
Abstract
Hemicerebellectomy results in remote delayed degeneration of precerebellar neurons. We have reported that such a lesion induces type 2 cannabinoid receptor (CB2) expression in precerebellar neurons and that stimulation of CB2, but not CB1, has neuroprotective effects. In this study, we found that in the same model, the CB2 agonist JWH-015 enhances neuronal nitric oxide synthase (nNOS) expression in axotomized neurons and that CB2-mediated neuroprotection is abrogated by pharmacological inhibition of nNOS. JWH-015 prevented the axotomy-induced upregulation of inducible NOS (iNOS) in astrocytes but had no effect on endothelial NOS (eNOS). In addition, we observed that JWH-015 significantly reduces hemicerebellectomy-induced neuroinflammatory responses and oxidative/nitrative stress. With regard to the signaling pathways of CB2/nNOS-mediated neuroprotection, we noted nNOS-dependent modulation of the expression of anti-oxidative (Hsp70) and anti-apoptotic (Bcl-2) proteins. These findings shed light on the interactions between the endocannabinoid and nitrergic systems after focal brain injury, implicating distinct functions of nNOS activation and iNOS inhibition in CB2 signaling, which protect neurons from axotomy-induced cell death.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Remote cell death, also known as remote damage, is neuronal degeneration in regions that are functionally linked to, but do not abut, the primary site of a lesion after focal brain injury [1, 2]. Such effects are associated with apoptotic [3] and inflammatory cascades [4] and are considered important predictors of outcome after brain injury [5]. Yet, the molecular changes and pathological mechanisms that are associated with remote damage have not been examined significantly.
We recently described the neuroprotective function of the endocannabinoid system (ECS) in hemicerebellectomy (HCb), a well-defined paradigm of cerebellar damage that is used to study remote cell death in axotomized neurons [6]. Using this model, we also found that axonal lesions induce the de novo synthesis of the type 2 cannabinoid (CB2) receptor, which is not expressed in neuronal cells under normal conditions, in approximately half of all lesioned neurons. Further, selective activation of CB2 impedes delayed apoptosis by signaling through the PI3K/Akt pathway [6].
These findings suggest that CB2 functions in an endogenous neuroprotective pathway that is induced by axotomy. Thus, selective agonists of CB2 are candidates for therapeutic, neuron-specific agents that do not have psychotropic effects in the management of secondary brain injury [7]. However, the components of this pathway and the mechanisms that underlie its neuroprotective effects have not been determined. Possible mediators of CB2 receptor signaling are members of the NO synthase (NOS) family—the neuronal (nNOS), endothelial (eNOS), and inducible (iNOS) isoforms [8–10].
All these three isoforms of NOS have been shown to be involved in the effects that are elicited by cannabinoid-based drugs under various pathophysiological conditions in the central and peripheral nervous systems, including neuromodulatory [11, 12], anti-excitotoxic [13], anti-inflammatory [14, 15], and vasodilatory [16] effects. Moreover, CB1 and nNOS co-localize in several areas of the brain [17]. Yet, the interactions between CB2 and the nitrergic system after brain injury have never been examined.
In this study, we investigated the function of the NOS/NO system in CB2-mediated neuroprotection by characterizing the interactions between CB2 and the nitrergic system in axotomized central neurons immunochemically, biochemically, and pharmacologically.
Following HCb, CB2 stimulation enhanced nNOS expression in neurons and concomitantly impaired the axotomy-induced up-regulation of iNOS in astrocytes. In addition, JWH-015 significantly decreased HCb-induced neuroinflammatory responses and oxidative/nitrative stress and increased the expression of proteins that mediate anti-oxidative (Hsp70) and anti-apoptotic (Bcl-2) mechanisms. Selective inhibition of nNOS expression/activity abrogated the neuroprotective effects of JWH-015 on neuronal survival and functional recovery, indicating that CB2-mediated neuroprotection requires nNOS activity.
Materials and methods
Animals and surgery
Adult male Wistar rats (200–250 g; Harlan, Italy) were used in the study. The experimental protocol used in this study was approved by the Italian Ministry of Health and was in agreement with the guidelines of the European Communities Council Directive of the 24 November 1986 (86/609/EEC) for the care and use of laboratory animals. All efforts were made to minimize the number of animals used and their suffering. For surgical procedures, the rats were deeply anesthetized by i.p. injections of tiletamine and zolazepam (50 mg/kg—Virbac, Milan, Italy) and xylazine (10 mg/kg—Bayer, Milan, Italy) and positioned in a stereotaxic apparatus. An incision was made in the skin on the skull, and the occipital bone was drilled and removed. Subsequently, the dura was incised to expose the cerebellum, and the right cerebellar hemisphere was removed by suction. The wound was sutured, and the animals were returned to their cages. For the unlesioned group, surgery was interrupted after the dura lesion was made, and after suturing, the animals were returned to their cages. The experimental groups are detailed in Table 1.
Reagents and drugs
All chemicals were of the purest analytical grade and purchased from Sigma Chemical Co. (Milan, Italy), unless stated otherwise. l-[2,3,4-3H]arginine (43 Ci/mmol) was purchased from Perkin Helmer (Milan, Italy). 7′-Dichlorodihydrofluorescein diacetate (DCFH-DA) was purchased from Invitrogen (Milan, Italy). The following CB2 receptor agonist and antagonist, respectively, were used: JWH-015 (3 mg /kg, i.p. for 7 days; Cayman Chemicals, Ann Arbor, Michigan , USA) and SR144528 (SR2; 3 mg /kg, i.p. for 7 days; Cayman Chemicals). Further, the nNOS inhibitor 3-bromo-7-nitroindazole (7NI; Sigma), dissolved in peanut oil vehicle, and the iNOS inhibitor 1400W (Cayman Chemicals) both at a dosage of 25 mg/kg body weight, were employed (Table 1).
Neurological evaluation
Neurological impairment was evaluated by the neurological severity score (NSS) [18]. The latter is a composite of motor, sensory, reflex, and balance tests, where for each test, one point is awarded for the inability to perform or for the lack of a tested reflex, and zero points are awarded for success. A NSS of 18 indicates severe injury, whereas a score of zero signifies healthy, uninjured rats. The NSS was evaluated at 24 h, 72 h, 5 days, and 7 days after damage by an investigator who was blinded to the experimental groups.
Antibodies
The following primary antibodies were used: rabbit anti-CB2 (1:200; Abcam, Cambridge, UK; #3561), mouse anti-nNOS (1:200; Sigma; #N2280), rabbit anti-iNOS (1:400; BD Bioscience, Oxford, UK; #610329), rabbit anti-eNOS (1:200; Sigma; #SAB4502013); mouse anti-GFAP (1:500; Sigma; #G3893) , mouse anti-NeuN (1:200; Millipore, Billerica, MA, USA; #MAB-377), mouse anti-3-nitrotyrosine (1:200; Abcam; #AB7048), rabbit anti-3-nitrotyrosine (1:200; Millipore; #06-284), mouse anti-cytochrome-c (1:1000; BD Pharmingen, #556433), rabbit polyclonal anti-MnSOD (1:1000; Stressgen, #SOD-110), rabbit anti-4-hydroxynonenal (4-HNE; 1:200; Alpha Diagnostic, TX, USA; #HNE-11S), and mouse anti-β-actin (1:2000; Sigma; #SAB3500350); rabbit anti-Hsp70 (1:500; Santa Cruz Biotechnologies; CA, USA; #sc-25837), or rabbit anti-Bcl-2 (1:500; Santa Cruz Biotechnologies; #sc-492). Secondary Cy3-, Cy2- and Cy5-conjugated antibodies were purchased from Jackson Immunoresearch (West Grove, PA, USA). The specificity of CB2 labeling was confirmed only by omitting the primary antibody, since in our previous study, protein content and mRNA levels for CB2 were simultaneously assessed to validate the specificity of the CB2 antibody [6].
