CSE which might fulfill similar functions as the other two gasotransmitters in the gut. A conjunction of all three gasotransmitters occurs in the carotid body, where nNOS is expressed in nerve fibers, HO2, and CSE colocalize in glomus cells and regulate carotid body afferent discharge in response to hypoxia [20].
H2S metabolism
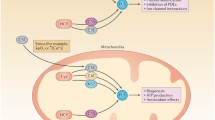
There has been much controversy over endogenous, mammalian levels of H2S, and the extent to which the gas derives from exogenous sources or reflects influences of biosynthetic enzymes (Figs. 1 and 2). Estimates for H2S concentrations have ranged from the high micromolar to the low nanomolar with recent appreciation that physiologic levels are probably relatively low [21]. A major confounding factor in H2S measurement has been the large endogenous stores of sulfane sulfur, which is artifactually reduced to H2S during assays [22, 23]. Another difficulty relates to sensitivity and specificity of the various techniques employed to measure H2S. Recently, several groups have developed fluorescent probes which may be substantially more sensitive and may permit imaging of H2S in intact cells [24, 25].

Pathways of H2S metabolism. Cysteine metabolism from methionine and dietary cyst(e)ine, which enter cells via specific transporters, leads to H2S production. H2S is derived from cysteine, cystine, and 3-mercaptopyruvate (3MP). 3-Mercaptopyruvate sulfurtransferase (3MST) and 2-cysteine aminotransferase (CAT) produce H2S and pyruvate from 3MP, which is formed from cysteine and α-ketogluterate produced by CAT. Cystathionine-β-Synthase (CBS) catalyzes the β-replacement of cysteine with homocysteine (Hcy) to generate H2S and the corresponding thiol ether (Hcy-S-Cys). Cystathionine-γ-lyase (CSE) catalyzes β-disulfide elimination on cystine, the product of which reacts with available thiols (Cys is shown) to generate H2S and a disulfide (Cys-S-S-Cys)

Structures of molecules involved in H2S physiology
CBS and CSE, the enzymes generally acknowledged as the principal sources of physiologic mammalian H2S, were both first known as participants in metabolism of cystathionine, which is formed by CBS via the condensation of homocysteine with serine to generate cystathionine as a thiol ether. The markedly elevated levels of homocysteine in patients with homocystinemia, a genetic deletion of Cbs, lead to substantial cardiovascular disability [26]. CBS forms H2S from cysteine or homocysteine with a combination of the two substrates providing maximal yields in vitro [27]. Inhibitors of CBS, such as hydroxylamine or amino-oxyacetate impair the generation of H2S from cysteine in the brain, but they are non-specific, affecting all pyridoxal phosphate enzymes. Moreover, because the K m of CBS for cysteine and homocysteine is 3–7 mM, high concentrations of these amino acids are employed in studies of H2S formation, whereas physiologic levels are less than 10% of the K m values [27].
The heme in CBS binds CO with high affinity, at least 100 times that of NO [28]. Hence, CO appears to be a physiologic inhibitor of CBS, which, as described below, may account for vasodilation of the cerebral circulation. CBS is also activated by S-adenosyl methionine, whose function is unclear but might reflect some relationship between signaling by H2S and biologic methylation [29].
CSE was first characterized as cystathionase, responsible for the pyridoxal phosphate dependent hydrolytic degradation of cystathionine [30]. CSE was proposed as a physiologic generator of H2S in peripheral tissues such as the liver, because inhibitors, such as propargylglycine and β-cyanoalanine, diminish H2S formation. While these inhibitors are relatively non-selective, more recent studies of CSE-deleted mice have definitively established that CSE is the predominant source of H2S in peripheral tissues [8]. Evidence supporting CSE as generating H2S for signaling purposes comes from the finding that CSE, like nNOS, eNOS, and HO2, is activated by calcium/calmodulin [8].
Less well characterized than CBS and CSE as a source of H2S in mammalian tissues is the enzyme 3-mercaptyopyruvate sulfotransferase (3-MST). Kimura and associates [23] developed evidence that 3-MST acts in conjunction with cysteine aminotransferase (CAT) to produce H2S from cysteine in the presence of α-ketoglutarate. The combination of 3-MST and CAT might be responsible for the generation of H2S in brain preparations from CBS-deleted mice (Table 1). Because 3-MST is maximally active at very high pH levels, it is not clear to what extent it is responsible for mammalian formation of H2S.
