
Abstract
Traumatic brain injury (TBI) is a serious health problem in the world. However, little is known about the pathogenesis and molecular mechanisms of TBI. Here, we show that TBI activates neuregulin 1 (NRG1)-ErbB4 signaling, with an increased expression of NRG1 and ErbB4 in the traumatic region. Specifically knocking out ErbB4 in parvalbumin-positive (PV+) interneurons exacerbates motor function deficits in mice after TBI. Consistently, PV-ErbB4−/− mice showed larger necrotic area and more edema when compared with PV-ErbB4+/+ mice. Replenishment of NRG1 through intranasal application of the recombinant protein in PV-ErbB4+/+ mice enhanced neurological function. Moreover, using an in vitro neuronal culture system, we found that NRG1–ErbB4 signaling protects neurons from glutamate-induced death, and such protective effects could be diminished by GABA receptor antagonist. These results indicate that NRG-ErbB4 signaling protects cortical neurons from TBI-induced damage, and such effect is probably mediated by promoting GABA activity. Taken together, these findings unveil a previously unappreciated role for NRG1-ErB4 signaling in preventing neuronal cell death during functional recovery after TBI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) represents a major cause of lifelong physical, behavioral, emotional and cognitive impairments globally, affecting more than 2 million patients per year in USA (Alexander 1995; Bigler 2003; Blennow et al. 2012; Smith et al. 2013; Taylor et al. 2017). TBI can lead to brain edema and hemorrhage through disruption of the blood brain barrier (BBB) (Smith et al. 2013; Brickler et al. 2018). These effects involve in primary and secondary damage via glutamate toxicity, oxidative stress, ischemia, neuroinflammation, and metabolic imbalance (Maas et al. 2008; Johnson et al. 2013; Bramlett and Dietrich 2015; Jassam et al. 2017).
Neuregulin 1 (NRG1), a growth factor, contains the epithelial growth factor (EGF)-like domain (Falls 2003; Mei and Xiong 2008; Buonanno 2010; Mei and Nave 2014). Alternative splicing produces more than 30 different NRG1 isoforms, which are types I, II and III (Falls 2003; Steinthorsdottir et al. 2004; Harrison and Law 2006; Bublil and Yarden 2007; Mei and Xiong 2008; Mei and Nave 2014). NRG1 isoforms differ in their patterns and levels of expression in different tissues including brain (Mei and Xiong 2008). Mice that carry mutations inactivating particular isoforms show different changes in neural development, suggesting that the different NRG1 isoforms have distinct functions (Mei and Xiong 2008). The ErbB receptors are a family that contains receptors of ErbB1 (or EGFR), ErbB2, ErbB3 and ErbB4 (Mei and Xiong 2008; Chen et al. 2010; Mei and Nave 2014). EGFR does not bind to NRG1, but can form heterodimers with ErbB4. ErbB2 functions as a co-receptor by forming heterodimers with other ligand-bound ErbBs. ErbB3 can bind to NRG1, but its homodimers are catalytically inactive. Among the ErbB proteins, ErbB4 is best characterized for its function in the CNS. It can both interact with NRG1 and become activated by NRG1 as a tyrosine kinase (Mei and Xiong 2008). ErbB4 undergoes tertiary structural changes when it binds to NRG1 and forms functional homodimers or heterodimers with ErbB1, ErbB2, or ErbB3 (Mei and Nave 2014). Phosphorylation of the ErbB4 receptor leads to the recruitment of adaptor molecules and activates numerous downstream signaling pathways that are crucial to neuronal development, axonal navigation, neuronal migration and synaptic function (Mei and Nave 2014). NRG1–ErbB4 signaling has been implicated in the assembly of the GABAergic circuitry (Fazzari et al. 2010). NRG1 and ErbB4 are necessary for the E/I balance by promoting or maintaining γ-Aminobutyric acid (GABA) release (Woo et al. 2007; Fazzari et al. 2010; Mei and Nave 2014).
NRG1–ErbB4 signaling protects neurons against ischemic injury, which may involve the inhibition of pro-inflammatory responses (Xu et al. 2004; Li et al. 2007, 2012). Increasing GABAergic neurotransmission is known to reduce cerebral ischemia-induced neuronal apoptosis (Costa et al. 2004; Zhou et al. 2008; DeFazio et al. 2009). It is interesting that ErbB4 is expressed specifically in GABAergic neurons (Fazzari et al. 2010), and NRG1 promotes GABA release via stimulating ErbB4 receptors (Fazzari et al. 2010). So it is possible that NRG1 neuroprotection may be mediated by ErbB4 receptors on GABAergic neurons.
In this study, we investigated NRG1–ErbB4 signaling during the pathological process of TBI. We demonstrate that TBI activates NRG1–ErbB4 signaling near the traumatic region of mice. In addition, we found that PV-ErbB4−/− mice showed aggravated motor function deficits when compared with PV-ErbB4+/+ littermates. Moreover, intranasal delivery of recombinant NRG1 after TBI improves the long-term functional recovery in mice, we also found that NRG1–ErbB4 signaling protects neurons from glutamate-induced death in vitro, and such protective effects could be diminished using a GABA receptor antagonist. Thus, this study demonstrated that NRG1–ErbB4 signaling may play a role in promoting neurological recovery after TBI.
Materials and methods
Animals
C57B6/J (male 8–12 weeks of age, and 25–30 g of body weight) were used. Wild-type (WT) mice were purchased from The Jackson Laboratory. Mice with targeted knockin transgenes ErbB4::CreERT2 and Rosa:: LSL-tdTomato (Madisen et al. 2010; Bean et al. 2014) were purchased from The Jackson Laboratory. Double-transgenic ErbB4::CreERT2; Rosa::LSL-tdTomato mice (here after referred as ErbB4-reporter mice) were generated by ErbB4::CreERT2 and Rosa::LSL-tdTomato mice. The administration of tamoxifen was done as previously described (Bean et al. 2014). Heart-rescued ErbB4 knockout mice (Tidcombe et al. 2003), LoxP-flanked ErbB4 mice (Garcia-Rivello et al. 2005) and PV-Cre mice were described previously (Wen et al. 2010). Parvalbumin (PV)-Cre mice were crossed with LoxP-flanked ErbB4 mice to generate PV-ErbB4−/− mice, with PV-ErbB4+/+ mice as a control. Mice were kept on a 12 h light/dark cycle with access to food and water ad libitum in a temperature-controlled room (~ 22 °C). All animal procedures were carried out in strict accordance with the principles of laboratory animal care and use approved by the Augusta University Animal Care and Use Committee guidelines.