Histology and immunohistochemistry
Rats were perfused transcardially with 250 mL of saline followed by 250 mL of 4% paraformaldehyde in a phosphate buffer (PB; 0.1 M; pH 7.4) under anesthesia induced by i.p. injections of sodium pentobarbital (60 mg/kg). Each brain was removed immediately, post-fixed in the same fixative for 2 h and, after three washes in PB, transferred to 30% sucrose in PB solution at 4°C until it sank. Brains were cut into four series of 30 μm-thick transverse sections by means of a freezing microtome and were collected in PB. To assess nNOS induction, one series of sections was processed for immunohistochemistry with a rabbit polyclonal antibody against neuronal nitric oxide synthase (nNOS; Sigma; 1:500). After quenching endogenous peroxidase with a 10-min incubation in 0.3% H2O2 in PB, the sections were incubated overnight with the primary antibody, diluted 1:500 in PB + 0.3% Triton X-100 and 4% normal donkey serum (Jackson Immunoresearch; West Grove, PA, USA), at 4°C. Immunohistochemical staining was performed using avidin-biotin-peroxidase with anti-rabbit biotinylated IgG as the secondary antibody and 3,3′-diaminobenzidine as the chromogen. Sections were mounted on chrome alum-gelatinized slides and air-dried for at least 24 h. Afterwards, the sections were counterstained using the Nissl method to assess the percentage of nNOS-expressing cells. Finally, sections were mounted on chrome alum-coated slides, then they were air dried, dehydrated with ethanol, cleared in xylene, and coverslipped. Bright field images were taken using a 20× objective of a light microscope (Zeiss, Axioskop 2) equipped with a digital camera (Nikon, Coolpix 990).
To investigate the cellular population that expressed nNOS, iNOS, 3-NT, 4-HNE, and CB2, one series of sections for each marker was incubated overnight with a cocktail of primary antibodies, including the above-mentioned antibodies and those against NeuN, GFAP, and OX-42. Sections were blocked in 5% normal donkey serum for 2 h at RT and incubated with a cocktail of primary antibodies. All primary antibody solutions were prepared in PB and 0.3% Triton X-100 and were incubated overnight. Each incubation step was followed by three, 5-min rinses in PB. Afterwards, sections were incubated 2 h at RT with a cocktail of secondary antibodies, including Cy3- or Cy2-conjugated donkey anti-rabbit IgG (1:200), Cy2- or Cy3-conjugated donkey anti-mouse IgG (1:200) and Cy5- conjugated donkey anti-goat (1:200). Sections were examined under a confocal laser scanning microscope (Leica SP5, Leica Microsystems, Wetzlar, Germany) equipped with four laser lines: violet diode emitting at 405 nm (for DAPI), argon emitting at 488 nm, and helium/neon emitting at 543 nm and 633 using a 40×/0.5 NA oil objective. Images were exported in JPEG format, contrast and brightness were adjusted, and final plates were composed with Adobe Illustrator CS3 or Corel Draw 9.
Qualitative and quantitative analyses
Qualitative and quantitative observations were limited to the pontine nuclei (Pn) of the experimental side that projects to the lesioned hemicerebellum. Using the Stereo Investigator System (MicroBrightField Europe e.K., Magdeburg, Germany), an optical fractionator, stereological design was applied to obtain unbiased estimates of total Nissl-stained and nNOS positive neurons. A stack of MAC 5000 controller modules (Ludl Electronic Products, Ltd. Hawthorne, NY, USA) was configured to interface an Olympus BX 50 microscope with a motorized stage and a HV-C20 Hitachi color digital camera with a Pentium II PC workstation. A three-dimensional optical dissector counting probe (x, y, z dimension of 30 × 30 × 10 μm, respectively) was applied. The Pn was outlined using the 4× objective, while the 100× oil immersion objective was used for marking the neuronal cells. Total Pn cell number was estimated according to the formula:
where ∑Q represents the total number of neurons counted in all optically sampled fields of the Pn, ssf is the section sampling fraction, asf is the area sampling fraction, and tsf is the thickness sampling fraction.
Quantitative analysis of nNOS/CB2, iNOS/GFAP, NeuN/3-NT, and NeuN-4HNE co-localization was performed off-line on confocal images acquired through the 20× objective at the 0.07 zoom factor. All double-labeled cells in three digital squared frames (250 μm per side) in five sections, regularly spaced throughout the caudo-rostral extent of the nucleus, were counted. Cellular co-localizations of double immunolabeled neurons and astrocytes were analyzed by counting and characterizing cell labeling off-line through the CLSM proprietary image analysis program (Zeiss; LSM software 2.3). Two digital images of the same optical section (one for each laser channel, green and red) were acquired and digitally merged in a third image, which was used for cell counting. The features of immunolabeled cells were analyzed by zooming on the cells and by serially excluding each channel (green and red) to better appreciate the cellular morphology. Double and single immunolabeled cells were then digitally marked, recorded, and the material stored in a data archive. All quantitative analyses were conducted blind to the animal's experimental group.
Densitometric analyses of fluorescence images
Quantification of the changes in the IR cytochrome c (cyt-c) signal in the experimental groups was performed on NeuN/cyt-c positive Pn neurons by densitometric analyses, as previously described [6]. After background subtraction, cyt-c cell-associated signals were quantified by manually outlining individual cells and measuring cell-associated fluorescence intensity with the ImageJ software (http://rsb.info.nih.gov/ij/). The F/A ratio defines mean fluorescence of individual cells (F) normalized to total cellular surface (A). Quantification was done on 250 cells per group (n = 5 rats per group).
Mitochondrial and cytosolic fraction
Pontine nuclei were isolated as reported previously [4] and were homogenized in buffer A (50 mM Tris–HCl (pH 7.4), 320 mM sucrose, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, with protease inhibitor cocktail—Sigma, #P8340) by 30 strokes with a glass Pyrex micro homogenizer. The homogenate was centrifuged at 1,000×g for 10 min and the resulting supernatant was centrifuged at 10,000×g for 20 min to obtain the mitochondrial pellet and the supernatant. The mitochondria-containing pellet was washed three times with buffer B (10 mM Tris–HCl (pH 7.4), 250 mM sucrose, 1 mM EGTA) by centrifugation for 10 min at 10,000×g. The supernatant was centrifuged at 100,000×g for 1 h to generate the cytosolic fraction.
Protein isolation and Western blotting
Pontine nuclei were isolated as reported previously [4] and were homogenized in lysis buffer (50 mM Tris–HCl (pH 7.5), 320 mM sucrose, 10% glycerol, 50 mM NaCl, 1% Triton X-100, 1 mM PMSF, with protease inhibitor cocktail (Sigma, #P8340)), incubated on ice for 30 min and centrifuged at 13,000×g for 20 min. The total protein content of resulting supernatant was determined. Proteins were applied to SDS-PAGE and electroblotted on a PVDF membrane. Immunoblotting analysis was performed using a chemiluminescence detection kit. The relative levels of immunoreactivity were determined by densitometry using the software ImageQuant 5.0. Samples were incubated with the specific primary antibodies. Densities of protein bands in the Western blots were measured, and mean ratios between proteins and β-actin were reported as percentage of control values.