CBS, CSE, and 3-MST appear to be highly conserved, with the sequences of bacterial forms of these enzymes fairly similar to mammalian isoforms. Very recently, Nudler and associates [31] have discovered that H2S is critical for the survival of bacteria and that a wide range of antibiotics, whose initial targets vary markedly, all act via H2S as a final, common pathway. Thus, bacteria with deletion of the H2S forming enzymes are markedly more sensitive to antibiotic killing. This discovery may portend a new class of antibiotic-sensitizing drugs that lower the bactericidal concentrations of antibiotics.
H2S signaling
Unlike NO and CO, H2S does not appear to stimulate guanylyl cyclase [32], even though it can bind with reasonably high affinity to heme containing domains like that found in guanylyl cyclase. H2S has been shown to signal via a mechanism analogous to nitrosylation whereby it forms a covalent linkage to the SH of cysteines, a process designated sulfhydration [11]. Sulfhydration was first detected by the biotin switch assay employed to monitor nitrosylation. In this procedure free thiols are blocked by methyl methane thiosulfonate (MMTS). The SH groups of nitrosylated cysteines can then be exposed by treatment with ascorbate and subsequently labeled and identified [33]. Even in the absence of ascorbate, some proteins are labeled by the biotin switch technique, which provided a clue to the existence of sulfhydration. Accordingly, sulfhydration can be detected in a modification of the biotin switch procedure with omission of the ascorbate step [11].
Recently, sulfhydration has been monitored by a new technique which overcomes concerns that some free thiols might not be blocked by MMTS [34]. The newer procedure employs a fluorescent maleimide derivative, which interacts selectively with sulfhydryl groups of cysteines, both sulfhdyrated and non-sulfhydrated. Treatment of samples with dithiothreitol selectively cleaves disulfide bonds, detaching the fluorescent signal from sulfhydrated but not non-sulfhydrated proteins and leading to decreased fluorescence [34]. This technique can be modified to simultaneously detect nitrosylation using a differently colored fluorescent maleimide after treatment with ascorbate to remove NO from nitrosylated cysteines, exposing previously nitrosylated SH groups [34].
Sulfhydration appears to be substantially more prevalent than nitrosylation. Whereas nitrosylation typically affects only about 1–5% of most proteins, 10–25% of endogenous glyceraldehyde-3-phosphate dehydrogenase (GAPDH), β-tubulin and actin are basally sulfhydrated [11].
Sulfhydration can influence protein function differently than nitrosylation. Nitrosylation provides an NO “cap” to reactive SH groups of cysteines typically inactivating proteins, though in some instances it has been shown to have an activating effect [35]. By contrast, in sulfhydration, an SH is converted to SSH which, with its lower pK a, is more reactive chemically than SH and may have greater exposure to the cellular environment. This notion is substantiated by the finding that sulfhydration of GAPDH increases catalytic activity 700%, and sulfhydration of actin similarly enhances biologic activity. Activation of GAPDH by sulfhydration is physiologically relevant, as total GAPDH activity of liver extracts is reduced about 25–30% in CSE deleted mice despite normal levels of GAPDH protein [11]. It appears that, as with nitrosylation, many, if not most, proteins are sulfhydrated.
Physiologic actions of H2S
Cardiovascular system
Like NO and CO, H2S dilates blood vessels. Studies with exogenous H2S largely report vascular relaxation, though under some conditions, such as high oxygen concentration, vasoconstriction is evident. NO was first elucidated as endothelial derived relaxing factor (EDRF). Investigations employing eNOS knock-out mice and NOS inhibitors reveal only a partial reduction of EDRF activity in certain vascular beds [8, 36, 37]. EDRF activity in HO2 knockout mice has not yet been reported. Studies of H2S in CSE knockout mice indicate a major contribution to EDRF activity [8]. Immunohistochemical analysis shows that CSE is highly localized to the endothelial layer of blood vessels. Cholinergic relaxation of the mesenteric artery is reduced by about 75–80% in homozygous CSE deleted mice and about 50% in heterozygotes. This cholinergic relaxation reflects EDRF activity being abolished by removal of the endothelium. CSE knockout mice develop age-dependent hypertension with maximal increases in blood pressure of about 20 mmHg, similar to levels of hypertension in eNOS knockouts [8].