Controlled cortical impact (CCI)
Unilateral TBI was induced in the right parietal lobe as described previously (Miller et al. 2015). A parietal craniotomy (diameter 3.5 mm) was centered 0.5 mm anterior and 2.0 mm lateral to bregma in anesthetized mice. CCI was induced using a 3-mm convex tip attached to an electromagnetic impactor (Leica) and a stereotaxic apparatus. The contusion depth was set to 1.5 mm from dura with a velocity of 3.0 m/s sustained for 150 ms. Sham mice underwent craniectomy surgeries but without the focal injury. After surgical procedures, the mice were seamed and received an intraperitoneal injection of warm saline to prevent dehydration, then the mice were transferred to a temperature-controlled chamber for recovery.
Rotarod test and beam walk test
Mice were habituated in the experimental room for 1 h prior to testing and equipment was cleaned with 70% ethanol between each test. The rotarod test was used to evaluate sensorimotor balance and coordination as previously described (Adelson et al. 2012). Briefly, animals were pre-trained for 4 days before CCI or sham injury. A baseline was collected on the fourth day of training, then again at 1 day, 2 days, 4 days, 8 days, 16 days and 32 days post-CCI or sham injury. Mice were placed on the rod and the starting velocity was set at 4 rpm and accelerating to 40 rpm in 300 s. Mice were given four trials each testing day with a 2-min intertrial interval. The time to fall was recorded for each mouse.
The beam walk test was used to assess fine motor coordination and balance as previously described (Merkler et al. 2001). Briefly, the square wood beam was 30 cm in height, 6 mm wide and 80 cm long. Mice were pre-trained to walk to a dark goal box for 4 days with four trials each day and the baseline total number of foot slips was collected on the fourth day of training, then again at 1 days, 2 days, 4 days, 8 days, 16 days and 32 days post-injury.
Evaluation of lesion volume
The brain section selection and lesion volume determination were performed as described previously (Brickler et al. 2018). In brief, lesion volume was assessed using Caveleri Estimator from StereoInvestigator (MicroBrightField, Williston, VT) and an upright Olympus BX51TRF motorized microscope (Olympus America, Center Valley, PA). Lesion volume was analyzed by estimating the area of tissue loss in the ipsilateral cortical hemisphere using five serial coronal sections (− 1.34 to + 1.11 mm posterior from Bregma). Nissl-stained sections were viewed under microscopy at a magnification of 4x. A random sampling scheme was used that estimates every 10th section from rostral to caudal, yielding five total sections to be analyzed. A randomly placed grid with 100 μm spaced points was placed over the ipsilateral hemisphere and the area of contusion was marked within each grid. Lesion boundaries were identified by loss of Nissl staining, pyknotic neurons and tissue hemorrhage. The marked areas, using grid spacing, was then used to estimate total tissue volume based on section thickness, section interval and total number of sections. Data are represented as volume of tissue loss or damage (mm3).
Brain water content analysis
To measure water content, brains were freshly dissected and weighed to obtain the wet weight (mg). They were then dried at 70 °C for 3 days to obtain the dry weight. Hemispheric water content was calculated as follows: % hemispheric water content = (wet weight − dry weight)/(wet weight) × 100 (Brickler et al. 2018).
Intranasal delivery of recombinant NRG1
The intranasal approach for NRG1 delivery into the brain was performed as described previously (Xia et al. 2018). Briefly, mice were randomly assigned to receive PBS or recombinant NRG1 treatment 2 h after TBI and subsequently to receive the similar treatment every other day for 14 days. The recombinant NRG1 was obtained as described previously (Woo et al. 2007). All mice were anesthetized and placed in a supine position with dorsal side down. NRG1 was freshly dissolved in PBS (1.0 μg/μL), delivered at a dose of 0.5 mg/kg, and applied into each nostril five times (~ 3 μL into each nostril for each trial), with an inter-treatment interval of 5 min. Animals in the control group received an equal volume of sterile PBS using the same regimen. For a 30 g mouse, approximately 15 μL of NRG1 or an equivalent volume of PBS was delivered over the course of ~ 25 min.
Neurological behavioral tests
Modified Neurological Severity Scale (mNSS) scores were used to evaluate the neurological function as previously described (Horita et al. 2006). The mNSS was a composite of motor, sensory, reflex, and balance tests. Neurological function was graded on a scale of 0–18 (normal 0; maximal deficit score 18). The higher the score, the more severe the injury was.
Body curl test for contralateral torso flexion
Neurological function was assessed after CCI using a modified elevated body swing test as previously described (Washington et al. 2012). Briefly, mice were hand-suspended by the tail by a blinded investigator and rated for degree of torso flexion from vertical toward the contralateral injury side. A numerical scoring system was developed based on the degree of contralateral body curl: absent (1), mild (2), moderate (3), and severe (4). Normal mice hang vertically without flexion and thus deviate 0o from vertical (rating of 1). Flexion of the torso 22.5° from vertical was rated 2 (mild), flexion between 22.5° and 45° from vertical was rated 3 (moderate), and flexion of the torso 45° or more and with/or without grasping of the hindlimb by forelimb was rated 4 (severe). Contralateral torso flexion was assessed 1, 2, 4, 8, 16 and 32-day after injury.
Immunohistochemistry and confocal microscopy
Tissue distributions of GFAP and Iba-1 were assessed by immunohistochemistry using an image analysis workstation (Yang et al. 2019). Mice were administered ketamine/xylazine (150 mg/kg and 10 mg/kg, respectively) and perfused with 0.1 M PB followed by ice-cold 4% paraformaldehyde (PFA). Brains were removed and postfixed in 4% PFA overnight at 4 °C, and cryoprotected in 10%, 20%, 30% (w/v) sucrose (Sigma) before freezing in OCT. Brains were cut into 20-μm sections.