Quantitative real-time PCR
One microgram of total RNA was used to generate cDNA with 10 U/μl SuperScript III reverse transcriptase, in the presence of 2 U/μl RNaseOUT, 1.25 μM oligos, 1.25 ng/μl random hexamers, 5 mM MgCl2, 0.5 mM dNTP mix, and DEPC-treated water. The reaction was performed using the following RT-PCR program: 25°C for 10 min, 42°C for 50 min, 85°C for 5 min, then, after addition of 0.1 U/μl of Escherichia coli RNase H, the product was incubated at 37°C for 20 min. For expression studies, the target transcripts were amplified in the ABI PRISM 7700 sequence detector (Applied Biosystems, Foster City, CA, USA), using the following primers: nNOS F1 (5′-TTCACTACACATCTGGAGACC-3′), nNOS R1 (5′-TTGAGCAGTTCATCATGTTCC-3′); iNOS F1 (5′-ATCTTTGCCACCAAGATGGC-3′), iNOS R1 (5′-ATTCCAGTTCCGGAAGTCATGC-3′); eNOS F1 (5′-AACTGCAGGTATTTGATGCTCG-3′) rat eNOS R1 (5′-ATACAGAGCTCAGTGATCTCC-3′); β-actin F1 (5′-ATCCTGACCCTGAAGTACCC-3′), and β-actin R1 (5′-AAGGTCTCAAACATGATCTGG-3′). One microliter of the first-strand cDNA product was used for amplification in triplicate. A twenty-five-microliter reaction solution contained 12.5 μl of Platinum SYBR Green qPCR SuperMix-UDG and 10 pmol of each primer. The following PCR program was used: 95°C for 10 min, 40 amplification cycles at 95°C for 30 s, 56°C for 30 s, and 72°C for 30 s. Serial dilutions (10, 100, 1,000, 10,000, and 100,000-fold) of each studied transcript were used to determine the amplification efficiency of each target and housekeeping gene. Quantitative RT-PCR (qRT-PCR) analyses of mRNA levels were performed comparing mRNA extracts from unlesioned and hemicerebellectomized rats using the 2−ΔΔCT method Viscomi et al. [6]. In the present study, data are presented as the fold-change in target gene expression normalized to the internal control gene and relative to the normal tissue control. The average threshold cycle (C T) was calculated for both the target genes and β-actin, and ΔC T was determined as [the mean of the triplicate C T values for the target gene] − [the mean of the triplicate C T values for β-actin]. ΔΔC T represented the difference between the paired tissue samples, as calculated by the formula ΔΔC T = (ΔC T of HCb-Eth − ΔC T of CTRL). The n-fold differential expression of the target gene of an HCb sample compared with the normal counterpart was expressed as 2−ΔΔCT.
Determination of nitrite and nitrate
The evaluation of nitrite and nitrate levels was performed by a colorimetric assay based on the Griess reaction. Briefly, the brain supernatants were deproteinizated by ultrafiltration through YM-10 Centricon tubes (molecular weight cut-off, 10 kDa) and then used for nitrite/nitrate determination: 100 μL of each sample were transferred into the wells of a 96-microplate in triplicate, then 100 μL of 8 mg/mL VCl3 (prepared in 1 M HCl) were added to each well (to reduce nitrate to nitrite), and this was rapidly followed by addition of 100 μL of the Griess reagent (50 μL of 2% sulphanamide, prepared in 5% orthophosphoric acid; 50 μL of 0.1% N-1-naphtyl-ethylendamin dihydrochlorid, prepared in milliQ water). Then, the reaction mixture was incubated at 37°C in the dark for 3 h, and absorbance was read at 540 nm in an ELISA microplate reader (Tecam, Mannedorf, Switzerland). Sodium nitrate solutions (0.05–10 μM) were used for preparing calibration curves.
NOS activity
NOS activity was determined in Pn homogenates by assessing the conversion of l-[3H]arginine into l-[3H]citrulline. Protein fractions were brought to 100 μL in a reaction mixture that contained 10 mM HEPES (pH 7.4), 0.75 mM ß-NADPH, 1.25 mM CaCl2, 20 μM l-arginine, 187 nM l-[2,3,4-3H]arginine, 5 μM tetrahydrobioterin, 50 nM calmodulin, 0.5 μM flavin mononucleotide, 0.5 μM flavin adenine dinucleotide, 0.5 mM dithiothreitol (final concentrations). To measure iNOS activity, CaCl2 was substituted by 1 mM EGTA. The reaction mixture was incubated for the indicated periods of time, and the reaction was terminated by the addition of 250 μL ice-cold stop buffer (50 mM HEPES (pH 5), 10 mM EGTA, 10 mM EDTA). l-[3H]Citrulline was separated by anion exchange chromatography and quantified by liquid scintillation spectroscopy. NOS enzyme activity was expressed as femtomoles of [3H]citrulline formed per minute per milligram of protein.
Determination of ROS
For quantification of ROS level, Pn homogenates were diluted in HEPES-based Locke's buffer (5 mM HEPES (pH 7.4), 154 mM NaCl, 5.6 mM KCl, 3.6 mM NaHCO3, 2.0 mM CaCl2, 10 mM glucose) to obtain a concentration of 5 mg tissue/mL. The homogenates (195 μL) were pipetted into NUNC 96-well black plate and allowed to warm to room temperature for 5 min. Then, 5 μL of DCFH-DA (10 μM final concentration) was added to each well and pre-incubated for 15 min at room temperature, in order to allow DCFH-DA incorporation into any membrane-bound vesicles and cleavage of diacetate group by esterases. After further 60 min of incubation, the conversion of DCFH-DA into the fluorescent product DCF was measured using the Viktor (PerkinElmer Life Sciences, Yokohama, Japan) with excitation at 485 nm and emission at 530 nm. ROS production was quantified using a DCF standard curve. Background fluorescence (conversion of DCFH-DA in the absence of homogenate) was corrected by including blanks.
Statistical analyses
All values were expressed as mean ± SEM. Differences between means were analyzed using one-way ANOVA with a Bonferroni post hoc test. All statistical analyses were performed using Prism-4 software (GraphPAD Software for Science, San Diego, CA, USA) with significance set at p < 0.05.
Results
Axotomy induces CB2 and nNOS coexpression in neurons and iNOS expression in astrocytes
We have observed that following HCb, nNOS- and CB2-dependent signaling pathways are induced in contralateral precerebellar stations 7 days postlesion. In particular, HCb-induced distal axotomy leads to the de novo synthesis of nNOS [19] and CB2 [6] in many Pn neurons. Laser confocal evidence of neuronal but not glial nNOS or CB2 labeling in this model have been reported in different studies [6, 19].
Before the lesion, no CB2 or nNOS positive cells were present (see CTRL in Fig. 1a). At 7 days after HCb, about 50% of surviving neurons express CB2 and about 80% of them express also nNOS, as shown by co-localization experiments. Only a small population of neurons were single positive presenting either nNOS (∼10%) or CB2 (∼10%) immunolabeling (see CTRL + Sal vs. HCb + Sal in Fig. 1a, b).

Hemicerebellectomy induces CB2/nNOS and iNOS/GFAP colocalization in pontine nuclei. a Double-labeled and merged confocal images of CB2 (red) and nNOS (green) in neurons in unlesioned (CTRL + Sal) and at 7 days after HCb (HCb + Sal). b Histogram of the percentages of CB2- and nNOS-positive neurons and CB2/nNOS colocalization in CTRL + Sal and HCb + Sal. c Histogram of the percentages of GFAP- and iNOS-positive astrocytes and GFAP/iNOS colocalization in CTRL + Sal and HCb + Sal. d Double-labeled and merged confocal images of GFAP (green) plus DAPI counterstaining (blue) and iNOS (red) in CTRL + Sal and HCb + Sal. e Histogram of the percentages of CB2- and nNOS-positive neurons in CTRL (0 days) and at various time points after HCb. f Histogram of the percentages of iNOS-positive astrocytes (GFAP positive) in CTRL (0 days) and at various time points after HCb. Scale bars: a, d = 20 μm
To determine the involvement of other NOS isoforms in HCb, we examined the expression of iNOS and eNOS before and after the lesion. Control animals (CTRL + Sal) were negative for iNOS, whereas eNOS-positive endothelial cells were observed (data not shown); HCb induced iNOS expression (Fig. 1c, d) without altering that of eNOS (data not shown). In lesioned animals, double immunofluorescence of iNOS and GFAP, a marker of activated astrocytes, demonstrated that iNOS was induced in reactive astrocytes (Fig. 1d), whereas double-labeling of iNOS with NeuN, a neuronal marker, or OX-42, a microglial marker, showed that neurons and microglial cells did not express iNOS (data not shown). At 7 days after HCb, in contralesional Pn, there was significant colocalization between GFAP and iNOS with only minor populations of cells that were positive for iNOS (∼3%) or GFAP (∼4.5%) only (Fig. 1c). These data suggest that HCb induces nNOS expression only in neurons and iNOS expression only in astrocytes.
