
Abstract
The ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) proteases are secreted enzymes that regulate extracellular matrix turnover by degrading specific matrix components. Roles for the proteases in inflammation and atherosclerosis have been suggested by a number of recent studies, and the role of ADAMTS-4 and -5 in the breakdown of aggrecan and subsequent degradation of cartilage during osteoarthritis has also been established. The ability of the ADAMTS proteases to degrade versican, the primary proteoglycan in the vasculature, is thought to be central to any hypothesized role for the proteases in atherosclerosis. In this review, we introduce the structure and function of the ADAMTS family of proteases and review the literature that links them with inflammation and atherosclerosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) proteases are a distinct group of zinc metalloproteases (MMPs) comprising 20 members, ADAMTS-1 to ADAMTS-20, that are structurally related to the ADAM and matrix MMP families of proteinases [1, 2]. ADAMTS proteases are non-membrane-bound enzymes that interact with components of the extracellular matrix (ECM) such as procollagen, hyalectans and cartilage oligomeric matrix protein to cause their degradation [2, 3]. The physiological roles of the ADAMTS proteases are only just beginning to be established, but a role in cartilage turnover has been described [1]. The ADAMTS proteases have been implicated in pathophysiological processes including osteoarthritis and cancer. A role for the ADAMTS proteases in inflammation and atherosclerosis has only recently come to light. This review will discuss the structure and function of the ADAMTS proteases and their potentially important roles in atherosclerosis.
The ADAMTS protease family
The mammalian ADAMTS proteases share between 20% and 40% homology with ADAMTS-13 being the most distinct. The ADAMTS family is usually subdivided into four classes based on structural and/or functional similarities. These are the proteoglycanases, the procollagen-n-peptidases, von Willebrand cleaving factor (ADAMTS-13) and the other ADAMTS proteases whose function is currently unclear (including ADAMTS-6, -7, -10, -12 and -16 to -19) [1, 4]. Targets of the ADAMTS proteases are summarized in Table 1.
The proteoglycanases
ADAMTS-1, -4, -5, -8, -9 and -15 are collectively known as the proteoglycanases as they cleave the proteoglycans aggrecan, versican and brevican to result in the degradation of the ECM [4]. A role for the ADAMTS proteases in ECM degradation was first suggested by knockout of ADAMTS-1 in mice where impaired formation of adipose tissue and accumulation of collagen within the skin tissue suggested that the processing of collagen and other ECM molecules was defective [5].
ADAMTS-1 was the first of the ADAMTS proteases to be characterized in 1997 [6] and was later identified as a potent inhibitor of angiogenesis [7] and then as an ‘aggrecanase’ [8]. Aggrecan is present within the articular cartilage where it is important for withstanding compressive deformation during joint movement and for protecting collagen from degradation. Aggrecan is cleaved by ADAMTS proteases at Glu–Ala bonds within the glycosaminoglycan region of the protein [4, 9, 10]. ADAMTS-4 and -5 show the highest aggrecanase activity and have been implicated in osteoarthritis and rheumatoid arthritis (RA) (reviewed by [11] and [12]).
Although the cleavage of aggrecan has been extensively researched, the cleavage of versican (a vascular proteoglycan structurally similar to aggrecan) by the ADAMTS proteases is of significant importance in atherosclerosis and inflammation. As detailed in this review, ADAMTS-1, -4 and -9 are able to cleave versican at Glu–Ala bonds, and this has particular significance to atherosclerosis as versican accumulation is observed in blood vessels susceptible to atherosclerosis [1, 13]. ADAMTS-4 is also able to cleave brevican, one of the primary proteoglycans in the central nervous system, and this action may be relevant to the pathophysiology of stroke [14]. Similar to versican and aggrecan, brevican can be cleaved by both MMPs and ADAMTS proteases at distinct sites. Interestingly, cleavage of brevican by ADAMTS-4 at a Glu–Ser bond has been linked to glioma invasion [15, 16].
The procollagen-n-proteinases
ADAMTS-2, -3 and -14 are termed procollagen-n-proteinases due to their involvement in the removal of N-terminal peptides from procollagen to form mature collagen. ADAMTS-2 is thought to play a role in the formation of skin, and similar to the other collagen C-proteinases, its expression can be upregulated by the cytokine transforming growth factor-β (TGF-β) [3]. More recently, the expression of ADAMTS-2 in monocytes and macrophages has been implicated in the regulation of inflammation and wound repair [17]. ADAMTS-3 and -14 are thought to function primarily in cartilage and tendon, respectively [2].
Von Willebrand cleaving factor
ADAMTS-13 has been identified as the protease that cleaves the multimeric glycoprotein, von Willebrand factor (VWF) [18]. This protein, present predominantly in the plasma, has a key role in coagulation. VWF is a carrier for clotting factor VIII and has a significant role in coagulation and thrombus formation as part of the thrombin pathway. In atherosclerosis, VWF binds to ECM molecules to promote platelet adhesion and aggregation at sites of damage to the vascular wall [1, 2]. ADAMTS-13 has been demonstrated to colocalize with VWF in thrombi of human coronary arteries [19]. Cleavage of VWF by ADAMTS-13 is thought to attenuate thrombus formation in atherosclerotic plaques, and a role for the disintegrin domain of ADAMTS-13 in this process has been identified [19]. In addition to this, a lack of ADAMTS-13 has been linked with thrombotic thrombocytopenic purpura where large multimers of VWF accumulate in the absence of ADAMTS-13 resulting in micro-thrombi and vascular obstructions [20].