The primary antibodies and their sources are listed in Table 1. The primary antibodies were diluted in blocking solution [0.1% (v/v) Triton x-100 and 10% fetal calf serum in 0.01 M PBS][and applied to brain sections overnight at 4 °C. Then, brain sections were washed and incubated with secondary antibodies for 2 h (for details see Table S1). Finally, brain sections were mounted on glass slides.
Mounted slides were then imaged under an inverted laser scanning confocal microscope (FV1000; Olympus). A total of 3–5 sections (− 1.06 to − 0.58 mm posterior from bregma) were examined per mouse, and 5–8 mice were analyzed for each data point. Confocal images represent projected stacks of 15–20 images which were collected at 0.5-μm steps. The cells were counted by FV10-ASW (Olympus) and Image J (NIH, http://imagej.nih.gov/ij).
Western blotting
Lysates were generated using RIPA buffer (Thermo Scientific) supplemented with 1% protease inhibitor cocktail set III EDTA-free (vol/vol, Calbiochem). Protein concentration was determined using the Thermo BCA Protein Assay Kit (Thermo Scientific) according to the manufacturer’s instructions. Samples were heated at 100 °C for 10 min, and 10 μg of total protein was loaded onto 12% acrylamide gel. Then, proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore) for 2 h at 56 V in transfer buffer. Membranes were blocked with 5% powdered milk in Tris-buffered saline (TBST) for 2 h at room temperature on an orbital shaker. Then, membranes were incubated with primary antibody overnight at 4 °C, washed thrice in TBST for 5 min, and incubated with horseradish peroxidase (HRP)-conjugated IgG secondary antibody for 2 h at room temperature. Chemiluminescent substrate detection reagent (Pierce™ ECL Western Blotting Substrate, Thermo Scientific) and autoradiography film processing was performed, followed by analysis with Image Lab (Bio-Rad).
Cell culture and treatment
Primary rat cortical neurons were prepared from 1-day-old Sprague–Dawley (SD) rats. Cells were cultured in Dulbecco’s modified Eagle’s medium/Ham’s F12 (DMEM/F12, 1:1, v/v) (Invitrogen) and 2% B27 (GIBCO) supplemented with 20 ng/ml basic fibroblast growth factor (bFGF) and 20 ng/ml epidermal growth factor (EGF) (Sigma) for 5 days. Cells were routinely maintained in DMEM containing at 37℃ in a humidified atmosphere of 5% CO2. After 24 h, cultures were subjected to treatment with different concentrations of glutamate. NRG1 was used as a recombinant polypeptide containing the entire EGF domain of the β-type NRG1 (rHRG β177–244) (Holmes et al. 1992). Ecto-ErbB4 was prepared from HEK293 cells as previously described (Woo et al. 2007).
Assay for cell viability and death
Cell viability was determined using the Dojindo Cell Counting Kit-8 (CCK-8, Dojindo Laboratories). Cells were seeded into a 96-well plate at a density of 1 × 104 cells per well, and CCK-8 solution (10 μl in each well containing 100 μl of medium) was added 24 h or 48 h later. The plate was incubated at 37 °C for 4 h, and the absorbance at 450 nm was then measured using a microplate reader. All experiments were done in triplicate and performed three times.
Cell viability (%) was calculated using the formula: (As − Ab)/(Ac − Ab)] × 100, where As is the experimental well absorbance (absorbance of cells, medium, CCK-8 and wells of the test compound), Ab is the blank well absorbance (absorbance of wells containing medium and CCK-8), and Ac is the control well absorbance (absorbance of wells containing cells, medium and CCK-8. Cell death ratio (%) was thus calculated using the formula: 100% cell viability.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6. Data are presented as mean ± SEM. When two groups were compared, a two-tailed unpaired Student’s t test was used unless otherwise noted. Two-way analysis of variance (ANOVA) followed by Bonferroni post-tests was used to analyze experiments with two variables.
Results
TBI induces persistent activation of NRG1–ErbB4 signaling in the cortex
To investigate whether TBI activates the NRG1–ErbB4 signaling in the injured cortex, we induced focal contusion injury using CCI in mice (Fig. 1a, b). Firstly, we collected the cortex ipsilateral to the injury (Fig. 1c) and quantified full-length NRG1, cleaved NRG1, ErbB4 and pErbB4 levels at 1, 2, 4, 8 and 16 days post-TBI (dpi) by western blotting analysis (Fig. 1d, e). Results showed that the full-length NRG1 level was significantly increased in the cortex of TBI animals at 1, 2, 4 and 8 days post-injury (dpi), and return to normal level at 16 dpi (Fig. 1f). The cleaved NRG1 level was significantly increased in the cortex of TBI mice at 1, 2, 4, 8 and 16 dpi (Fig. 1g). Furthermore, ErbB4 level was decreased in the cortex of TBI mice at 1 and 2 and 4 dpi, then return to normal level at 8 dpi, and increased in the cortex of TBI mice at 16 dpi (Fig. 1h). pErbB4 level was increased in the cortex of TBI mice at 2, 4, 8 and 16 dpi (Fig. 1i).

TBI triggers a persistent activation of NGR1–ErBb4 signaling in the cortex. a The procedures for CCI. b The brain of mice after sham or TBI. Scale bar: 4 mm. c Diagram of coronal mice brain section showing relationship of lesion cavity (red) to regions used for western blot analysis (red squares). d, e Representative Western blot bands of full-length NRG1, cleaved NRG1, ErbB4 and p-ErbB4 from the ipsilateral hemisphere after sham or TBI. f–i Quantitative analyses of endogenous full-length NRG1, cleaved NRG1, ErbB4 and p-ErbB4 at 1, 2, 4, 8, 16 dpi. Data are represented as mean ± SEM, *p < 0.05, **p < 0.01, n = 5 animals per group
As shown, the total ErbB4 level first decreased and then increased after TBI. It was possible that the death of neurons (including the ErbB4+ cell) occurred at the beginning of TBI, or the rapid production of pErbB4 leaded to the decrease in ErbB4 level. Taken together, these results indicate that TBI triggers a persistent activation of the NRG1–ErbB4 signaling near the injured site.
