
Abstract
Colon cancer affects millions of individuals in Western countries. Cannabidiol, a safe and non-psychotropic ingredient of Cannabis sativa, exerts pharmacological actions (antioxidant and intestinal antinflammatory) and mechanisms (inhibition of endocannabinoid enzymatic degradation) potentially beneficial for colon carcinogenesis. Thus, we investigated its possible chemopreventive effect in the model of colon cancer induced by azoxymethane (AOM) in mice. AOM treatment was associated with aberrant crypt foci (ACF, preneoplastic lesions), polyps, and tumour formation, up-regulation of phospho-Akt, iNOS and COX-2 and down-regulation of caspase-3. Cannabidiol-reduced ACF, polyps and tumours and counteracted AOM-induced phospho-Akt and caspase-3 changes. In colorectal carcinoma cell lines, cannabidiol protected DNA from oxidative damage, increased endocannabinoid levels and reduced cell proliferation in a CB1-, TRPV1- and PPARγ-antagonists sensitive manner. It is concluded that cannabidiol exerts chemopreventive effect in vivo and reduces cell proliferation through multiple mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colon cancer is a major cause of morbidity and mortality in Western countries. In 2011, an estimated 101,340 new cases of colorectal cancer were diagnosed in the USA, with 49,380 estimated deaths [1]. Colon cancer is thought to arise as the result of a series of histopathological and molecular changes that transform normal colonic epithelial cells into a colorectal carcinoma, with aberrant crypt foci (ACF) and polyps as intermediate steps in this process [2]. This multi-step process spans 10–15 years, thereby providing an opportunity for prevention [3].
Δ9-tetrahydrocannabinol (Δ9-THC), the main psychotropic ingredient of the marijuana plant Cannabis sativa, binds two Gi/o coupled membrane receptors, named cannabinoid receptors (CB1 and CB2), which are also activated by endogenous ligands [anandamide and 2-arachidonoylglycerol (2-AG)] [4, 5]. Endocannabinoids are biosynthesised ‘on demand’ from membrane phospholipids and are inactivated through a reuptake process (facilitated by a putative endocannabinoid membrane transporter), followed by enzymatic degradation catalysed by the fatty acid amide hydrolase (FAAH, in the case of anandamide and, to some extent, 2-AG) or monoacylglycerol lipase (MAGL, in the case of 2-AG) [4]. Cannabinoids—via direct or indirect activation of CB1 and/or CB2 receptors—exert protective effects in well-established models of colon cancer [6–8].
In addition to Δ9-THC, the plant Cannabis also contains non-psychotropic cannabinoids with potential therapeutic interest. The best studied among these compounds is cannabidiol, which, unlike Δ9-THC, has very low affinity for both CB1 and CB2 receptors [5, 9]. Cannabidiol has an extremely safe profile in humans [9] and exerts a number of pharmacological actions (e.g. analgesic/anti-inflammatory, antioxidant, neuroprotective) of potential clinical interest [9]. Few studies have investigated the effect of cannabidiol in the gut. Specifically, cannabidiol has been shown to reduce intestinal contractility [10, 11] and, more importantly, to exert antinflammatory effects [12, 13], a relevant observation in the light of the well-known association existing between intestinal inflammation and colorectal cancer [14]. In addition, cannabidiol may inhibit FAAH [15] and exerts antioxidant action in colorectal carcinoma cell lines [12]. Both FAAH inhibition [6, 16] and antioxidant effects [17] are potentially beneficial for colon carcinogenesis.
Because cannabidiol exerts pharmacological effects (e.g. antioxidant, intestinal antinflammatory) and mechanisms (e.g. inhibition of endocannabinoids enzymatic degradation) potentially beneficial for colon carcinogenesis, we investigated the potential chemopreventive effect of this non-psychotropic marijuana component in an experimental model of colon cancer. The possible mode of action was evaluated in colorectal carcinoma cell lines.
Materials and methods
Animals
Male ICR mice (Harlan Italy, S. Pietro al Natisone UD, Italy) weighting 26–30 g were used in conformity to the Italian DL no. 116 of 27 January 1992 and associated guidelines in the European Communities Council (86/609/ECC and 2010/63/UE).
Cell cultures
For in vitro experiments, two human colon adenocarcinoma cell lines (i.e. Caco-2 and HCT116 cells, ATCC from LGC Standards, Milan, Italy), with a different genetic profile (APC gene mutated in Caco-2 cells, K-RAS mutated in HCT116 cells) [18, 19] were used. Both cell lines were cultured in Dulbecco’s modified Eagle’s medium containing 10% foetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin, 1% non-essential amino acids, and 2 mM l-glutamine, in conformity with the manufacturer’s protocols.