Time course analyses of the expression of these proteins upon lesion demonstrated that CB2 induction preceded that of nNOS and iNOS (Fig. 1e, f). In fact, significant increment of CB2+ neurons was observed starting from 4 days after HCb, while nNOS in neurons and iNOS in astrocytes appeared only at 7 days (Fig. 1e, f). This time frame was also confirmed by CB2/nNOS co-localization quantitative analyses. At 4 days, only 2% of CB2 neurons presented also nNOS expression, while at 7 days, this percentage raised to 80% (compare the panels b and e in Fig. 1).
Stimulation of CB2 has contrasting effects on nNOS and iNOS activity and expression
To determine the effect of CB2 signaling on the NOS/NO system following HCb, the expression and activity profiles of the NOS isoforms were measured in the Pn of unlesioned and lesioned rats that were treated chronically with a selective CB2 agonist (JWH-015) or antagonist (SR144528) for 7 days (Table 1). Pn samples from unlesioned and lesioned animals were analyzed by qRT-PCR, immunoblotting, immunohistochemistry, enzymatic assay, and spectrophotometry of stable NO metabolites.
In the absence of pharmacological treatment, consistent with our morphological observations, HCb enhanced nNOS (HCb + Sal vs. CTRL + Sal, p < 0.001) and iNOS (HCb + Sal vs. CTRL + Sal, p < 0.001) but not eNOS (HCb + Sal vs. CTRL + Sal, p > 0.05) mRNA and protein levels (Fig. 2a, b). These increases corresponded to rises in the tissue concentrations of stable NO metabolites—nitrite and nitrate (NOx, Fig. 2c)—and in specific enzymatic activity, which climbed from 355 ± 20 fmol/min per milligram protein to 450 ± 20 fmol/min per milligram protein (CTRL + Sal vs. HCb + Sal; p < 0.05; Table 2).

Pharmacological manipulation of CB2 influences nNOS and iNOS expression and activity. a nNOS, iNOS, and eNOS mRNA expression in HCb + Sal, CTRL + JWH, HCB + JWH, HCB + JWH + SR2 and HCb + SR2 animals, expressed as percentage of CTRL + Sal. b nNOS, iNOS, and eNOS protein levels in HCb + Sal, CTRL + JWH, HCB + JWH, HCB + JWH + SR2, and HCb + SR2 animals, expressed as percentage of CTRL + Sal. c NOx levels in HCb + Sal, CTRL + JWH, HCB + JWH. HCB + JWH + SR2 and HCb + SR2 animals expressed as percentage of CTRL + Sal, showing the effectiveness of JWH in increasing NOx levels (HCb + JWH vs. HCb + Sal). d Histograms of stereological counts of Nissl- and nNOS-positive neurons and nNOS/Nissl ratios in CTRL + Sal, HCb + Sal, CTRL + JWH, HCb + JWH, HCB + JWH + SR2, and HCb + SR2. Data are expressed as mean ± SEM (n = 5/group). One-way ANOVA followed by Bonferroni multiple comparison test. *p < 0.05 vs. HCb + Sal; **p < 0.01 vs. HCb + Sal; ***p < 0.001 vs. HCb + Sal; # p < 0.001 vs. CTRL + Sal; ## p < 0.001 vs. CTRL + Sal; ### p < 0.001 vs. CTRL + Sal
Notably, chronic treatment with JWH-015 altered HCb-induced nNOS and iNOS expression. Compared with the pattern of HCb animals, CB2 stimulation upregulated nNOS mRNA and protein and downregulated iNOS (HCb + JWH; Fig. 2a, b and Table 2). Conversely, the antagonism of CB2 by SR144528 completely reversed the effects of JWH-015 on nNOS and iNOS activity and expression (HCb + JWH + SR2; Fig. 2a, b and Table 2). Interestingly, chronic blockade of CB2 with SR144528 caused a small but significant reduction in nNOS expression and activity but not that of the other NOS isoforms (HCb + SR2; Fig. 2a, b and Table 2). Consistent with the absence of CB2 under basal conditions [7], JWH-015 did not affect the transcriptional or translational profiles of the NOS isoforms in controls (CTRL + JWH; Fig. 2a, b).
The influence of CB2 activation on nNOS expression was confirmed by immunohistochemistry (Fig. 2d). By post hoc comparison, the total number of nNOS-positive neurons and the nNOS/Nissl ratio were significantly higher in HCb + JWH animals than in the HCb + Sal group (Fig. 2d; p < 0.001). Again, the blockade of CB2 by SR144528 completely reversed the effects of JWH-015 on both nNOS-positive neurons and nNOS/Nissl ratio (HCb + JWH + SR2 vs. HCb + JWH, p < 0.01; Fig. 2d). Further, the total number of nNOS-positive neurons and the percentage of nNOS-expressing neurons fell significantly with SR144528 (HCb + SR2 vs. HCb + Sal, p < 0.01).
Stimulation of CB2 impairs HCb-induced astrocytic and microglial responses
By immunohistochemistry, HCb was accompanied by a marked inflammatory reaction, characterized by astrocytic and microglial activation (HCb + Sal vs. CTRL + Sal, p < 0.001; Fig. 3), as reported [4]. Pharmacological activation of CB2 attenuated the postlesional activation of astrocytes, as demonstrated by the reduction in the total number of GFAP-positive astrocytes (HCb + JWH vs. HCb + Sal, p < 0.001; Fig. 3a, c) and in iNOS/GFAP ratio (HCb + JWH vs. HCb + Sal, p < 0.001; Fig. 3a). Similarly, the total number of OX-42-positive microglial cells fell significantly with JWH-015 (HCb + JWH vs. HCb + Sal, p < 0.001; Fig. 3b, c). The blockade of CB2 by SR144528 completely reversed the inhibitory effects of JWH-015 on astrocytic and microglial activation, as well as on iNOS/GFAP ratio (HCb + JWH + SR2; Fig. 3a, b).

Pharmacological manipulation of CB2 impairs astrocytic and microglial responses. a Histograms of the number of GFAP- and iNOS-positive cells and iNOS/GFAP ratios in CTRL + Sal, HCb + Sal, CTRL + JWH, HCb + JWH, HCB + JWH + SR2, and HCb + SR2. b Histograms of the number of OX-42-positive cells in CTRL + Sal, HCb + Sal, CTRL + JWH, HCb + JWH, HCB + JWH + SR2, and HCb + SR2. Data are expressed as mean ± SEM (n = 5/group). One-way ANOVA followed by Bonferroni multiple comparison test. c Double-labeled and merged confocal images of GFAP (green) (blue) and OX-42 (red) plus DAPI-counterstaining in lesioned animals treated with saline (HCb + Sal) or JWH-015 (HCb + JWH). *p < 0.05 vs. HCb + Sal; **p < 0.01 vs. HCb + Sal; ***p < 0.001 vs. HCb + Sal; # p < 0.001 vs. CTRL + Sal; ## p < 0.01 vs. CTRL + Sal; ### p < 0.001 vs. CTRL + Sal. Scale bars: 20 μm
CB2-mediated neuroprotection requires active nNOS
CB2 stimulation protects neurons from axotomy-induced apoptosis and enhances nNOS expression and activity. To determine whether CB2-mediated neuroprotection is nNOS-dependent, we blocked nNOS activity with 7-nitroindazole (7NI), a highly selective nNOS inhibitor. Various experimental HCb groups were treated with JWH-015 and 7NI, alone or in combination (Table 1), and neuroprotection was established by assessing neuronal survival, cytochrome c (cyt-c) release from mitochondria, and the degree of functional recovery, as stated by neurological severity score (NSS).
nNOS inhibition by 7NI was measured by analyzing nNOS enzymatic activity and NOx levels in Pn homogenates. Compared with CTRL + Sal animals, CTRL + 7NI-treated rats experienced a small non-significant reduction in Ca2+-dependent NOS activity (Table 2) and in stable NO metabolites (−20%; Fig. 4a). On the other hand, after HCb, a much larger effect of 7NI on nNOS activity was observed. Compared with HCb + Sal animals, HCb + 7NI-treated rats experienced a significant reduction in Ca2+-dependent NOS activity (−50%; p < 0.01; Table 2) and in stable NO metabolites −64%; p < 0.001; Fig. 4a).