PV-ErbB4−/− mice display increased cortical lesion volume and neuron death after TBI
ErbB4 expression in the cortex is largely restricted to GABAergic interneurons (Fazzari et al. 2010; Mei and Nave 2014). In particular, it is expressed by a majority of PV+ cells, one specific class of GABAergic interneurons (Fazzari et al. 2010; Wen et al. 2010). Here, our results also showed that ErbB4 is mainly expressed in PV+ cortical interneurons (Fig. S1A-C), which is consistent with previous studies. Thus, we used the PV-ErbB4−/− mice in which ErbB4 is ablated specifically in PV-positive interneurons to address the potential role of NRG1–ErbB4 signaling in the outcomes of TBI. Interestingly, we also found that there was a significant increase in the number of ErbB4+ neurons in the cortex of TBI mice as compared to sham mice at 16 dpi (Fig. S1D, E). To evaluate whether PV-ErbB4 knockout influences the acute and sub-acute histological outcome following cortical trauma, we evaluated lesion formation at 4 dpi in PV-ErbB4+/+ mice and PV-ErbB4−/− littermates. Tetrazolium chloride (TTC) stains (Fig. 2a) and Nissl-stained sections (Fig. 2b) were used to assess for cortical tissue damage. We found that PV-ErbB4−/− mice displayed larger lesion volumes (Fig. 2a–c) and more water content (Fig. 2d) in the ipsilateral cortex compared to PV-ErbB4+/+ mice at 4 dpi. These findings correlated with decreased Neun+ cells (Fig. 2e, f) in the ipsilateral cortex of PV-ErbB4−/− mice compared to PV-ErbB4+/+ mice. Taken together, these findings suggest that PV-ErbB4 knockout increases cortical lesion volumes and neuron death after TBI.

Analysis of cortical lesion volume and cell death after TBI. a TTC staining was performed on PV-ErbB4−/− mice and PV-ErbB4+/+ mice that had received TBI. Scale bar: 3 mm. b Nissl staining was performed on PV-ErbB4−/− mice and PV-ErbB4+/+ mice that had received TBI. Scale bar: 1 mm. c Quantified representation of lesion volume (mm3) shows that PV-ErbB4−/− mice have increased cortical loss at 4 dpi compared to PV-ErbB4+/+ mice. d PV-ErbB4−/− mice showed a significant increase in the overall water content of the brain. e Confocal immunofluorescence staining for NeuN in the ipsilateral cortex of PV-ErbB4+/+ mice and PV-ErbB4−/− mice at 4 dpi after TBI. Scale bar: 40 μm. f Quantification of the number of NeuN+ cells in the ipsilateral cortex at 4 dpi shows a significant reduction in the number of neuron in the cortex of PV-ErbB4−/− mice compared to PV-ErbB4+/+ mice. Data are represented as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, n = 5 animals per group
The activation of astrocytes and microglia is enhanced in PV-ErbB4−/− mice following TBI
TBI can induce an inflammatory response characterized by infiltration of leukocytes and monocytes, production of inflammatory cytokines, and activation of resident glial cells (Jassam et al. 2017; Simon et al. 2017). Thus, we evaluated the number of astrocytes and microglia in the site of injury. We found that PV-ErbB4−/− mice exhibit significantly increased astrocyte (GFAP+) in the injured ipsilateral cortex when compared with PV-ErbB4+/+ littermates (Fig. 3a, b). Then, we used western blot analysis to determine the level of GFAP in the injured ipsilateral cortex, which was consistent with the results of immunohistochemistry (Fig. 3c, d). Next, we found a significant increase in the number of microglia (Iba1+) cells in the injured ipsilateral cortex of PV-ErbB4−/− mice when compared with PV-ErbB4+/+ mice (Fig. 3e, f).

Knocking out of ErbB4 in PV+ neuron enhances astrocytic and microglial response. a Representative images of astrocytes immunostained for GFAP at 4 dpi. Scale bars: 1 mm. Rectangle indicates cortical area used for cell counting. Scale bars: 50 μm. b Quantification of GFAP+ cells in PV-ErbB4+/+ mice versus PV-ErbB4−/− mice at 4 dpi. c Representative Western blot bands GFAP from the ipsilateral hemisphere of PV-ErbB4+/+ mice and PV-ErbB4−/− mice at 4 dpi. d Quantitative analyses of GFAP at 4 dpi. e Representative images of astrocytes immunostained for Iba1 at 4 dpi. Scale bars: 40 μm. f Quantification of Iba1+ cells in PV-ErbB4+/+ mice versus PV-ErbB4−/− mice at 4 dpi. Data are represented as mean ± SEM, *p < 0.05, **p < 0.01, n = 5 animals per group
Taken together, these results indicate that a specific knockout of ErbB4 in PV+ interneuron enhances the activation of astrocytes and microglia in the TBI site. These observations suggest that the astrocytic and microglial response after TBI partly relies on NRG1–ErbB4 signaling.
ErbB4 knockout influences functional recovery following cortical impact injury
Cortical impact injury usually results in significant behavioral deficits in mice (Zhao et al. 2012). To evaluate whether ErbB4 knockout influences behavioral recovery after TBI, we assessed changes in gross and fine motor function using rotarod test and beam walk test, respectively, at 1, 2, 4, 8, 16 and 32 dpi in PV-ErbB4+/+ mice and PV-ErbB4−/− littermates. PV-ErbB4−/− mice showed significant less time on the rotarod compared to PV-ErbB4+/+ littermates at 4, 8, 16 and 32 dpi (Fig. S2A). Furthermore, PV-ErbB4−/− mice showed more foot slips compared to PV-ErbB4+/+ littermates at 1, 2, 4, 8 and 16 dpi in beam walk test (Fig. S2B). In addition, neurological performance was evaluated by mNSS. The higher scores indicate the more severe injury. As shown in Fig. S2C, higher scores in PV-ErbB4−/− mice were observed compared to PV-ErbB4+/+ littermates at 4, 8 and 16 dpi, suggesting that ErbB4 knockout in PV+ interneuron exacerbates neurological deficits in mice after TBI. Moreover, abnormal posturing was quantified using the contralateral torso flexion test on multiple days after injury showing a significant increase in the scale in PV-ErbB4−/− mice compared to PV-ErbB4+/+ littermates at 2, 4, 8, 16 and 32 dpi (Fig. S2D). These results suggest that NRG1–ErbB4 signaling plays an important role in the behavioral function recovery after TBI.