In vivo treatments and tumour evaluation
Mice were randomly divided into the following groups: Group 1 (control) was treated with vehicles; group 2 was treated with azoxymethane (AOM) plus the vehicle used to dissolve cannabidiol; group 3 was treated with AOM plus cannabidiol (1 mg/kg); and group 4 was treated with AOM plus cannabidiol (5 mg/kg). AOM (40 mg/kg in total, IP) was administered in four single doses of 10 mg/kg, at the beginning of the first, second, third, and fourth week. Cannabidiol was given (IP) three times per week for the whole duration of the experiment starting 1 week before the first administration of AOM [6]. All animals were euthanised by asphyxiation with CO2 3 months after the first injection of AOM. Based on our laboratory experience, this time (at the dose of AOM used) was associated with the occurrence of a significant number of aberrant crypt foci (ACF), polyps and tumours. Colons were examined as previously reported [6] using a light microscope at 20× magnification (Leica Microsystems, Milan, Italy). Only foci with four or more crypts (which are best correlated with the final tumour incidence) were evaluated. ACF were distinguished from surrounding normal crypts by greater size, larger and elongated luminal opening, thicker lining, and compression of the surrounding epithelium. The criterion to distinguish polyps from tumors was established considering the main characteristic features of these two lesions (i.e. crypt distortion around a central focus and increased distance from luminal to basal surface of cells for polyps and high grade of dysplasia with complete loss of crypt morphology for tumors) [20].
Preparation of cytosolic lysates
Lysates from full-thickness colons were obtained as previously described [12]. Briefly, tissues were homogenised using a buffer solution (1:2, w/v) containing 0.5 M β-glycerophosphate, 20 mM MgCl2, 10 mM EGTA, 100 mM dithiothreitol, 100 mM dimethylsulfonyl fluoride, 2 mg/ml apronitin, 2 mM leupeptin and 10 mM Na3VO4.
For lysate preparations from Caco-2 cells, 8 × 105 cells were seeded in Petri dishes, brought to subconfluence (∼70%) and, after 24-h exposure to cannabidiol, collected using the following buffer: 50 mM Tris–HCl, pH 7.4, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EGTA, 1 mM NaF, 1% NP-40, 1 mM PMSF, 1 mM Na3VO4 plus a complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). The protein concentration was determined on supernatant (following centrifugation at 16,200×g for 15 min) using the Bradford method.
Western blot analysis
Western blot analyses were performed ex vivo in full-thickness colonic tissues of animals treated or not with AOM (alone or with cannabidiol 1 mg/kg) to investigate the expression of inducible nitric oxide synthase (iNOS), cycloxygenase (COX-2), phospho-Akt and caspase-3. We also investigated the expression of phospho-Akt in Caco-2 cells, treated with cannabidiol (10 μM). Protein lysates (50–70 μg) were separated on sodium dodecyl sulfate polyacrylamide gels, and membranes were incubated with anti-iNOS, anti-COX-2 (BD Biosciences from Becton Dickinson, Buccinasco, Italy), anti-β-actin (Sigma, Milan, Italy), anti-phosho-Akt or anti-Akt and anti-cleaved-caspase-3 (fragment p17) or anti-uncleaved caspase-3 (fragment p30) (Cell Signaling from Euroclone, Milan, Italy). Signals were visualised using ImageQuant 400 equipped with Quantity One Software 4.6.3 (GE Healthcare, Milan, Italy).
Cytotoxicity studies: neutral red (NR) uptake
Caco-2 and HCT116 cells were seeded in 96-well plates [1 × 104 cells per well (Caco-2) and 2.5 × 103 cells per well (HCT116)] and allowed to adhere for 48 h; after this period, cells were incubated with cannabidiol (0.01–10 μM) for 24 h and subsequently with NR dye solution (50 μg/ml) for 3 h. Cells were lysed with 1% acetic acid, and the absorbance was read at 532 nm (iMarkTM microplate absorbance reader, BioRad). Dimethyl sulphoxide (DMSO) (20%) was used as a positive control. The results are expressed as percentage of cell viability (n = 3 experiments including 8–10 replicates for each treatment).
Proliferation studies: MTT assay and 3 H-thymidine incorporation
Caco-2 and HCT116 cells were seeded, allowed to adhere and starved by serum deprivation for 24 h. For the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, cells were treated with cannabidiol (0.01–10 μM) for 24 h and incubated with MTT (250 μg/ml) for 1 h at 37°C. The mitochondrial reduction of MTT to formazan was then quantitated at 490 nm (iMarkTM microplate reader, BioRad, Italy).