The EDRF activity associated with NO is most evident in large vessels such as the aorta, while in the resistance vessels that are the primary determinants of blood pressure, actions of NO are less prominent. In the mesenteric artery, a resistance vessel, H2S is predominant [8]. Relative roles of H2S, NO, and CO in various vascular beds may be elucidated by systematic comparison of mice with deletion of HO2, eNOS, or CSE.
NO and H2S differ markedly in mechanisms whereby they influence blood vessels. NO and CO stimulate cyclic GMP levels while recent studies indicate that H2S vasodilation largely reflects hyperpolarization elicited by opening ATP-sensitive potassium channels (K ATP) [38–40]. While vasorelaxation by exogenous H2S has long been known to involve such channels, recent work establishes that physiologic vasorelaxation is mediated by H2S. Thus, glibenclamide, a potent and selective inhibitor of the K ATP channel, reduces effects of H2S and diminishes cholinergic hyperpolarization of mesenteric arterial smooth muscle by about 70% while not affecting relaxation elicited by NO donors [40].
H2S stimulates K ATP channels by sulfhydrating them at cysteine-43. These channels are activated physiologically when bound by phosphatidylinositol(4,5)bisphosphate (PIP2). The binding of PIP2 to K ATP channels is abolished in cells devoid of CSE or containing a catalytically inactive form of the enzyme. Moreover, H2S donors substantially enhance the binding of PIP2 to K ATP channels, and PIP2 binding occurs at the sulfhydrated cysteine-43 [40].
The observation that H2S physiologically acts by sulfhydrating and activating the K ATP channel supports the notion that H2S is a major if not predominant mediator of EDRF activity. Numerous investigators have found much if not most EDRF activity involves cGMP-independent blood vessel hyperpolarization [37] implying that EDRF is primarily dependent upon an endothelial-derived hyperpolarizing factor whose activity is largely attributable to H2S .
The major role of H2S in regulating the peripheral circulation suggests that it may be the principal vasoactive gasotransmitter, implying therapeutic relevance. This notion is supported by the limited success of studies devoted to inhibiting or enhancing NO formation respectively to combat endotoxic shock or to treat hypertension [41].
H2S may also impact the cerebral circulation. Hypoxia is well known to stimulate cerebral blood flow, but underlying molecular mechanisms have been elusive. Very recently, CSE has been identified as a major regulatory factor for cerebral arteriolar vasodilation, acting in conjunction with CO formed by HO2 [42, 43], similar to that seen in the carotid body [20]. HO2 is an established physiologic O2 sensor, especially in the carotid body where it is exquisitely sensitive to oxygen and is inhibited by hypoxia in a precisely graded fashion [44]. At physiologic concentrations, CO inhibits CBS, the predominant generator of H2S in the cerebral circulation [29]. Thus, by inhibiting HO2, hypoxia would lead to activation of CBS and generation of H2S as a vasorelaxant.
Before endogenous H2S was shown to regulate blood vessels, exogenous H2S had been shown to exert beneficial cardiovascular actions. Many studies have dealt with myocardial ischemia, which is substantially diminished by administration of H2S donors during ischemia/reperfusion of the heart [45–47]. Numerous mechanisms had been proposed for these cardioprotective actions [48, 49]. Particularly promising is evidence that H2S acts by inhibiting apoptosis, as H2S donors reproducibly diminish poly(ADP-ribose) polymerase cleavage, as well as cleavage of caspase-3 [50]. H2S also preserves mitochondrial structure and function in response to myocardial ischemia. H2S may also be cardioprotective by decreasing the “work” of the heart, analogous to beta-blockers, through diminishing contractility of cardiac myocytes, largely by inhibiting L-type calcium channels [51].
Because of the promising cardiovascular actions of H2S a variety of drugs have been developed based on this gasotransmitter. Some are simple H2S donors, such as GYY4137, while others combine an H2S donating structure with an anti-inflammatory drug such as diclofenac or a classical vasodilator such as sildenafil [52–54].