Intranasal delivery of recombinant NRG1 rescues neurological functions after TBI
As the absence of endogenous NRG1–ErbB4 signaling worsens neurological impairments and brain injury after TBI, we hypothesized that intracerebral delivery of exogenous NRG1 to TBI-treated mice would ameliorate neurological deficits after TBI. To test this, we employed the intranasal approach for NRG1 delivery into the brain (Kozlovskaya et al. 2014). In a pilot dose–response study, we tested the effects of various intranasal NRG1 dosages (0, 0.5, 1, and 1.5 mg/kg) on behavioral performance up to 32 d after TBI using the rotarod test. The results showed that 0.5 mg/kg was the lowest dose among the tested doses to improve post-TBI performance in the rotarod test significantly (Fig. 4A). Higher doses did not exhibit further protection (data not shown). Therefore, 0.5 mg/kg was selected as the optimal dose for all subsequent studies. PV-ErbB4+/+ mice were subjected to TBI, and equal volumes of PBS or NRG1 (0.5 mg/kg, freshly dissolved in PBS at 1 μg/μL) were delivered into their nostrils 2 h after the injury and then once every other day for 32 days. After post-TBI intranasal NRG1 administrations, the motor function of PV-ErbB4+/+ mice was significantly improved in rotarod test and beam walk test (Fig. 4a, b). Furthermore, NRG1 treatment alleviates neurological deficits (Fig. 4c) and abnormal posturing (Fig. 4d). In contrast, NRG1 has no effects on the motor function improvement in PV-ErbB4−/− mice after TBI (Fig. 4e–h). These results suggest that intranasal administration of recombinant NRG1 improves most of the neurological functions examined in PV-ErbB4+/+ mice after TBI.

Intranasal delivery of exogenous, recombinant NRG1 facilitates motor function recovery after TBI in mice. a–d PV-ErbB4+/+ mice were subjected to unilateral TBI in the right hemisphere. Recombinant NRG1 (0.5 mg/kg) or an equivalent volume of vehicle (PBS) was delivered intranasally 2 h after TBI and every other day up to 32 dpi. Rotarod test (a), beam walk test (b), neurological function test (c) and contralateral torso flexion test (d) were used to assess motor functions in mice. NGR1 improves motor function recovery in the PV-ErbB4+/+ mice after TBI. e–h PV-ErbB4−/− mice were subjected to unilateral TBI in the right hemisphere. Recombinant NRG1 (0.5 mg/kg) or an equivalent volume of vehicle (PBS) was delivered intranasally 2 h after TBI and every other day up to 32 dpi. Rotarod test (e), beam walk test (f), neurological function test (g) and contralateral torso flexion test (h) were used to assess motor functions in mice. NGR1 does not affect motor function in the PV-ErbB4−/− mice after TBI. *p < 0.05, **p < 0.01, ***p < 0.001, n = 12–14 animals per group
NRG1–ErbB4 signaling-mediated neuroprotection involves GABAergic transmission
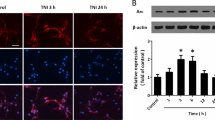
TBI induces deleterious effects involving in primary and secondary damages via glutamate toxicity (Bramlett and Dietrich 2015). TBI-induced upregulation of glutamate release in the extracellular space cannot be cleared effectively and result in neuronal cell death (Faden et al. 1989; Kierans et al. 2014; Dorsett et al. 2017). Increasing GABAergic neurotransmission is known to reduce cerebral ischemia-induced neuronal apoptosis (DeFazio et al. 2009). So it is possible that NRG1 neuroprotection may be mediated by ErbB4 receptors on GABAergic neurons via the regulation of GABA transmission. To test this hypothesis, we established a primary cortical neurons culture system in the present of 4 mM glutamate in vitro (Takada et al. 2003; Kim et al. 2016). Different concentrations of NRG1 were added to the primary neuronal culture media. After 24 h, the percentages of cell death were determined by CCK-8 assay. As shown in Fig. 5A, NRG1 treatment can significantly decrease the percentages of cell death compared to vehicle treatment. These data suggested that NRG1 reduced cell death in the presence of glutamate in a dose-dependent manner.

The neuroprotective effect of NRG1–ErbB4 signaling in TBI may involve GABAergic transmission. a NRG1 significantly decreased cell death. b Ecto-ErbB4 can block the neuroprotective effect of NRG1, and AG1478 can block the neuroprotective effect of NRG1. c The NRG1 neuroprotection against glutamate-induced cell death is abolished in PV-ErbB4−/− neuron. d GABA receptor agonist rescues neuron from glutamate-induced death. e GABA receptor agonists decrease cell death but do not have a synergistic effect with NRG1. f The neuroprotective effects of NRG1 are partially blocked by GABA receptor antagonists. Mus muscimol, Bac Baclofen. Data are represented as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 8 samples per group
To confirm the specificity of NRG1′s neuroprotective effect, we expressed and purified ecto-ErbB4, which binds to and prevents NRG1 from interacting with ErbB4 receptor kinases. Cortical neuronal cultures were treated with 4 nM NRG1 in the presence of 2 μg/ml ecto-ErbB4, or with 4 nM NRG1 alone. As shown in Fig. 5b, the percentage of cell death in the NRG1-treated group with ecto-ErbB4 was higher than that in the NRG1 only. The results show that ecto-ErbB4 can neutralize the neuroprotective effect of NRG1. In addition, AG1478, an ErbB4 inhibitor, was used to explore the involvement of ErbB4 in NRG1’s neuroprotection. As shown in Fig. 5b, AG1478 neutralizes the neuroprotective effect of NRG1, and the percentage of cell death was increased compared to the group treated with NRG1 only. Then, we tested the effect of PV-ErbB4 knockout on neuroprotection of NRG1. As shown in Fig. 5c, the percentage of cell death in the PV-ErbB4−/− group treated with NRG1 was significantly higher than that of PV-ErbB4+/+ group. Taken together, these results provide evidence that the neuroprotective effect of NRG1 is mediated by ErbB4.