Using this assay, the antiproliferative effect of cannabidiol (10 μM) was evaluated (in Caco-2 cells) in the presence of rimonabant (0.1 μM, CB1 receptor antagonist), AM251 (10 μM, CB1 receptor antagonist), SR144528 (0.1 μM, CB2 receptor antagonist), AM630 (0.1 μM, CB2 receptor antagonist), capsazepine [1 μM, transient receptor potential vanilloid 1 (TRPV1) antagonist] and GW9662 [10 μM, peroxisome proliferator-activated receptor γ (PPARγ) antagonist], all incubated 30 min before cannabidiol.
For 3H-thymidine incorporation assay [21], cells were incubated with cannabidiol (0.01–10 μM) in the presence of [methyl-3H]-thymidine (1 μCi/well) for 24 h and collected with 1 N NaOH for β-counting (PerkinElmer, Milan, Italy).
Identification and quantification of endocannabinoids (anandamide and 2-AG), PEA and OEA
Endocannabinoids (anandamide and 2-AG), palmitoylethanolamide (PEA) and oleoylethanolamide (OEA) levels were measured in Caco-2 cells. Cells were exposed to cannabidiol (10 μM) for 24 h and harvested in 70 % methanol before cell processing, subsequently extracted, purified and analysed by isotope dilution liquid chromatography-atmospheric pressure-chemical ionisation mass spectrometry [6, 12].
Genotoxicity studies: comet assay
Genotoxicity studies were performed by single cell electrophoresis assay (Comet assay) [21] Following 24-h exposure to cannabidiol (10 μM), Caco-2 cells were incubated with 75 μM H2O2 (damaging stimulus) or phosphate-buffered saline PBS (undamaging stimulus) for 5 min. After centrifugation at 1,000×g for 5 min, pellets were mixed with 0.85% low melting point agarose and added to 1% normal melting point agarose gels. Gels were then suspended in 2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris and 1% Triton X-100, pH 10 at 4°C for 1 h and electrophoresed in alkaline buffer (300 mM NaOH, 1 mM Na2EDTA, pH 12) at 26 V, 300 mA for 20 min. After neutralisation in 0.4 M Tris–HCl (pH 7.5), gels were stained with 2 μg/ml ethidium bromide. Images were analysed using a Leica microscope equipped with a Casp software.
Drugs
AOM, MTT, 3-amino-7-dimethylamino-2-methylphenazine hydrochloride (NR) were purchase from Sigma (Milan, Italy); AM251, AM630, capsazepine and GW9662 were obtained from Tocris Cookson (Bristol, UK). Rimonabant and SR144528 (N-[-1S-endo-1,3,3-trimethyl bicyclo [2.2.1] heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-pyrazole-3-carboxamide) were from Sanofi-Aventis (Montpellier, France). Methyl-[3H]-thymidine was purchased from PerkinElmer (Monza, Italy). Cannabidiol (purity by high-performance liquid chromatography, 99.76%) was kindly supplied by GW Pharmaceuticals (Porton Down, Wiltshire, UK). All reagents for Western blot analyses, and cell cultures were obtained from Sigma (Milan, Italy), Bio-Rad Laboratories (Milan, Italy) and Microtech Srl (Naples, Italy). The vehicles for in vivo experiments (10% ethanol, 10% Tween-20, 80% saline and 0.2 ml/kg) and in vitro experiments (0.1% DMSO v/v in cell media) had no effect on the response under study.
Statistics
Data are expressed as the mean ± standard error (SE mean) of n experiments. To determine statistical significance, Student’s t test was used for comparing a single treatment mean with a control mean, and an one-way analysis of variance followed by a Tukey–Kramer multiple comparisons test was used for analysis of multiple treatment means. The chi-square test was used to evaluate the significance between the number of mice with or without ACF, polyps or tumours. P’s < 0.05 were considered significant. The IC50 value, i.e. the concentration of cannabidiol able to produce 50% of maximal inhibition of cell proliferation (geometric mean ± 95% CL), was calculated with the aid of a computer programme (GraphPad Prism 4).
Results
Effect of cannabidiol on the formation of ACF, polyps and tumours
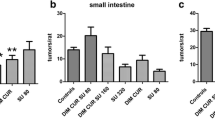
The carcinogenic agent AOM given alone induced the expected appearance of ACF (Fig. 1a), polyps (Fig. 1b) and tumours (Fig. 1c) after 3 months of treatment. The percentage of mice bearing ACF containing four or more crypts, polyps and tumours observed after the treatment with AOM is shown in Table 1.