Inhibition of nNOS blocks CB2-mediated neuroprotection. a NOx levels, expressed as percentage of CTRL + Sal, in HCb + Sal, CTRL + 7NI, and HCb + 7NI animals, showing the effectiveness of the nNOS inhibitor 7NI in reducing NOx levels. b Histograms of stereological neuronal counts in CTRL + Sal, HCb + Sal, HCb + JWH, CTRL + 7NI, HCb + 7NI, and HCb + 7NI + JWH. c Representative immunoblots and densitometric graphs of the cytosolic levels of cytochrome-c in pontine nuclei of CTRL + Sal, HCb + Sal, HCb + JWH, CTRL + 7NI, HCb + 7NI, and HCb + 7NI + JWH. d Histograms of densitometric values of cytochrome-c release, expressed as mean fluorescence of individual cells, normalized to total cellular surface (F/A; n = 250 cells/group) in CTRL + Sal, HCb + Sal, HCb + JWH, CTRL + 7NI, HCb + 7NI, and HCb + 7NI + JWH. e Time course of neurological recovery (NSS) in the CTRL + 7NI, HCb + Sal, HCb + JWH, CTRL + 7NI, HCb + 7NI, and HCb + 7NI + JWH groups. Data are expressed as mean ± SEM (n = 5/group). One-way ANOVA followed by Bonferroni multiple comparison test. **p < 0.01 vs. HCb + Sal; ***p < 0.001 vs. HCb + Sal; ## p < 0.01 vs. HCb + JWH; ### p < 0.001 vs. HCb + JWH
Consistent with our results [6], JWH-015 protected Pn neurons from HCb-induced degeneration. Compared with HCb + Sal animals, neuronal survival improved (p < 0.001; Fig. 4b), cyt-c release declined (p < 0.001; Fig. 4c, d), and functional recovery was greater (p < 0.001; Fig. 4e) in the HCb + JWH group. Conversely, 7NI exacerbated the negative effects only after lesion, as demonstrated in the comparisons between CTRL + Sal and CTRL + 7NI and between HCb + Sal and HCb + 7NI animals with regard to neuronal survival (p < 0.01; Fig. 4b), cyt-c release (p < 0.01; Fig. 4c, d), and functional recovery (p < 0.01; Fig. 4e). Notably, in combination with 7NI, JWH-015 was not neuroprotective, comparable with the effects with 7NI alone (HCb + 7NI + JWH vs. HCb + JWH, p < 0.001; Fig. 4b–e).
CB2-mediated neuroprotection is paralleled by iNOS inhibition
CB2 activation not only enhances nNOS but also inhibits iNOS activity (Fig. 2 and Table 2). Thus, it is possible that CB2 signaling promotes neuronal survival and recovery by regulating nNOS and modulating iNOS. To examine this hypothesis, we treated various groups of lesioned animals with 1400W, a specific iNOS inhibitor, alone or in combination with JWH-015 (Table 1) and measured their response to iNOS blockade, comparing neuronal survival, cyt-c release, and neurological outcome. iNOS inhibition by 1400W was assessed based on enzymatic activity and NOx levels in Pn homogenates.
Compared with HCb + Sal, 1400W inhibited Ca2+-independent NOS activity, which was attributable to iNOS [11], although tissue levels of nitrite and nitrate remained constant (HCb + Sal vs. HCb + 1400W, p > 0.05; Fig. 5a).

Inhibition of iNOS parallels CB2-mediated neuroprotection. a NOx levels, expressed as percentage of CTRL + Sal, in HCb + Sal and HCb + 1400W animals, showing that the iNOS inhibitor 1400W does not affect NOx levels (HCb + 1400W vs. HCb + Sal). b Histograms of stereological neuronal counts in HCb + Sal, HCb + JWH, HCb + 1400W, and HCb + 1400W + JWH. c Representative immunoblots and densitometric graphs of the cytosolic levels of cytochrome-c in pontine nuclei of CTRL + Sal, HCb + Sal, HCb + JWH, HCb + 1400W, and HCb + 1400W + JWH. d Histograms representing the densitometric values of cytochrome-c release, expressed as mean fluorescence of individual cells, normalized to total cellular surface (F/A; n = 250 cells/group), in HCb + Sal, HCb + JWH, HCb + 1400W, and HCb + 1400W + JWH. e Time course of neurological recovery (NSS) in the HCb + Sal, HCb + JWH, HCb + 1400W, and HCb + 1400W + JWH groups. Data are expressed as mean ± SEM (n = 5/group). One-way ANOVA followed by Bonferroni multiple comparison test. **p < 0.01 vs. HCb + Sal; ***p < 0.001 vs. HCb + Sal
Selective iNOS inhibition resulted in neuroprotection from HCb-mediated cell death, as evidenced by increased neuronal survival (p < 0.001; Fig. 5b), decreased cyt-c release (p < 0.001; Fig. 5c), and improved functional recovery (p < 0.001; Fig. 5d) (compare HCb + Sal vs. HCb + 1400W). These results suggest that iNOS is detrimental in the development of remote damage following HCb. No synergy occurred when 1400W was administered with JWH-015 (compare HCb + JWH vs. HCb + 1400W + JWH in Fig. 5b–d), suggesting that both drugs act by inhibiting iNOS.
CB2 stimulation and iNOS inhibition attenuate oxidative/nitrative stress in axotomized neurons
Accumulating evidence suggests that oxidative/nitrative stress—caused by excess production of reactive oxygen species (ROS) and NO at the site of injury—is an important mediator of remote damage. In fact, simultaneous increases in ROS and NO production may facilitate the formation of the highly reactive peroxynitrite, which effects oxidative damage to various neuronal components, including proteins and lipids; peroxynitrite has been implicated in most of the adverse effects of excessive NO generation [20].
To determine the function of oxidative/nitrative stress in HCb, ROS levels, peroxynitrite-mediated protein nitration, and lipid peroxidation were measured. By spectrofluorometry of ROS generation in Pn samples from control and lesioned animals, axotomy induced an approximately threefold increase in ROS levels (HCb + Sal vs. CTRL + Sal, p < 0.001; Fig. 6a). The involvement of oxidative/nitrative stress during HCb was confirmed by the presence of 3-nitrotyrosine (3-NT) and 4-hydroxynonenal (4-HNE), two end products of peroxynitrite, only in the Pn of lesioned animals (HCb + Sal), but not in unlesioned ones (CTRL + Sal; Fig. 6b).

CB2 stimulation modulates oxidative/nitrative indices in axotomized neurons. a Histograms of ROS level, expressed as percentage of CTRL + Sal in HCb + Sal, HCb + JWH, HCb + SR2, HCb + 1400W, HCb + 1400W + JWH, HCb + 7NI, and HCb + 7NI + JWH. b Histograms of the percentage of NeuN + neurons expressing 3-nitrotyrosine (3-NT) and 4-hydroxynonenal (4-HNE) in CTRL + Sal, HCb + Sal, HCb + JWH, HCb + SR2, HCb + 1400W, HCb + 1400W + JWH, HCb + 7NI, and HCb + 7NI + JWH. c Double-labeled and merged confocal images of 3-NT (red), and NeuN (green) in neurons of the pontine nuclei at 7 days after HCb in HCb + Sal. d Double-labeled and merged confocal images of 3-NT (red) and 4-HNE (green) in neurons of the pontine nuclei at 7 days after HCb in HCb + Sal. Data are expressed as mean ± SEM (n = 5/group). One-way ANOVA followed by Bonferroni multiple comparison test. ***p < 0.001 vs. HCb + Sal; ###p < 0.001 vs. CTRL + Sal; §§§p < 0.001 vs. HCb + JWH. Scale bars: c 15 μm; d 25 μm
By double immunofluorescence with NeuN, both 3-NT and 4-HNE were demonstrated in neurons after HCb (Fig. 6c). Further, 3-NT and 4-HNE colocalized in approximately 60% of axotomized Pn neurons (Fig. 6b, d).