NRG1 can promote GABA release via ErbB4 receptors. Increased GABA neurotransmission exerts neuroprotective effects to reduce cerebral ischemia-induced neuronal death (DeFazio et al. 2009). To test whether the neuroprotective effects of NRG1–ErbB4 signaling are mediated by a GABAergic mechanism, we first examined the effect of co-application of NRG1 and GABA receptor agonists in the cortical neuronal cultures. As shown in Fig. 5d, co-application of the GABAA receptor agonist Mus and GABAB receptor agonist Bac had a neuroprotective effect, which is consistent with the findings of a previous investigation (Guan et al. 2015). Then, we examined the effect of co-application of NRG1 and the GABA receptor agonists muscimol and baclofen. As shown in Fig. 5e, co-application of muscimol and baclofen with 4 nM NRG1 markedly diminished neuronal death, but the effect was similar to that induced by 4 nM NRG1 alone. This result indicates that the GABA receptor agonists and NRG1 do not have synergistic effects, suggesting that NRG1-mediated neuroprotection may involve GABAergic release. Next, we tested the effect of blockade of GABA receptors, treated with GABA receptor antagonists BMI and CGP, on neuroprotection conferred by NRG1. As shown in Fig. 5f, the percentage of cell death in the group treated with 1 μM BMI, 10 μM CGP and in the group treated with 4 nM NRG1 was significantly higher than that in the group treated with 4 μM NRG1 alone. These results suggest that the neuroprotective effects of NRG1 were partly blocked by GABA receptor antagonists, providing further support for the hypothesis that NRG1-mediated neuroprotection is via increasing GABAergic release. Taken together, these results demonstrate that the neuroprotective effects of NRG1–ErbB4 signaling are mediated, at least in part, via increasing GABAergic transmission.
Discussion
There are four major findings in the present study. First, TBI activates NRG1–ErbB4 signaling in the cortex following TBI. Second, specific ablation of ErbB4 in PV+ interneurons impaired functional recovery following TBI. Third, NRG1 reduced cortical neuronal cell death in vitro and in vivo. Fourth, the neuroprotective effect of NRG1–ErbB4 signaling against glutamate-induced neuronal apoptosis may involve GABAergic transmission. Together, the results indicate that NRG1-mediated neuroprotection involves enhanced GABAergic transmission via ErbB4 receptors.
ErbB4 is specifically expressed in GABAergic interneurons and is important for normal CNS function (Mei and Nave 2014). Previous studies reported that NRG1 was neuroprotective in rat models of ischemia (Li et al. 2012; Lu et al. 2016), and ErbB receptors are upregulated in neurons following ischemic stroke and may be involved in neuroprotection and repair (Xu and Ford 2005; Lu et al. 2016). In addition, closed head injury also induces up-regulation of ErbB-4 receptor at the site of injury (Erlich et al. 2000). Therefore, we are interested in whether there is a neuroprotection effect of NRG1–ErbB4 signaling in the model of TBI. Using a well-established focal contusion model of TBI, we found that TBI elevates the expression of NRG1 and ErbB4 in the injured site. Furthermore, we found that cortical lesion formation was significantly increased post-CCI injury in PV-ErbB4−/− mice when compared with PV-ErbB4+/+ littermates which correlated with reduced numbers of NeuN-positive cells in the cortex, increased motor deficits and increased numbers of astrocytes and microglia. However, it is still unknown how NRG1–ErbB4 signaling modulates immune system. Future studies are needed to dissect the relative contribution of glia cells to outcomes of TBI. Then, we found that NRG1 rescues cortical neuron death induced by glutamate treatment, then we used this in vitro cortical neuron culture model to clarify the molecular mechanisms of NRG1’s neuronal protection. Our study demonstrated that the ErbB4 inhibitor AG1478 blocked NRG1 protection against glutamate-induced apoptosis. These observations demonstrate that NRG1 is working via ErbB4, and all the neuroprotective effect of NRG1 was abolished in PV-ErbB4−/− neurons.
GABAergic inhibitory interneurons are the only source of GABA (Guan et al. 2015). Although GABAergic cells constitute 10–20% of the neuronal population, they serve important roles in CNS. An increasing number of studies have demonstrated that enhanced GABAergic transmission is neuroprotective using in vivo and in vitro models of ischemia (Mayor and Tymianski 2018). Previous studies showed that NRG1 regulates GABAergic transmission via ErbB4 receptors, which are expressed in GABAergic presynaptic terminals in the cerebral cortex (Fazzari et al. 2010; Mei and Nave 2014). In this study, GABA receptor agonists Mus and Bac abolish the neuroprotection effects of NRG1. Furthermore, NRG1 treatment does not show neuroprotection against glutamate-induced death in the neurons with a specific ablation of ErbB4 in PV+ GABAergic interneurons. The result supports that the NRG1-mediated neuroprotection against brain damage are mediated via ErbB4 by enhancing GABA release. These findings indicate that GABAergic transmission contributes to the neuroprotective effects of NRG1–ErbB4 signaling, but NRG1–ErbB4 signaling-mediated neuroprotection against glutamate- or TBI-induced neuronal death may also involve some other mechanisms.
The surprising results presented here have yet to be extended from mouse models to human physiology. We also found that NRG1 treatment can restore the motor function defects in vivo. Therefore, a better understanding of these neuroprotective pathways in TBI may broader therapeutic potential, especially when the window for treating acute injuries has passed. We are hopeful that our findings can offer a promising therapeutic avenue for patients who are suffering from functional deficits associated with TBI and other neurodegenerative disorders.
In conclusion, our findings suggest that the roles of the pleiotropic NRG1–ErbB4 signaling extend to protection of neuronal network communication and improvement of functional outcomes in acute and chronic phases of TBI. If similar mechanisms operate in humans, they might partly contribute to the normal life expectancy of TBI patients. Therefore, recombinant forms of NRG1 should continue to be tested for their potential to preserve or rescue neurons and improve long-term functional recovery after acute brain injuries.