Aberrant crypt foci with four or more crypts (ACF ≥ 4/mouse) (a), polyps (b) and tumours (c) induced in the mouse colon by AOM: effect of cannabidiol (CBD, non-psychotropic cannabinoid, 1 and 5 mg/kg). AOM (40 mg/kg in total, IP) was administered, at the single dose of 10 mg/kg, at the beginning of the first, second, third and fourth week. CBD was given (IP) three times a week for the whole duration of the experiment starting 1 week before the first administration of AOM. Measurements were performed 3 months after the first injection of AOM. Insets: representative images of an ACF (a), polyp (b) and tumour (c) captured at ×20, ×10 and ×5 magnifications, respectively. Each bar represents the mean ± SE mean of 9–11 mice. *P < 0.05 and **P < 0.01 vs vehicle
Cannabidiol (1 mg/kg) significantly reduced AOM-induced ACF (67% inhibition) (Fig. 1a), polyps (57% inhibition) (Fig. 1b) and tumours (66% inhibition) (Fig. 1c) as well as the percentage of mice bearing polyps (Table 1). In addition, 1 mg cannabidiol showed a strong trend towards the inhibition of the percentage of mice bearing tumours (Table 1), although a conventional statistical significance was not fully achieved (P = 0.06). Cannabidiol (5 mg/kg) significantly reduced the formation of polyps (Fig. 1b) and the percentage of mice bearing polyps only (Table 1).
COX-2, iNOS, phospho-Akt and caspase-3 expression in colonic tissues
Western blot analysis revealed the expression of COX-2, iNOS, phospho-Akt and caspase-3 (Fig. 2a–d) in colonic tissues of both healthy and AOM-treated animals. The densitometric analysis indicated a significant increase in the expression of COX-2 (Fig. 2a), iNOS (Fig. 2b) and phospho-Akt (Fig. 2c) in the colons of AOM-treated mice. Cannabidiol (1 mg/kg) did not cause significant changes in the expression of COX-2 and iNOS in AOM-treated animals (Fig. 2a, b) but significantly reduced AOM-induced Akt protein phosphorylation (Fig. 2c). AOM treatment caused a significant down-regulation of cleaved caspase-3 expression, which was restored by cannabidiol (Fig. 2d).

Cyclooxygenase-2 (COX-2) (a), inducible nitric oxide synthase (iNOS) (b), phospho-Akt (c) and cleaved caspase-3 (active fragment p17) (d) expression in colonic tissues of mice treated or not with AOM: effect of cannabidiol (CBD, 1 mg/kg). AOM (40 mg/kg in total, IP) was administered, at the single dose of 10 mg/kg, at the beginning of the first, second, third and fourth week. CBD was administered (IP) three times a week for the whole duration of the experiment starting 1 week before the first administration of AOM. Each bar represents the mean ± SE mean of four to five independent experiments. *P < 0.05 and **P < 0.01 vs control; # P < 0.05 vs AOM
Cytotoxicity (neutral red) assays in human colon adenocarcinoma cells
The effect of cannabidiol on cell viability, using the neutral red assay, was evaluated in both Caco-2 and HCT116 cell lines (Table 2). Cannabidiol, at the concentration ranging from 0.01 to 10 μM, did not affect Caco-2 or HCT116 cell viability after 24-h exposure. DMSO (20% v/v), used as positive control, significantly (P < 0.001) reduced both Caco-2 and HCT116 cell viability (Table 2).
Antiproliferative effect of cannabidiol
The effect of non-cytotoxic concentrations of cannabidiol (CBD, 0.01–10 μM) was evaluated on cell proliferation in both Caco-2 and HCT116 cells using two different techniques. In both cell lines and using MTT assay and 3H-thymidine incorporation, cannabidiol exerted a significant antiproliferative effect (Fig. 3) [IC50 (95% CL) in the 3H-thymidine incorporation assay: 0.67 (0.0145–31.4) μM]. Using the MTT assay, we found that the effect of CBD (10 μM) on Caco-2 cell proliferation was counteracted by rimonabant (0.1 μM) and AM251 (1 μM) (two CB1 receptor antagonists), capsazepine (1 μM, a TRPV1 receptor antagonist) and GW9662 (10 μM, a PPARγ receptor antagonist) (Fig. 4). By contrast, the effect of cannabidiol was not significantly changed by SR144528 (10 μM) and AM630 (1 μM) (CB2 receptor antagonists) (Fig. 4). All receptor antagonists employed in this set of experiments were not cytotoxic and did not affect, per se, cell proliferation (data not shown).