H2S and inflammation
The literature on NO, CO, and H2S has been plagued with conflicting claims for their effects. Nowhere has this been most evident than with H2S and inflammation. Prominent pro-inflammatory effects have been reported in association with increased formation of sulfide in neutrophils as well as activation of these cells [55]. Administration of H2S donors has been reported to accentuate inflammatory factors associated with burns, while burn injuries were reduced by treatment by the CSE inhibitor propargylglycine [56] (Table 1). Lung injury elicited by bacterial sepsis can be alleviated by treatment with propargylglycine and worsened with H2S donors [55]. By contrast, there are numerous reports of anti-inflammatory effects for H2S donors as described below. A consensus has emerged in recent years that the apparently contradictory findings largely reflect variations in dose–response relationships. At relatively low, physiologic concentrations H2S appears to be anti-inflammatory, while high concentrations elicit inflammation, a pattern reminiscent of NO, which is anti-inflammatory in low concentrations and pro-inflammatory at high levels. CO, well known to be lethal in high doses, is also often beneficial when administered in low doses [57, 58].
What physiologic mechanisms underlie influences of H2S on inflammation? One of the best characterized involves the disposition of leukocytes, especially their adherence to vascular endothelium as well as their extravasation. H2S donors and sulfide salts diminish lymphocyte and neutrophil infiltration in models of inflammation, whereas inhibitors of H2S biosynthesis increase leukocyte adherence [59]. H2S donors diminish edema, presumably due to inhibition of plasma exudation, while CBS and CSE inhibitors increase the formation of edema in response to inflammatory stimuli [59]. A molecular mechanism underlying anti-inflammatory roles of H2S may include its scavenging peroxynitrite, a toxic derivative of NO, as well as other oxidants [60].
H2S has been shown to exert beneficial influences in disorders of joints, including resolving synovitis in rodents [61] and alleviating the pathology of carrageenen-associated arthritis [62]. H2S donors also have been extensively explored in intestinal disorders, with beneficial effects in several models of colitis [63].
H2S may participate in some actions of tumor necrosis factor alpha (TNF-α). While TNF-α is regarded as pro-inflammatory, it does display anti-apoptotic actions mediated via nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB). The anti-apoptotic actions of NF-κB appear to be mediated by H2S generated by CSE [34]. TNF-α treatment (10 ug/kg for 4 h) in peritoneal macrophages triples H2S generation by stimulating the binding of the transcription factor SP1 to the CSE promoter increasing CSE protein levels. The H2S generated by CSE enhances the binding of NF-κB to promoters of downstream genes, whose signaling is markedly diminished in CSE knockout mice. H2S acts by sulfhydrating the p65 subunit of NF-κB, which promotes its binding to the co-activator ribosomal protein S3. The anti-apoptotic influences of NF-κB are substantially reduced in CSE-deleted mice [34].
The anti-inflammatory influences of H2S have led to efforts to develop therapeutic agents. Classic non-steroidal anti-inflammatory drugs (NSAIDs) often cause gastric irritation by inhibiting the formation of prostaglandins, which are physiologic cytoprotectants of the gastric mucosa. H2S, on the other hand, reduces mucosal inflammation, protects the gastrointestinal mucosa from injury and also augments tissue repair. In direct comparisons of naproxen and its H2S-linked derivative, the latter exerted comparable therapeutic efficacy with reduced gastric damage [64, 65]. Several other NSAIDs have been combined with H2S donors. Mechanistic studies have been conducted with some of these drugs, with particularly extensive investigations utilizing S-diclofenac [52, 66, 67]. S-diclofenac has been shown to inhibit cell proliferation [68, 69] and to protect against ischemia-reperfusion injury in perfused hearts [53].
New directions
Evidence for H2S as a physiologic gasotransmitter has lagged behind CO and NO, but H2S is rapidly catching up. Therapeutic applications may emerge in the not-too-distant future, especially in the area of anti-inflammatory drugs. Definitive understanding of how H2S participates in inflammatory processes may come from studies of inflammation in mice with deletion of CSE and/or CBS. In the gastrointestinal system and liver, CSE levels greatly exceed those of CBS. Because many major proteins are physiologically sulfhydrated, it is possible that many metabolic functions of the liver are determined in notable part by the actions of H2S, as is evident by the substantial decrease in GAPDH activity in livers of CSE knockout mice, due to the loss of the activating influence of GAPDH sulfhydration [11].
One area not addressed in this review is the role of H2S in the brain, which was discussed in a previous review [70]. Studies with mice lacking CBS and CSE suggest that the majority of H2S in the brain derives from CBS rather than CSE. The limited immunohistochemical studies thus far performed reveal CBS predominantly in glia [14]. CSE may have neuronal as well as glial localizations so that even if it generates a smaller amount of H2S, this enzyme might be the source of a neurotransmitter pool [15]. In the intestine, there is evidence that CSE is localized to the myenteric plexus of neurons and may exert physiologic influences on intestinal motility [15, 71]. Conceivably, neuronal CSE in the gut occurs in the same neurons known to possess HO2 and nNOS, which are co-localized in neuronal populations [72].