References
Adelson JD, Barreto GE, Xu L et al (2012) Neuroprotection from stroke in the absence of MHCI or PirB. Neuron 73:1100–1107. https://doi.org/10.1016/j.neuron.2012.01.020
Alexander MP (1995) Mild traumatic brain injury: pathophysiology, natural history, and clinical management. Neurology 45:1253–1260
Bean JC, Lin TW, Sathyamurthy A, Liu F, Yin DM, Xiong WC, Mei L (2014) Genetic labeling reveals novel cellular targets of schizophrenia susceptibility gene: distribution of GABA and non-GABA ErbB4-positive cells in adult mouse brain. J Neurosci 34:13549–13566. https://doi.org/10.1523/JNEUROSCI.2021-14.2014
Bigler ED (2003) Neurobiology and neuropathology underlie the neuropsychological deficits associated with traumatic brain injury. Arch Clin Neuropsychol 18:595–621
Blennow K, Hardy J, Zetterberg H (2012) The neuropathology and neurobiology of traumatic brain injury. Neuron 76:886–899. https://doi.org/10.1016/j.neuron.2012.11.021
Bramlett HM, Dietrich WD (2015) Long-Term Consequences of Traumatic Brain Injury: current Status of Potential Mechanisms of Injury and Neurological Outcomes. J Neurotrauma 32:1834–1848. https://doi.org/10.1089/neu.2014.3352
Brickler TR, Hazy A, Guilhaume Correa F et al (2018) Angiopoietin/Tie2 axis regulates the age-at-injury cerebrovascular response to traumatic brain injury. J Neurosci 38:9618–9634. https://doi.org/10.1523/JNEUROSCI.0914-18.2018
Bublil EM, Yarden Y (2007) The EGF receptor family: spearheading a merger of signaling and therapeutics. Curr Opin Cell Biol 19:124–134. https://doi.org/10.1016/j.ceb.2007.02.008
Buonanno A (2010) The neuregulin signaling pathway and schizophrenia: from genes to synapses and neural circuits. Brain Res Bull 83:122–131. https://doi.org/10.1016/j.brainresbull.2010.07.012
Chen YJ, Zhang M, Yin DM et al (2010) ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc Natl Acad Sci USA 107:21818–21823. https://doi.org/10.1073/pnas.1010669107
Costa C, Leone G, Saulle E, Pisani F, Bernardi G, Calabresi P (2004) Coactivation of GABA(A) and GABA(B) receptor results in neuroprotection during in vitro ischemia. Stroke 35:596–600. https://doi.org/10.1161/01.STR.0000113691.32026.06
DeFazio RA, Raval AP, Lin HW, Dave KR, Della-Morte D, Perez-Pinzon MA (2009) GABA synapses mediate neuroprotection after ischemic and epsilonPKC preconditioning in rat hippocampal slice cultures. J Cereb Blood Flow Metab 29:375–384. https://doi.org/10.1038/jcbfm.2008.126
Dorsett CR, McGuire JL, DePasquale EA, Gardner AE, Floyd CL, McCullumsmith RE (2017) Glutamate neurotransmission in rodent models of traumatic brain injury. J Neurotrauma 34:263–272. https://doi.org/10.1089/neu.2015.4373
Erlich S, Shohami E, Pinkas-Kramarski R (2000) Closed head injury induces up-regulation of ErbB-4 receptor at the site of injury. Mol Cell Neurosci 16:597–608. https://doi.org/10.1006/mcne.2000.0894
Faden AI, Demediuk P, Panter SS, Vink R (1989) The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244:798–800
Falls DL (2003) Neuregulins: functions, forms, and signaling strategies. Exp Cell Res 284:14–30
Fazzari P, Paternain AV, Valiente M et al (2010) Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 464:1376–1380. https://doi.org/10.1038/nature08928
Garcia-Rivello H, Taranda J, Said M et al (2005) Dilated cardiomyopathy in Erb-b4-deficient ventricular muscle. Am J Physiol Heart Circ Physiol 289:H1153–H1160. https://doi.org/10.1152/ajpheart.00048.2005
Guan YF, Wu CY, Fang YY et al (2015) Neuregulin 1 protects against ischemic brain injury via ErbB4 receptors by increasing GABAergic transmission. Neuroscience 307:151–159. https://doi.org/10.1016/j.neuroscience.2015.08.047
Harrison PJ, Law AJ (2006) Neuregulin 1 and schizophrenia: genetics, gene expression, and neurobiology. Biol Psychiat 60:132–140. https://doi.org/10.1016/j.biopsych.2005.11.002
Holmes WE, Sliwkowski MX, Akita RW et al (1992) Identification of heregulin, a specific activator of p185erbB2. Science 256:1205–1210
Horita Y, Honmou O, Harada K, Houkin K, Hamada H, Kocsis JD (2006) Intravenous administration of glial cell line-derived neurotrophic factor gene-modified human mesenchymal stem cells protects against injury in a cerebral ischemia model in the adult rat. J Neurosci Res 84:1495–1504. https://doi.org/10.1002/jnr.21056
Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J (2017) Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron 95:1246–1265. https://doi.org/10.1016/j.neuron.2017.07.010
Johnson VE, Stewart W, Smith DH (2013) Axonal pathology in traumatic brain injury. Exp Neurol 246:35–43. https://doi.org/10.1016/j.expneurol.2012.01.013
Kierans AS, Kirov II, Gonen O et al (2014) Myoinositol and glutamate complex neurometabolite abnormality after mild traumatic brain injury. Neurology 82:521–528. https://doi.org/10.1212/WNL.0000000000000105
Kim JJ, Kang YJ, Shin SA et al (2016) Phlorofucofuroeckol improves glutamate-induced neurotoxicity through modulation of oxidative stress-mediated mitochondrial dysfunction in PC12 Cells. PLoS ONE 11:e0163433. https://doi.org/10.1371/journal.pone.0163433
Kozlovskaya L, Abou-Kaoud M, Stepensky D (2014) Quantitative analysis of drug delivery to the brain via nasal route. J Control Release 189:133–140. https://doi.org/10.1016/j.jconrel.2014.06.053
Li Y, Xu Z, Ford GD, Croslan DR, Cairobe T, Li Z, Ford BD (2007) Neuroprotection by neuregulin-1 in a rat model of permanent focal cerebral ischemia. Brain Res 1184:277–283. https://doi.org/10.1016/j.brainres.2007.09.037