Antiproliferative effects of cannabidiol (0.01–10 μM, 24-h exposure) in Caco-2 (a, b) and HCT116 (c, d) cells. Proliferation rate was studied using two different techniques: the MTT assay (a, c) and the 3H-thymidine incorporation (b, c). Each bar represents the mean ± SE mean of three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 vs control

Antiproliferative effects, evaluated by MTT assay, of cannabidiol (CBD, 10 μM, 24 h-exposure) alone or in the presence of rimonabant (0.1 μM) and AM251 (1 μM) (two selective CB1 receptor antagonists), SR144528 (10 μM) and AM630 (1 μM) (two selective CB2 receptor antagonists), capsazepine (1 μM) (a TRPV1 antagonist) and GW9662 (10 μM) (a PPARγ antagonist). The antagonists were incubated 30 min before CBD. Each bar represents the mean ± SE mean of three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 vs control; # P < 0.05 and ## P < 0.01 vs CBD
Endocannabinoids, palmitoylethanolamide and oleoylethanolamide levels in Caco-2 cells
The exposure to CBD (0.1–10 μM) for 24 h induced an increase in 2-AG levels (Fig. 5b) in subconfluent Caco-2 cells. The effect was significant for the 0.1 μM concentration. No significant differences were observed in anandamide, palmitoylethanolamide and oleoylethanolamide levels following cannabidiol (0.1–10 μM) incubation for 24 h (Fig. 5a, c and d).

Levels of anandamide (a), 2-arachidonoylglycerol (2-AG, b), palmitoylethanolamide (PEA, c) and oleoylethanolamide (OEA, d) in Caco-2 cells exposed to cannabidiol (0.1–10 μM, 24 h). Each bar represents the mean ± SE mean of three independent experiments. *P < 0.05 vs control
Phospho-Akt expression in Caco-2 cells
Figure 6 shows the effect of cannabidiol on Akt phosphorylation in Caco-2 cells. Cannabidiol (10 μM for 24 h) was able to significantly reduce the expression of phospho-Akt in proliferating Caco-2 cells.

Effect of cannabidiol (CBD, 10 μM for 24 h) on phospho-Akt expression in proliferating Caco-2 cells. Each bar represents the mean ± SE mean of three independent experiments. ***P < 0.001 vs control
Genotoxicity assay
Compared to the control cells (a), cannabidiol (10 μM) alone did not significantly affect DNA damage after 24-h exposure (c), suggesting the absence of a genotoxic effect (Fig. 7). Exposure of Caco-2 cells to hydrogen peroxide (75 μM) produced a significant increase in the percentage of DNA in comet tails (b), whereas a pre-treatment with cannabidiol (10 μM) (D) for 24 h significantly reduced the H2O2-induced DNA damage (Fig. 7).

Effect of cannabidiol (CBD, 10 μM for 24 h) on hydrogen peroxide (H2O2)-induced DNA damage evaluated by the comet assay. The DNA damage was induced in Caco-2 cells by 75 μM H2O2 (b) and compared with PBS-treated (undamaged) cells (a). The effect of CBD was studied in presence (d) or absence (c) of H2O2. a–d Representative comets. Each bar represents the mean ± SE mean of three independent experiments where at least 75 cells per gel in triplicate were scored. ### P < 0.001 vs undamaged cells (a, PBS) and ***P < 0.001 vs damaged cells (b, H2O2). DNA damage, expressed as percentage of fluorescence in the comet tail (% DNA tail) was quantified using at least 75 cells per gel were scored and each sample was evaluated in triplicate (n = 3 independent experiments)
Discussion
Although cannabidiol has been shown to kill glioma cells [22], to inhibit cancer cell invasion [23] and to reduce the growth of breast carcinoma and lung metastases in rodents [24, 25], its effect on colon carcinogenesis has not been evaluated to date. This is an important omission, since colon cancer affects millions of individuals in Western countries [1–3]. In the present study, we have shown that cannabidiol exerts (1) protective effects in an experimental model of colon cancer and (2) antiproliferative actions in colorectal carcinoma cells.
Effect of cannabidiol in the AOM model of colon cancer
We have shown that cannabidiol exerted beneficial effects in AOM-treated mice. More specifically, we found that cannabidiol, at the dose of 1 mg/kg, exerted an optimal chemopreventive effect, by being able to significantly reduce ACF, polyps and tumours. At the higher 5 mg/kg dose, it prevented the formation of polyps only. The lack of a dose-related effect of cannabidiol has been also previously documented in several in vivo pharmacological assays [9]. Because an optimal pharmacological effect was observed at the 1 mg/kg, ex vivo intestinal biochemical changes (i.e. caspase-3, phospho-Akt, iNOS, COX-2 evaluations) were evaluated in animals treated with this dose of cannabidiol. We found that the protective effect of cannabidiol on colon carcinogenesis was associated with up-regulation of the active fragment of caspase-3, i.e. one of the major final effectors of the apoptotic process [26]. Pro-apoptotic mechanisms induced by cannabidiol have been previously documented in human breast carcinoma and glioma cells [22, 24].