References
Lloyd D (2006) Hydrogen sulfide: clandestine microbial messenger? Trends Microbiol 14:456–462
Szabó C (2007) Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov 6:917–935
Kimura H, Nagai Y, Umemura K, Kimura Y (2005) Physiological roles of hydrogen sulfide: synaptic modulation, neuroprotection, and smooth muscle relaxation. Antioxid Redox Signal 7:795–803
Davis KL, Martin E, Turko IV, Murad F (2001) Novel effects of nitric oxide. Annu Rev Pharmacol Toxicol 41:203–236
Hetrick EM, Schoenfisch MH (2009) Analytical chemistry of nitric oxide. Annu Rev Anal Chem (Palo Alto Calif) 2:409–433
Bredt DS (2003) Nitric oxide signaling specificity—the heart of the problem. Journal of Cell Science 116:9–15
Bredt DS, Snyder SH (1990) Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A 87:682–685
Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S et al (2008) H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322:587–590
Rapoport RM, Draznin MB, Murad F (1983) Endothelium-dependent relaxation in rat aorta may be mediated through cyclic GMP-dependent protein phosphorylation. Nature 306:174–176
Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J (1992) S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A 89:444–448
Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH (2009) H2S signals through protein S-sulfhydration. Science Signaling 2:ra72
Bredt DS, Hwang PM, Snyder SH (1990) Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 347:768–770
Ishii I, Akahoshi N, Yu X-N, Kobayashi Y, Namekata K, Komaki G, Kimura H (2004) Murine cystathionine gamma-lyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem J 381:113–123
Enokido Y, Suzuki E, Iwasawa K, Namekata K, Okazawa H, Kimura H (2005) Cystathionine beta-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. The FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology 19:1854–1856
Linden DR, Sha L, Mazzone A, Stoltz GJ, Bernard CE, Furne JK, Levitt MD, Farrugia G, Szurszewski JH (2008) Production of the gaseous signal molecule hydrogen sulfide in mouse tissues. J Neurochem 106:1577–1585
Boehning D, Sedaghat L, Sedlak TW, Snyder SH (2004) Heme oxygenase-2 is activated by calcium-calmodulin. J Biol Chem 279:30927–30930
Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH (1993) Carbon monoxide: a putative neural messenger. Science 259:381–384
Battish R, Cao GY, Lynn RB, Chakder S, Rattan S (2000) Heme oxygenase-2 distribution in anorectum: colocalization with neuronal nitric oxide synthase. Am J Physiol Gastrointest Liver Physiol 278:G148–G155
Zakhary R, Gaine SP, Dinerman JL, Ruat M, Flavahan NA, Snyder SH (1996) Heme oxygenase 2: endothelial and neuronal localization and role in endothelium-dependent relaxation. Proc Natl Acad Sci U S A 93:795–798
Peng Y-J, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR (2010) H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci 107:10719–10724
Furne J, Saeed A, Levitt MD (2008) Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am J Physiol Regul Integr Comp Physiol 295:R1479–R1485
Kajimura M, Fukuda R, Bateman RM, Yamamoto T, Suematsu M (2010) Interactions of multiple gas-transducing systems: hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxid Redox Signal 13:157–192
Kimura H (2011) Hydrogen sulfide: its production, release and functions. Amino Acids 41:113–121
Peng H, Cheng Y, Dai C, King AL, Predmore BL, Lefer DJ, Wang B (2011) A fluorescent probe for fast and quantitative detection of hydrogen sulfide in blood. Angew Chem Int Ed Engl 50:9672–9675
Lippert AR, New EJ, Chang CJ (2011) Reaction-based fluorescent probes for selective imaging of hydrogen sulfide in living cells. J Am Chem Soc 133:10078–10080
McCully KS (1969) Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 56:111–128
Singh S, Padovani D, Ra L, Chiku T, Banerjee R (2009) Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem 284:22457–22466
Puranik M, Weeks CL, Lahaye D, Kabil O, Taoka S, Nielsen SB, Groves JT, Banerjee R, Spiro TG (2006) Dynamics of carbon monoxide binding to cystathionine beta-synthase. J Biol Chem 281:13433–13438