HCb-induced oxidative/nitrative stress was significantly attenuated by JWH-015 (HCb + JWH vs. HCb + Sal, p < 0.001; Fig. 6a, b), similar to chronic treatment with 1400W (HCb + 1400W vs. HCb + Sal, p < 0.001; Fig. 6a, b). Simultaneous administration of JWH-015 and 1400W did not enhance protection, suggesting that both drugs act on the same pathway (Fig. 6a, b). Conversely, neither nNOS inhibition nor CB2 blockade affected ROS production or the number of 3-NT/4-HNE-positive neurons (Fig. 6a, b). Coadministration of JWH-015 with 7NI failed to have antioxidant effects, comparable with the effects of 7NI alone (HCb + 7NI + JWH vs. HCb + JWH, p < 0.001; Fig. 6a, b).
CB2 stimulation increases Bcl-2 and Hsp70 expression in an nNOS-dependent manner
By immunoblotting, we measured the expression of Bcl-2, heat shock protein (Hsp) 70, and copper/zinc and manganese superoxide dismutases (SODs), which are modulated by the nitrergic system and protect neurons from oxidative stress and apoptosis [21]. HCb upregulated Bcl-2 and Hsp70 slightly (HCb + Sal vs. CTRL + Sal, p < 0.05; Fig 7a) but not the SODs (data not shown). JWH-015 increased the expression of Bcl-2 and Hsp70 (HCb + JWH vs. HCb + Sal, p < 0.01; Fig. 7a). In contrast, the inhibition of nNOS by 7NI in HCb rats decreased the HCb-induced expression of Bcl-2 (HCb + 7NI vs. HCb + Sal, p < 0.001; Fig. 7a) and Hsp70 (HCb + 7NI vs. HCb + Sal, p < 0.05; Fig. 7a).

CB2 stimulation increases the expression of Bcl-2 and Hsp70 in an nNOS-dependent manner. a Pontine nuclei from the experimental groups were homogenized and assayed by Western blot with anti-Bcl-2 or anti-Hsp70. Densitometric values (right) are shown. Data are expressed as mean ± SEM (n = 5/group). One-way ANOVA followed by Bonferroni multiple comparison test. *p < 0.05 vs. HCb + Sal; **p < 0.01 vs. HCb + Sal; ***p < 0.001 vs. HCb + Sal; # p < 0.001 vs. CTRL + Sal. §§§ p < 0.001 vs. HCb + JWH
Consistent with the function for nNOS in CB2-dependent signaling, the administration of 7NI and JWH-015 abolished the JWH-015-mediated induction of Bcl-2 (HCb + 7NI + JWH vs. HCb-JWH, p < 0.001; Fig. 7a) and Hsp70 (HCb + 7NI + JWH vs. HCb + JWH, p < 0.01; Fig. 7a). Conversely, inhibition of iNOS by 1400W did not affect their expression (HCb + 1400W vs. HCb + Sal; Fig. 7a). On iNOS inhibition, in combination with CB2 stimulation, 1400W did not alter the JWH-015-mediated Bcl-2 response but inhibited the JWH-015-mediated Hsp70 response (HCb + 1400W + JWH vs. HCb + JWH; Fig. 7a).
Discussion
In this study, we examined the cellular and molecular mechanisms of CB2-dependent neuroprotection in a clinically relevant rodent model of remote brain damage. Mechanisms of CB2 up-regulation has been only partly analyzed in a previous study [6], while in the present study, we focused on functional interactions between CB2 and NOS signaling. CB2 stimulation influenced the balance between nNOS and iNOS in the axotomized precerebellar stations. Specifically, nNOS increased in neurons and iNOS decreased in astrocytes, on chronic stimulation of CB2, improving molecular and neurological outcomes after cerebellar ablation.
Selective CB2 agonism attenuates axotomy-induced oxidative/nitrative stress in neurons and prevents astrocytic and microglial activation. Such actions are abolished by nNOS inhibition, indicating that nNOS products are required for CB2-dependent neuroprotection. Yet, CB2 activation is accompanied by iNOS inhibition, suggesting that CB2 neuroprotection also inhibits the neurotoxic activity of NO, driven by iNOS in reactive astrocytes.
The interaction between the endocannabinoid and nitrergic systems has been well documented under various pathophysiological states and appears to be complex. Several in vitro and in vivo studies have reported that the activation of CB1 and CB2 can stimulate and inhibit NO production, disparately impacting the expression and activity of the NOS isoforms [12, 16, 22].
Time course analyses of the expression of CB2 and nNOS/iNOS showed that CB2 temporally precedes the induction of both NOS isoforms, indicating the importance of CB2 signaling in the observed changes in nNOS/iNOS system. In particular, we observed that CB2 stimulation enhances nNOS expression in neurons and simultaneously reverses the axotomy-induced upregulation of iNOS in astrocytes. Our immunohistochemical data demonstrate that axotomy selectively induces the de novo expression of CB2 and nNOS in Pn neurons, possibly as part of a neuronal adaptive response to axotomy-induced damage. The role played by CB2 on JWH-015 effects was confirmed by their complete reversion upon SR144528 administration. On the other hand, in the absence of CB2 stimulation, CB2 antagonist has small effect on neurodegeneration. This evidence is intriguing and would require specific analyses to be clarified. At present, it can be hypothesized that CB2 expression per se has a little role and that CB2 function is probably in relation to the local availability of endocannabinoids. Although in primary CNS lesion sites, the concentration of endogenous cannabinoids has been reported to be enhanced, more data on remote degeneration areas need to be collected.
The relationship between CB2 and nNOS was confirmed on stimulation of CB2 with JWH-015, which increased the expression and activity of nNOS in neurons and the percentage of neurons that coexpressed CB2 and nNOS (Fig. 1)—novel evidence of an anatomical and functional link between CB2 and nNOS in adult neurons.
The effect of CB2 on iNOS appears to be more complex. In our experimental lesion, we detected CB2 in injured neurons but not in reactive astrocytes or reactive microglial cells. This observation contrasts those of other groups, who have noted robust induction of CB2 in activated microglial cells after pathological neuroinflammatory insults [23, 24]. However, because the glial responses in remote neurodegeneration differ from those observed in other models of neurodegeneration [1], such disparities must be considered with caution. CB2 stimulation has profound effects on astrocytic and microglial activation that could be explained as an indirect consequence of its neuroprotective potential. In fact in our model, by directly promoting the survival of damaged neurons, JWH-015 may attenuate the inflammatory response mainly by reducing the altered cross-talk between dying/damaged neurons and glial cells. However, we cannot conclusively rule out that glial cells may have low but functional amounts of CB2 or that infiltrating leukocytes contribute to the CB2-dependent effects on iNOS expression and subsequent inflammatory response.
Another important difference between the neuroinflammatory responses in primary and remote damage is the contrasting functions of the two principal types of glial cells: astrocytes and microglia. We demonstrated that reactive astrocytes, but not microglial cells, mediate neurodegenerative events in axotomy-induced remote neuronal death [4, 25]. In the current study, we observed that axotomy-induced iNOS in reactive astrocytes but not in reactive microglia, implicating astrocytes as key mediators of remote damage.