When we investigated the potential role of the phosphoinositide 3-kinase (PI3K)/Akt pathway, which is crucial for the regulation of cell growth, migration, differentiation, and apoptosis [27, 28], we found that cannabidiol counteracted AOM-induced up-regulation of the phosphorylated form of Akt protein (and it also down-regulated Akt phorsphorylation in Caco-2 cells). These data are suggestive of an involvement of the PI3K-Akt survival signalling cascade in cannabidiol-induced protective effect. Interestingly, Greenbough and colleagues found that the psychotropic cannabinoid ∆9-THC, via CB1 activation, induced apoptosis in colorectal cancer cells and that its protective effect also involved inhibition of the PI3K-Akt survival signalling cascade [29].
Finally, we found that cannabidiol did not change the over-expression of COX-2 and iNOS, two key enzymes involved in colon carcinogenesis [30, 31]. Likewise, the protective effect of cannabidiol against glioma in vivo was not associated with changes in COX-2 activity in glioma tumour tissues [26]. We have previously shown that the antinflammatory effect of cannabidiol in the gut is associated with down-regulation of iNOS, but not COX-2, expression [12].
Effect of cannabidiol on colorectal carcinoma cell lines
In order to identify the potential receptor(s) underlying the antitumoural action of cannabidiol in the gut, we investigated the effect of this non-psychotropic cannabinoid on colorectal carcinoma cell lines. Cannabidiol is known to exert antiproliferative effects in different tumour cell lines [21, 24]. In the present study, we have shown that this compound, at non-cytotoxic concentrations, exerts antiproliferative effects in two different colorectal carcinoma cell lines, i.e. Caco-2 and HCT116 cells. A complete concentration–response effect was observed using the 3H-thymidine incorporation—but not the MTT—method. This difference probably reflects the diverse sensitivity of the two methods, being the 3H-thymidine incorporation assay more sensitive than the MTT assay [32].
To evaluate the target(s) downstream the in vitro effect of cannabidiol, we investigated, in Caco-2 cells, the potential involvement of: (1) cannabinoid receptors because cannabidiol may increase endocannabinoid levels [33, 15], which, in turn, may exert antiproliferative effects in vitro via CB1 and, only in part, CB2 receptor activation [34]; (2) TRPV1 because cannabidiol may directly activate this receptor [15]; in addition, anandamide, an endogenous TRPV1 ligand [15], is elevated in the AOM model of colon cancer [6], as well as in biopsies of patients with colon cancer [34]; (3) PPARγ because cannabidiol may activate PPARγ [35] and PPARγ agonists exert protective effect in colon carcinogenesis [36]. Our data show that the antiproliferative effect of cannabidiol was counteracted by rimonabant and AM251 (two CB1 receptor antagonists), capsazepine (a TRPV1 receptor antagonist) and GW9662 (a PPARγ receptor antagonist), thus suggesting that this non-psychotropic phytocannabinoid may exert anti-cancer effects in vitro through multiple mechanisms. In line with our results, it has been recently demonstrated that cannabidiol reduces intestinal permeability in Caco-2 cells in a CB1 and TRPV1 antagonist sensitive manner [37]. Interestingly, it has been previously demonstrated that the TRPV1 agonist capsaicin induces apoptosis in colorectal carcinoma cell lines by activating PPARγ [38]. Because cannabidiol does not bind CB1 receptors with high affinity, the reversal by rimonabant could be explained by indirect activation of such receptors, e.g. via enhancement of endocannabinoid(s) in colorectal carcinoma cell lines. Indeed, we have here demonstrated that cannabidiol was able to increase 2-AG levels in Caco-2 cells. In addition, anandamide levels appeared to be increased with this concentration of cannabidiol although in a non-statistically significant manner. Although FAAH is not the primary enzyme involved in 2-AG metabolism [4], we have previously demonstrated, in both Caco-2 cells and colon of AOM-treated mice [6, 33], that arachidonoyl serotonin, another FAAH inhibitor, increases the content of both anandamide and 2-AG.