Banerjee R, Zou C-G (2005) Redox regulation and reaction mechanism of human cystathionine-beta-synthase: a PLP-dependent hemesensor protein. Arch Biochem Biophys 433:144–156
Chatagner F (1969) Biosynthesis of cystathionine from homoserine and cysteine by rat liver cystathionase. FEBS Lett 4:231–233
Shatalin K, Shatalina E, Mironov A, Nudler E (2011) H2S: a universal defense against antibiotics in bacteria. Science 334:986–990
Zhao W, Zhang J, Lu Y, Wang R (2001) The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J 20:6008–6016
Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH (2001) Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nature cell biology 3:193–197
Sen N, Paul BD, Gadalla MM, Nakamura T, Mustafa AK, Tanusree S, Kim S, Snyder SH (2011) Hydrogen sulfide-linked sulfhydration of NF-kB mediates its anti-apoptotic actions. Molecular Cell 45:13–24
Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, Lipton SA (2011) S-nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc Natl Acad Sci U S A 108:14330–14335
Brandes RP, Schmitz-Winnenthal FH, Feletou M, Godecke A, Huang PL, Vanhoutte PM, Fleming I, Busse R (2000) An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proc Natl Acad Sci U S A 97:9747–9752
Félétou M, Vanhoutte PM (2007) Endothelium-dependent hyperpolarizations: past beliefs and present facts. Ann Med 39:495–516
Zhao W, Wang R (2002) H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol 283:H474–H480
Jiang B, Tang G, Cao K, Wu L, Wang R (2010) Molecular mechanism for H(2)S-induced activation of K(ATP) channels. Antioxid Redox Signal 12:1167–1178
Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R et al (2011) Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 109:1259–1268
Petros A, Lamb G, Leone A, Moncada S, Bennett D, Vallance P (1994) Effects of a nitric oxide synthase inhibitor in humans with septic shock. Cardiovasc Res 28:34–39
Ishikawa M, Kajimura M, Adachi T, Maruyama K, Makino N, Goda N, Yamaguchi T, Sekizuka E, Suematsu M (2005) Carbon monoxide from heme oxygenase-2 Is a tonic regulator against NO-dependent vasodilatation in the adult rat cerebral microcirculation. Circ Res 97:e104–e114
Morikawa T, Kajimura M, Nakamura T, Hishiki T, Nakanishi T, Yukutake Y, Nagahata Y, Ishikawa M, Hattori K, Takenouchi T, et al. (2012) Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc Natl Acad Sci U S A (in press)
Prabhakar NR, Dinerman JL, Agani FH, Snyder SH (1995) Carbon monoxide: a role in carotid body chemoreception. Proc Natl Acad Sci U S A 92:1994–1997
Johansen D, Ytrehus K, Baxter GF (2006) Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury—evidence for a role of K ATP channels. Basic research in cardiology 101:53–60
Elsey DJ, Fowkes RC, Baxter GF (2010) l-cysteine stimulates hydrogen sulfide synthesis in myocardium associated with attenuation of ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther 15:53–59
Sivarajah A, McDonald MC, Thiemermann C (2006) The production of hydrogen sulfide limits myocardial ischemia and reperfusion injury and contributes to the cardioprotective effects of preconditioning with endotoxin, but not ischemia in the rat. Shock 26:154–161
Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C et al (2007) Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A 104:15560–15565
Minamishima S, Bougaki M, Sips PY, Yu JD, Minamishima YA, Elrod JW, Lefer DJ, Bloch KD, Ichinose F (2009) Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3-dependent mechanism in mice. Circulation 120:888–896
Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM, Szabo C, Sellke FW (2008) The effects of therapeutic sulfide on myocardial apoptosis in response to ischemia-reperfusion injury. Eur J Cardiothorac Surg 33:906–913
Sun YG, Cao YX, Wang WW, Ma SF, Yao T, Zhu YC (2008) Hydrogen sulphide is an inhibitor of L-type calcium channels and mechanical contraction in rat cardiomyocytes. Cardiovasc Res 79:632–641
Li L, Rossoni G, Sparatore A, Lee LC, Del Soldato P, Moore PK (2007) Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic Biol Med 42:706–719
Rossoni G, Sparatore A, Tazzari V, Manfredi B, Del Soldato P, Berti F (2008) The hydrogen sulphide-releasing derivative of diclofenac protects against ischaemia-reperfusion injury in the isolated rabbit heart. Br J Pharmacol 153:100–109