The NOS/NO system is beneficial or detrimental under pathological conditions of the CNS [26]. The observed changes in nNOS and iNOS expression and activity, following HCb and on CB2 activation, correlate inversely with the severity of neurodegenerative events in the Pn, indicating that NO signaling via nNOS and iNOS acts oppositely to the physiopathology of the remote neuronal damage (Table 2). The inhibition of nNOS activity negatively affects neuronal survival, nitrative/oxidative stress, and neurological improvement, consistent with the neuroprotective function of nNOS-derived NO signaling in axotomized neurons. Further, pharmacological blockade of iNOS in astrocytes has beneficial effects on neuronal survival in lesioned animals, indicating that iNOS-derived NO from reactive astrocytes is cytotoxic to injured neurons. Although difficult to evaluate because of the opposite sign of the effect, the relative contribution of the two mechanisms is slightly in favor of the role of nNOS-derived NO. In fact, considering for instance, neuronal degeneration, the block of nNOS activity induces an increment of neuronal degeneration of ∼40%, whereas that of iNOS activity induces a survival improvement of about ∼30% (compare data from Fig. 4 and Fig. 5).
These data extend previous findings, in which the selective inhibition of nNOS and iNOS has neurotoxic and neuroprotective effects, respectively, in various in vivo models of axonal damage [27–29]. Further, genetic ablation of nNOS effects the apoptosis of spinal cord neurons after peripheral nerve transection, whereas axotomy-induced neuronal death is significantly lower in iNOS−/− and eNOS−/− mice [30, 31]. Improved neuronal recovery has been also been observed in iNOS-deficient mice following spinal cord trauma [32].
However, the specific roles of nNOS and iNOS in postaxotomy processes are unknown. In vivo studies have shown that functional iNOS is necessary for injured tissues to be repaired [33], whereas nNOS mediates neuronal death and retrograde axonal degeneration following axotomy, alone or with iNOS [34, 35]. Martin et al. [20] reported that axotomy-induced apoptosis of mouse cortical neurons declines significantly in nNOS−/− mice but not in iNOS−/− mice. These conflicting findings might be attributed to differences in the pathophysiology of the specific neuronal lesion, the type of neuronal population that is affected, and the species and age of the animals.
In this study, peroxynitrite, a highly reactive oxidant that forms from the reaction between nitric oxide and superoxide anion, was selectively accumulated in axotomized Pn neurons within 7 days after axotomy, leading to oxidative damage of proteins (3-NT) and lipids (4-HNE) in the same neurons (Fig. 5). Further, inhibition of iNOS expression/activity by 1400W and JWH-015 significantly reduced peroxynitrite formation in neurons and protected them from mitochondrial apoptosis, demonstrating that oxidative/nitrative stress and iNOS-derived NO are important mediators in HCb-induced delayed neuronal death. Considering the importance of peroxynitrite in triggering apoptosis, it could be argued that CB2 activation may also affect ROS generation independently of NOS/NO. However, this possibility is ruled out by the clear block of all CB2-mediated neuroprotective effects upon nNOS blockage (Figs. 4 and 5).
Our data also link the severity of neuronal oxidative stress to the neuroinflammatory response that is likely triggered by iNOS-expressing GFAP-positive astrocytes. Consistent with these findings, we have observed that neuronal degeneration in the Pn after cerebellar ablation can be delayed—but not suppressed—by impairing the postlesional activation of astrocytes with glucocorticoids, whereas inhibiting microglial activation fails to mitigate neuronal loss, suggesting that astrocytes are important effector cells in mediating axotomy-induced death of Pn neurons [4, 25].
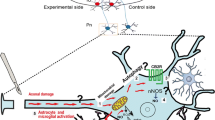
Considering all of these findings, we propose a mechanism in which oxidative/nitrative stress is the result of a vicious cycle between injured neurons and reactive astrocytes (Fig. 8). Factors that are released from injured neurons could trigger persistent astrocytic activation. Once activated, astrocytes release amounts of NO chronically, via iNOS. At high local concentrations and during the pro-oxidant state in injured neurons, NO is toxic (primarily via peroxynitrite formation), exacerbating the mitochondrial damage that ultimately kills neurons through cytochrome-c-dependent pathways.

Schematic of the hypothetical function of CB2 in modulating axotomy-induced neurodegeneration. Details in “Discussion”. Single asterisk means that ROS sources are probably not limited to the mitochondria; double asterisk means that other pathways not here analyzed might be of relevance in determining neuronal survival; triple asterisk means that the crosstalk between neuronal degeneration and astrocytic activation still wait clarification
Thus, such altered crosstalk between activated astrocytes and damaged neurons establishes a positive feedback loop that accelerates progressive neurodegeneration after development of a focal cerebellar lesion in the Pn. Under these conditions, CB2 might break the neurodegenerative/neuroinflammatory cycle by promoting neuronal survival and recovery. Specifically, because CB2 stimulation induces important mediators of neuroprotective responses to oxidative and mitochondrial stress, such as Akt [6], nNOS, Bcl-2, and Hsp70 (this study), we propose that CB2-mediated neuroprotection against axotomy is linked to the reestablishment of the physiological redox state in injured neurons. In this nontoxic redox state, nNOS-derived NO is a pro-survival signal, stimulating, beyond the already present postlesional enhancement, the expression of antioxidant (Hsp70) and antiapoptotic (Bcl-2) systems, thus possibly limiting axotomy-induced damage in neurons. On the other hand, it is important to note that, due to the complexity and redundancy of cannabinoid signaling, it is likely that Hsp70 and Bcl-2 are only a small part of the picture and many other pathways not here analyzed might be of relevance.
One might wonder why in our model NO from iNOS is toxic while NO from nNOS is protective. Different NOS isoforms have different cellular distribution and kinetic properties. nNOS is restricted in neurons and the rate of NO production by this enzyme is regulated by changes in intracellular calcium levels, while iNOS is selectively expressed in reactive astrocytes and NO production by iNOS is both independent of calcium levels and occurs at much higher rates than with the nNOS. Thus, nNOS in damaged neurons may produce transient and low amounts (in a nanomolar range) of NO that can act through cGMP/protein kinase G- or S-nitrosylation to trigger neuroprotective signaling, including pro-survival pathways (e.g., Akt, Bcl-2, and Hsp70) [26]. Locally, much larger concentrations of NO (in the micromolar range) could be achieved by means of the iNOS within reactive astrocytes. In this context, NO produced by iNOS can become neurotoxic through the generation of the peroxynitrite via its reaction with superoxide [26]. It is conceivable that the highest level of peroxynitrite can be achieved in the microenvironment created by the reactive astrocytes adherent to the damaged neurons, in which high local concentrations of both NO (mainly by iNOS) and ROS are generated.
With regard to major sources ROS in our model, we have no direct data. Tentatively, it can be argued that because dysfunctional mitochondria produce large amount of ROS [3, 20], cytochrome c-releasing mitochondria in axotomized neurons could be an important and perhaps dominant source of ROS in our model. However, we cannot exclude that also astrocytes and microglial cells may contribute to the oxidative stress by producing large amount of ROS from mitochondria, as well as by different enzymatic pathways, such as NADPH oxidase, xanthine oxidase, and cyclooxygenase.
In conclusion, in our experimental model of remote neurodegeneration, we found that the stimulation of CB2 signaling triggers a cascade of molecular and cellular events that limit the severity of the neurodegeneration that is associated with the late stages of axotomy. After axotomy, CB2 agonism: (1) increases nNOS expression and activity in neurons, (2) reduces iNOS expression and activity in astrocytes, (3) attenuates oxidative/nitrative stress in neurons, (4) increases the levels of proteins that mediate anti-oxidative (Hsp70) and anti-apoptotic (Bcl-2) mechanisms, and (5) mitigates neuroinflammation. Notably, selective inhibition of nNOS expression/activity negated the neuroprotective effects of JWH-015 on neuronal survival and functional recovery, indicating that CB2-mediated neuroprotection requires nNOS activity.