Finally, using the single cell electrophoretic assay (Comet assay), a widely accepted tool for investigating DNA damage, we have demonstrated that cannabidiol was unable to induce DNA damage and, more importantly, whereas it exerted protective effects against hydrogen peroxide-induced DNA damage. These results are of interest because DNA mutation is a crucial step in carcinogenesis and oxidatively derived DNA lesions have been observed in many tumours, where they are strongly implicated in the aetiology of colon cancer.
Conclusions
In conclusion, we have here demonstrated here that the non-psychotropic phytocannabinoid cannabidiol exerts chemopreventive effects in an experimental model of colon cancer, an effect that is associated with down-regulation of phospho-Akt and up-regulation of caspase-3. Studies on colorectal carcinoma cells suggest that cannabidiol protects DNA damage caused by an oxidative insult and exerts antiproliferative effects through multiple mechanisms, including involvement of CB1 receptors, TRPV1 and PPARγ. In the light of its safety records and considering that cannabidiol is already currently used in patients with multiple sclerosis [9], our findings suggest that cannabidiol might be worthy of clinical consideration in colon cancer prevention.
References
Siegel R, Ward E, Brawley O, Jemal A (2011) Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 61:212–236
Markowitz SD, Bertagnolli MM (2009) Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 361:2449–2460
Half E, Arber N (2009) Colon cancer: preventive agents and the present status of chemoprevention. Expert Opin Pharmacother 10:211–219
Di Marzo V (2008) Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov 7:438–455
Pertwee RG (2009) Emerging strategies for exploiting cannabinoid receptor agonists as medicines. Br J Pharmacol 156:397–411
Izzo AA, Aviello G, Petrosino S, Orlando P, Morsicano G, Lutz B, Borrelli F, Capasso R, Nigam S, Capasso F et al (2008) Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J Mol Med 86:89–98
Cianchi F, Papucci L, Schiavone N, Lulli M, Magnelli L, Vinci MC, Messerini L, Manera C, Ronconi E, Romagnani P et al (2008) Cannabinoid receptor activation induces apoptosis through tumor necrosis factor alpha-mediated ceramide de novo synthesis in colon cancer cells. Clin Cancer Res 14:7691–7700
Wang D, Wang H, Ning W, Backlund MG, Dey SK, DuBois RN (2008) Loss of cannabinoid receptor 1 accelerates intestinal tumor growth. Cancer Res 68:6468–6476
Izzo AA, Borrelli F, Capasso R, Di Marzo V, Mechoulam R (2009) Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol Sci 30:515–527
Capasso R, Borrelli F, Aviello G, Romano B, Scalisi C, Capasso F, Izzo AA (2008) Cannabidiol, extracted from Cannabis sativa, selectively inhibits inflammatory hypermotility in mice. Br J Pharmacol 154:1001–1008
Cluny NL, Naylor RJ, Whittle BA, Javid FA (2011) The effects of cannabidiolic acid and cannabidiol on contractility of the gastrointestinal tract of Suncus murinus. Arch Pharm Res 34:1509–1517
Borrelli F, Aviello G, Romano B, Orlando P, Capasso R, Macello F, Guadagno F, Petrosino S, Capasso F, Di Marzo V et al (2009) Cannabidiol, a safe and non-psychotropic ingredient of the marijuana plant Cannabis sativa, is protective in a murine model of colitis. J Mol Med 87:1111–1121
Jamontt JM, Molleman A, Pertwee RG, Parsons ME (2010) The effects of delta-tetrahydrocannabinol and cannabidiol alone and in combination on damage, inflammation and in vitro motility disturbances in rat colitis. Br J Pharmacol 160:712–723
Terzić J, Grivennikov S, Karin E, Karin M (2010) Inflammation and colon cancer. Gastroenterology 138:2101–2114
De Petrocellis L, Ligresti A, Moriello AS, Allarà M, Bisogno T, Petrosino S, Stott CG, Di Marzo V (2011) Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol 163:1479–1494
Izzo AA, Sharkey KA (2010) Cannabinoids and the gut: new developments and emerging concepts. Pharmacol Ther 126:21–38
Klaunig JE, Wang Z, Pu X, Zhou S (2011) Oxidative stress and oxidative damage in chemical carcinogenesis. Toxicol Appl Pharmacol 254:86–99
Fukuyama R, Niculaita R, Ng KP, Obusez E, Sanchez J, Kalady M, Aung PP, Casey G, Sizemore N (2008) Mutated in colorectal cancer, a putative tumor suppressor for serrated colorectal cancer, selectively represses beta-catenin-dependent transcription. Oncogene 27:6044–6055