Li Y, Lein PJ, Liu C et al (2012) Neuregulin-1 is neuroprotective in a rat model of organophosphate-induced delayed neuronal injury. Toxicol Appl Pharmacol 262:194–204. https://doi.org/10.1016/j.taap.2012.05.001
Lu YM, Gao YP, Tao RR et al (2016) Calpain-dependent ErbB4 cleavage is involved in brain ischemia-induced neuronal death. Mol Neurobiol 53:2600–2609. https://doi.org/10.1007/s12035-015-9275-2
Maas AI, Stocchetti N, Bullock R (2008) Moderate and severe traumatic brain injury in adults. Lancet Neurol 7:728–741. https://doi.org/10.1016/s1474-4422(08)70164-9
Madisen L, Zwingman TA, Sunkin SM et al (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. https://doi.org/10.1038/nn.2467
Mayor D, Tymianski M (2018) Neurotransmitters in the mediation of cerebral ischemic injury. Neuropharmacology 134:178–188. https://doi.org/10.1016/j.neuropharm.2017.11.050
Mei L, Nave KA (2014) Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 83:27–49. https://doi.org/10.1016/j.neuron.2014.06.007
Mei L, Xiong WC (2008) Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci 9:437–452. https://doi.org/10.1038/nrn2392
Merkler D, Metz GA, Raineteau O, Dietz V, Schwab ME, Fouad K (2001) Locomotor recovery in spinal cord-injured rats treated with an antibody neutralizing the myelin-associated neurite growth inhibitor Nogo-A. J Neurosci 21:3665–3673
Miller DM, Singh IN, Wang JA, Hall ED (2015) Nrf2-ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp Neurol 264:103–110. https://doi.org/10.1016/j.expneurol.2014.11.008
Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM (2017) The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol 13:171–191. https://doi.org/10.1038/nrneurol.2017.13
Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 9:211–221. https://doi.org/10.1038/nrneurol.2013.29
Steinthorsdottir V, Stefansson H, Ghosh S et al (2004) Multiple novel transcription initiation sites for NRG1. Gene 342:97–105. https://doi.org/10.1016/j.gene.2004.07.029
Takada Y, Yonezawa A, Kume T, Katsuki H, Kaneko S, Sugimoto H, Akaike A (2003) Nicotinic acetylcholine receptor-mediated neuroprotection by donepezil against glutamate neurotoxicity in rat cortical neurons. J Pharmacol Exp Ther 306:772–777. https://doi.org/10.1124/jpet.103.050104
Taylor CA, Bell JM, Breiding MJ, Xu L (2017) Traumatic brain injury-related emergency department visits, hospitalizations, and deaths—United States, 2007 and 2013. MMWR Surveill Summ 66:1–16. https://doi.org/10.15585/mmwr.ss6609a1
Tidcombe H, Jackson-Fisher A, Mathers K, Stern DF, Gassmann M, Golding JP (2003) Neural and mammary gland defects in ErbB4 knockout mice genetically rescued from embryonic lethality. Proc Natl Acad Sci USA 100:8281–8286. https://doi.org/10.1073/pnas.1436402100
Washington PM, Forcelli PA, Wilkins T, Zapple DN, Parsadanian M, Burns MP (2012) The effect of injury severity on behavior: a phenotypic study of cognitive and emotional deficits after mild, moderate, and severe controlled cortical impact injury in mice. J Neurotrauma 29:2283–2296. https://doi.org/10.1089/neu.2012.2456
Wen L, Lu YS, Zhu XH et al (2010) Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proc Natl Acad Sci USA 107:1211–1216. https://doi.org/10.1073/pnas.0910302107
Woo RS, Li XM, Tao Y et al (2007) Neuregulin-1 enhances depolarization-induced GABA release. Neuron 54:599–610. https://doi.org/10.1016/j.neuron.2007.04.009
Xia Y, Pu H, Leak RK et al (2018) Tissue plasminogen activator promotes white matter integrity and functional recovery in a murine model of traumatic brain injury. Proc Natl Acad Sci USA 115:E9230–E9238. https://doi.org/10.1073/pnas.1810693115
Xu Z, Ford BD (2005) Upregulation of erbB receptors in rat brain after middle cerebral arterial occlusion. Neurosci Lett 375:181–186. https://doi.org/10.1016/j.neulet.2004.11.039
Xu Z, Jiang J, Ford G, Ford BD (2004) Neuregulin-1 is neuroprotective and attenuates inflammatory responses induced by ischemic stroke. Biochem Biophys Res Commun 322:440–446. https://doi.org/10.1016/j.bbrc.2004.07.149
Yang Y, Zhong Z, Wang B et al (2019) Early-life high-fat diet-induced obesity programs hippocampal development and cognitive functions via regulation of gut commensal Akkermansia muciniphila. Neuropsychopharmacology. https://doi.org/10.1038/s41386-019-0437-1
Zhao Z, Loane DJ, Murray MG 2nd, Stoica BA, Faden AI (2012) Comparing the predictive value of multiple cognitive, affective, and motor tasks after rodent traumatic brain injury. J Neurotrauma 29:2475–2489. https://doi.org/10.1089/neu.2012.2511
Zhou C, Li C, Yu HM, Zhang F, Han D, Zhang GY (2008) Neuroprotection of gamma-aminobutyric acid receptor agonists via enhancing neuronal nitric oxide synthase (Ser847) phosphorylation through increased neuronal nitric oxide synthase and PSD95 interaction and inhibited protein phosphatase activity in cerebral ischemia. J Neurosci Res 86:2973–2983. https://doi.org/10.1002/jnr.21728
Funding
This research was supported by the National Natural Science Foundation of China (31971035, 81471116, 31771182, 81560196), and the Natural Science Foundation of Jiangxi Province (20171ACB20002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no financial conflict of interest regarding the publication of this article.
Additional information
Communicated by Thomas Deller.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Deng, W., Luo, F., Li, Bm. et al. NRG1–ErbB4 signaling promotes functional recovery in a murine model of traumatic brain injury via regulation of GABA release. Exp Brain Res 237, 3351–3362 (2019). https://doi.org/10.1007/s00221-019-05680-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-019-05680-2