Sidhapuriwala J, Li L, Sparatore A, Bhatia M, Moore PK (2007) Effect of S-diclofenac, a novel hydrogen sulfide releasing derivative, on carrageenan-induced hindpaw oedema formation in the rat. Eur J Pharmacol 569:149–154
Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath RD, Wang ZJ, Anuar FB, Whiteman M, Salto-Tellez M, Moore PK (2005) Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. The FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology 19:1196–1198
Zhang J, Sio SWS, Moochhala S, Bhatia M (2010) Role of hydrogen sulfide in severe burn injury-induced inflammation in mice. Mol Med (Cambridge) 16:417–424
Fujita T, Toda K, Karimova A, Yan SF, Naka Y, Yet SF, Pinsky DJ (2001) Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med 7:598–604
Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, Usheva A, Stachulak C, Bodyak N, Smith RN, Csizmadia E et al (2003) Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med 9:183–190
Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL (2006) Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. The FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology 20:2118–2120
Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong B-S, Cheung NS, Halliwell B, Moore PK (2004) The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite “scavenger”? J Neurochem 90:765–768
Ekundi-Valentim E, Santos KT, Camargo EA, Denadai-Souza A, Teixeira SA, Zanoni CI, Grant AD, Wallace J, Muscara MN, Costa SK (2010) Differing effects of exogenous and endogenous hydrogen sulphide in carrageenan-induced knee joint synovitis in the rat. Br J Pharmacol 159:1463–1474
Bhatia M, Sidhapuriwala J, Moochhala SM, Moore PK (2005) Hydrogen sulphide is a mediator of carrageenan-induced hindpaw oedema in the rat. Br J Pharmacol 145:141–144
Fiorucci S, Orlandi S, Mencarelli A, Caliendo G, Santagada V, Distrutti E, Santucci L, Cirino G, Wallace JL (2007) Enhanced activity of a hydrogen sulphide-releasing derivative of mesalamine (ATB-429) in a mouse model of colitis. Br J Pharmacol 150:996–1002
Wallace JL, Caliendo G, Santagada V, Cirino G (2010) Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346). Br J Pharmacol 159:1236–1246
Fiorucci S, Antonelli E, Distrutti E, Rizzo G, Mencarelli A, Orlandi S, Zanardo R, Renga B, Di Sante M, Morelli A et al (2005) Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology 129:1210–1224
Lee M, Sparatore A, Del Soldato P, McGeer E, McGeer PL (2010) Hydrogen sulfide-releasing NSAIDs attenuate neuroinflammation induced by microglial and astrocytic activation. Glia 58:103–113
Wallace JL, Caliendo G, Santagada V, Cirino G, Fiorucci S (2007) Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulfide-releasing diclofenac derivative in the rat. Gastroenterology 132:261–271
Elsey DJ, Fowkes RC, Baxter GF (2010) Regulation of cardiovascular cell function by hydrogen sulfide (H(2)S). Cell biochemistry and function 28:95–106
Wang M-J, Cai W-J, Zhu Y-C (2010) Mechanisms of angiogenesis: role of hydrogen sulphide. Clin Exp Pharmacol Physiol 37:764–771
Kimura H (2010) Hydrogen sulfide: from brain to gut. Antioxid Redox Signal 12:1111–1123
Teague B, Asiedu S, Moore PK (2002) The smooth muscle relaxant effect of hydrogen sulphide in vitro: evidence for a physiological role to control intestinal contractility. Br J Pharmacol 137:139–145
Xue L, Farrugia G, Miller SM, Ferris CD, Snyder SH, Szurszewski JH (2000) Carbon monoxide and nitric oxide as coneurotransmitters in the enteric nervous system: evidence from genomic deletion of biosynthetic enzymes. Proc Natl Acad Sci U S A 97:1851–1855
Acknowledgments
This work has been supported by National Institutes of Health Medical Scientist Training Program Award (T32 GM007309) to M.S.V. and US Public Health Service Grants (MH018501) to S.H.S.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vandiver, M.S., Snyder, S.H. Hydrogen sulfide: a gasotransmitter of clinical relevance. J Mol Med 90, 255–263 (2012). https://doi.org/10.1007/s00109-012-0873-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-012-0873-4