Dunn EF, Iida M, Myers RA, Campbell DA, Hintz KA, Armstrong EA, Li C, Wheeler DL (2011) Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene 30:561–574
Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, Besselsen DG, Godfrey VL, Doetschman T, Dove WF et al (2003) Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 124:762–777
Aviello G, Rowland I, Gill CI, Acquaviva AM, Capasso F, McCann M, Capasso R, Izzo AA, Borrelli F (2010) Anti-proliferative effect of rhein, an anthraquinone isolated from Cassia species, on Caco-2 human adenocarcinoma cells. J Cell Mol Med 14:2006–2014
Massi P, Vaccani A, Bianchessi S, Costa B, Macchi P, Parolaro D (2006) The non-psychoactive cannabidiol triggers caspase activation and oxidative stress in human glioma cells. Cell Mol Life Sci 63:2057–2066
Ramer R, Merkord J, Rohde H, Hinz B (2010) Cannabidiol inhibits cancer cell invasion via upregulation of tissue inhibitor of matrix metalloproteinases-1. Biochem Pharmacol 79:955–966
Ligresti A, Moriello AS, Starowicz K, Matias I, Pisanti S, De Petrocellis L, Laezza C, Portella G, Bifulco M, Di Marzo V (2006) Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J Pharmacol Exp Ther 318:1375–1387
Shrivastava A, Kuzontkoski PM, Groopman J, Prasad A (2011) Cannabidiol induces programmed cell death in breast cancer cells by coordinating the crosstalk between apoptosis and autophagy. Mol Cancer Ther 10:1161–1172
Kim R (2005) Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer 103:1551–1560
Sheng H, Shao J, Townsend CM Jr, Evers BM (2003) Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut 52:1472–1478
Wang Q, Wang X, Hernandez A, Kim S, Evers BM (2001) Inhibition of the phosphatidylinositol 3-kinase pathway contributes to HT29 and Caco-2 intestinal cell differentiation. Gastroenterology 120:1381–1392
Greenhough A, Patsos HA, Williams AC, Paraskeva C (2007) The cannabinoid delta(9)-tetrahydrocannabinol inhibits RAS-MAPK and PI3K-AKT survival signalling and induces BAD-mediated apoptosis in colorectal cancer cells. Int J Cancer 121:2172–2180
Rao CV (2004) Nitric oxide signaling in colon cancer chemoprevention. Mutat Res 555:107–119
Wu WK, Sung JJ, Lee CW, Yu J, Cho CH (2010) Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: an update on the molecular mechanisms. Cancer Lett 295:7–16
Wiepz GJ, Edwin F, Patel T, Bertics PJ (2006) Methods for determining the proliferation of cells in response to EGFR ligands. Methods Mol Biol 327:179–187
Izzo AA, Camilleri M (2009) Cannabinoids in intestinal inflammation and cancer. Pharmacol Res 60:117–125
Ligresti A, Bisogno T, Matias I, De Petrocellis L, Cascio MG, Cosenza V, D’argenio G, Scaglione G, Bifulco M, Sorrentini I et al (2003) Possible endocannabinoid control of colorectal cancer growth. Gastroenterology 125:677–687
O’Sullivan SE, Sun Y, Bennett AJ, Randall MD, Kendall DA (2009) Time-dependent vascular actions of cannabidiol in the rat aorta. Eur J Pharmacol 612:61–68
Osawa E, Nakajima A, Wada K, Ishimine S, Fujisawa N, Kawamori T, Matsuhashi N, Kadowaki T, Ochiai M, Sekihara H et al (2003) Peroxisome proliferator-activated receptor gamma ligands suppress colon carcinogenesis induced by azoxymethane in mice. Gastroenterology 124:361–367
Alhamoruni A, Lee AC, Wright KL, Larvin M, O’Sullivan SE (2010) Pharmacological effects of cannabinoids on the Caco-2 cell culture model of intestinal permeability. J Pharmacol Exp Ther 335:92–102
Kim CS, Park WH, Park JY, Kang KH, Kim M, Kawada T, Han I, Yu R (2004) Capsaicin, a spicy component of hot pepper, induces apoptosis by activation of the peroxisome proliferator-activated receptor gamma in HT-29 human colon cancer cells. J Med Food 7:267–273
Acknowledgements
GA is grateful to Nexus award “Marcello Tonini”.
Conflict of interest
This work was partly supported by GW Pharmaceuticals (UK).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Aviello, G., Romano, B., Borrelli, F. et al. Chemopreventive effect of the non-psychotropic phytocannabinoid cannabidiol on experimental colon cancer. J Mol Med 90, 925–934 (2012). https://doi.org/10.1007/s00109-011-0856-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-011-0856-x