References
Block F, Dihne M, Loos M (2005) Inflammation in areas of remote changes following focal brain lesion. ProgNeurobiol 75:342–365
Viscomi MT, Florenzano F, Latini L, Molinari M (2010) Remote cell death in cerebellar system. Cerebellum 8:184–91
Martin LJ, Price AC, McClendon KB, Al-Abdulla NA, Subramaniam JR, Wong PC, Liu Z (2003) Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J Neurochem 85:234–247
Viscomi MT, Florenzano F, Latini L, Amantea D, Bernardi G, Molinari M (2008) Methyl prednisolone treatment delays remote cell death after focal brain lesion. Neuroscience 154:1267–1282
Coleman MP, Perry VH (2002) Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci 25:532–537
Viscomi MT, Oddi S, Latini L, Pasquariello N, Florenzano F, Bernardi G, Molinari M, Maccarrone M (2009) Selective CB2 receptor agonism protects central neurons from remote axotomy-induced apoptosis through the PI3K/Akt pathway. J Neurosci 29:4564–4570
Viscomi MT, Oddi S, Latini L, Bisicchia E, Maccarrone M, Molinari M (2010) The endocannabinoid system: a new entry in remote cell death mechanisms. Exp Neurol 224:56–65
Guix FX, Uribesalgo I, Coma M, Munoz FJ (2005) The physiology and pathophysiology of nitric oxide in the brain. ProgNeurobiol 76:126–152
Dawson TM, Snyder SH (1994) Gases as biological messengers: nitric oxide and carbon monoxide in the brain. J Neurosci 14:5147–5159
Bredt DS (1999) Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic Res 31:577–596
Makara JK, Katona I, Nyiri G, Nemeth B, Ledent C, Watanabe M, de Vente J, Freund TF, Hajos N (2007) Involvement of nitric oxide in depolarization-induced suppression of inhibition in hippocampal pyramidal cells during activation of cholinergic receptors. J Neurosci 27:10211–10222
Hillard CJ, Muthian S, Kearn CS (1999) Effects of CB(1) cannabinoid receptor activation on cerebellar granule cell nitric oxide synthase activity. FEBS Lett 459:277–281
Kim SH, Won SJ, Mao XO, Jin K, Greenberg DA (2006) Molecular mechanisms of cannabinoid protection from neuronal excitotoxicity. Mol Pharmacol 69:691–696
Molina-Holgado F, Pinteaux E, Moore JD, Molina-Holgado E, Guaza C, Gibson RM, Rothwell NJ (2003) Endogenous interleukin-1 receptor antagonist mediates anti-inflammatory and neuroprotective actions of cannabinoids in neurons and glia. J Neurosci 23:6470–6474
Maccarrone M, Bari M, Battista N, Finazzi-Agrò A (2002) Endocannabinoid degradation, endotoxic shock and inflammation. Curr Drug Targets Inflamm Allergy 1:53–63
Maccarrone M, Fiori A, Bari M, Granata F, Gasperi V, De Stefano ME, Finazzi-Agrò A, Strom R (2006) Regulation by cannabinoid receptors of anandamide transport across the blood–brain barrier and through other endothelial cells. ThrombHaemost 95:117–127
Fusco FR, Martorana A, Giampa C, De March Z, Farini D, D'Angelo V, Sancesario G, Bernardi G (2004) Immunolocalization of CB1 receptor in rat striatal neurons: a confocal microscopy study. Synapse 53:159–167
Watanabe T, Okuda Y, Nonoguchi N, Zhao MZ, Kajimoto Y, Furutama D, Yuhawa H, Shibata MA, Otsuki Y, Kuroiwa T et al (2004) Postischemic intraventricular administration of FGF-2 expressing adenoviral vectors improves neurologic outcome and reduces infarct volume after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab 24:1205–1213
Viscomi MT, Florenzano F, Conversi D, Bernardi G, Molinari M (2004) Axotomy dependent purinergic and nitrergic co-expression. Neuroscience 123:393–404
Martin LJ, Adams NA, Pan Y, Price A, Wong M (2011) The mitochondrial permeability transition pore regulates nitric oxide-mediated apoptosis of neurons induced by target deprivation. J Neurosci 31:359–370
Calabrese V, Boyd-Kimball D, Scapagnini G, Butterfield DA (2004) Nitric oxide and cellular stress response in brain aging and neurodegenerative disorders: the role of vitagenes. In Vivo 18:245–267
Gonzalez C, Herradon E, Abalo R, Vera G, Perez-Nievas BG, Leza JC, Martin MI, Lopez-Miranda V (2011) Cannabinoid/agonist WIN 55,212-2 reduces cardiac ischaemia-reperfusion injury in Zucker diabetic fatty rats: role of CB2 receptors and iNOS/eNOS. Diabetes Metab Res Rev 27:331–340
Stella N (2009) Endocannabinoid signaling in microglial cells. Neuropharmacology 56(Suppl 1):244–253
Benito C, Nunez E, Tolon RM, Carrier EJ, Rabano A, Hillard CJ, Romero J (2003) Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer's disease brains. J Neurosci 23:11136–11141
Viscomi MT, Latini L, Florenzano F, Bernardi G, Molinari M (2008) Minocycline attenuates microglial activation but fails to mitigate degeneration in inferior olive and pontine nuclei after focal cerebellar lesion. Cerebellum 7:401–405
Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM (2007) Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. NatRevNeurosci 8:766–775
Thippeswamy T, Jain RK, Mumtaz N, Morris R (2001) Inhibition of neuronal nitric oxide synthase results in neurodegenerative changes in the axotomised dorsal root ganglion neurons: evidence for a neuroprotective role of nitric oxide in vivo. Neurosci Res 40:37–44
Hamada Y, Ikata T, Katoh S, Tsuchiya K, Niwa M, Tsutsumishita Y, Fukuzawa K (1996) Roles of nitric oxide in compression injury of rat spinal cord. Free Radic Biol Med 20:1–9
Jafarian-Tehrani M, Louin G, Royo NC, Besson VC, Bohme GA, Plotkine M, Marchand-Verrecchia C (2005) 1400 W, a potent selective inducible NOS inhibitor, improves histopathological outcome following traumatic brain injury in rats. Nitric Oxide 12:61–69
Keilhoff G, Fansa H, Wolf G (2002) Neuronal nitric oxide synthase is the dominant nitric oxide supplier for the survival of dorsal root ganglia after peripheral nerve axotomy. J Chem Neuroanat 24:181–187
Keilhoff G, Fansa H, Wolf G (2004) Neuronal NOS deficiency promotes apoptotic cell death of spinal cord neurons after peripheral nerve transection. Nitric Oxide 10:101–111
Isaksson J, Farooque M, Olsson Y (2005) Improved functional outcome after spinal cord injury in iNOS-deficient mice. Spinal Cord 43:167–170
Levy D, Kubes P, Zochodne DW (2001) Delayed peripheral nerve degeneration, regeneration, and pain in mice lacking inducible nitric oxide synthase. J Neuropathol Exp Neurol 60:411–421
Koeberle PD, Ball AK (1999) Nitric oxide synthase inhibition delays axonal degeneration and promotes the survival of axotomized retinal ganglion cells. Exp Neurol 158:366–381
Ikeda K, Iwasaki Y, Kinoshita M (1998) Neuronal nitric oxide synthase inhibitor, 7-nitroindazole, delays motor dysfunction and spinal motoneuron degeneration in the wobbler mouse. J Neurol Sci 160:9–15
Acknowledgements
This study was supported in part by grants from Ministero della Salute to M. Molinari. Financial support from Fondazione TERCAS (grant 2009-2012) to M.M. is gratefully acknowledged. We thank Prof. G. Bernardi for his continuous support and encouragement. The professional editorial work of Blue Pencil Science is also acknowledged.
Conflict of interest
The authors have no conflicting financial interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
S. Oddi, L. Latini, and M. T. Viscomi contributed equally to this work.
M. Molinari and M. Maccarrone are equal senior authors.
Rights and permissions
About this article
Cite this article
Oddi, S., Latini, L., Viscomi, M.T. et al. Distinct regulation of nNOS and iNOS by CB2 receptor in remote delayed neurodegeneration. J Mol Med 90, 371–387 (2012). https://doi.org/10.1007/s00109-011-0846-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-011-0846-z