The domain structure of ADAMTS proteases
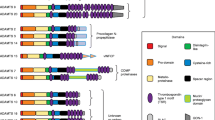
The ADAMTS proteases have a domain structure (Fig. 1) that is conserved throughout the family. ADAMTS proteases are synthesized as proproteins which are cleaved to become active through the removal of the prodomain by a furin proprotein convertase within the trans-Golgi [21, 22]. The catalytic (metalloproteinase) domain of ADAMTS proteases is comprised of a catalytic zinc coordinated by three histidine residues. This active site motif is followed by a highly conserved methionine residue, a pattern also observed in the ADAM and MMP families [2]. The catalytic domain is followed by a smaller, disintegrin-like domain. This region shares 25–45% homology with the snake venom disintegrins but does not interact with integrins. Following this region are the thrombospondin type 1-like repeats (TSR). These sequences are unique to the ADAMTS proteases amongst the matrix protease families and are homologous to the type 1 repeats found within thrombospondins 1 and 2. One important function of this domain is to bind to the ECM and to glycosaminoglycans such as heparin [1, 2]. The highly conserved cysteine-rich domain follows the TSR region and contains ten cysteine residues. The crystal structure of ADAMTS-1 revealed that this was stacked against the active site of the protease [23]. It is followed by a spacer region that varies in length and sequence but usually contains several hydrophobic residues towards the N-terminal end. Between 1 and 14 TSRs follow the spacer region in all ADAMTS proteases except ADAMTS-4 [2, 4].

Archetypal domain structure for the ADAMTS proteases. The ADAMTS proteases contain a signal sequence that is followed by a prodomain of varying length and a catalytic metalloproteinase domain. This protease domain region is highly conserved between ADAMTS members with ADAMTS-13 being the exception, having an unusually short prodomain. The ancillary domain is made up of a disintegrin-like domain, a thrombospondin type I repeat, a cysteine-rich domain and a spacer domain. The remainder of the protein is variable according to the different family members. The variable sections are made up of a number of thrombospondin type I repeats and other domains. See text for more details
The ability of the ADAMTS proteases to bind to the extracellular matrix is one important property that distinguishes the ADAMTS family from the ADAM and MMP families [21]. The proteases bind to sulphated glycosaminoglycans present in the ECM including heparin and aggrecan [1]. Both the catalytic and the non-catalytic regions of the ADAMTS proteases are required for full proteolytic activity and substrate binding. This is achieved through post-translational processing (C-terminal truncation) of mature ADAMTS proteases within the spacer region and may be mediated by MMPs [24].
Studies using murine ADAMTS-1 domain-deletion constructs and C-terminally processed forms of human ADAMTS-4 have identified the TSP type 1 motifs and the cysteine-rich and spacer regions in the C-terminal domain as crucial for ECM binding, with truncation of this region resulting in a loss of ECM binding [1, 25]. In addition, the spacer and cysteine-rich regions together with the thrombospondin motifs are required for substrate recognition and cleavage by the proteases [26, 27]. Cleavage of the cysteine-rich and spacer regions reduces the affinity of ADAMTS-4 for heparin and reduces its ability to cleave aggrecan [2, 28].
Regulation of ADAMTS proteases
Regulation of the ADAMTS proteases remains a poorly understood area. One regulatory property that is known is that the activity of ADAMTS can be regulated by the tissue inhibitors of metalloproteinases (TIMPs) [1, 29]. TIMPs-1, -2, -3 and -4 are endogenous inhibitors of the MMPs [30]. However, TIMP-3 has been found to be an important specific inhibitor of ADAMTS-4 and -5. In addition, TIMP-2 and TIMP-3 can inhibit ADAMTS-1 aggrecanase activity [31]. Hashimoto et al. [32] incubated ADAMTS-4 with TIMPs to demonstrate the inhibitory action of these molecules on the ability of ADAMTS-4 to cleave aggrecan. Analysis of the IC50 values revealed that the activity of TIMP-3 was 50-fold higher than that observed with TIMPs-1 and -2 and approximately 250-fold higher than the activity of TIMP-4, suggesting that this inhibitory action on ADAMTS-4 is specific to TIMP-3 [32]. Although the mechanism of inhibition has not been fully elucidated, a study by Kashiwagi et al. [33] demonstrated that the N-terminal domain of TIMP-3 was able to inhibit the aggrecanase activity of ADAMTS-4 and -5 suggesting that the N-terminal end of TIMP-3 may interact with ADAMTS. A recent study has demonstrated that this interaction is enhanced by the C-terminal domain of ADAMTS-4 and ADAMTS-5. Binding of TIMP-3 may prevent interaction between the ADAMTS and aggrecan by altering the active site to favour interaction with TIMP-3 rather than the aggrecan substrate [33, 34].
Inflammation and atherosclerosis
Atherosclerosis, the principal cause of heart attacks and stroke is the one of the leading causes of death in Western societies. Atherosclerosis is a progressive inflammatory disease triggered by damage to the vascular endothelium by various risk factors including genetic predisposition, hypertension, smoking, hypercholesterolemia and type 2 diabetes mellitus [35–38]. The disease is characterized by the formation of a plaque composed of cholesterol, lipids, inflammatory cells and debris resulting from cellular apoptosis. In the primary stages of atherosclerosis, monocytes and other inflammatory cells are attracted to the site of damage by the elevated expression of cell surface adhesion molecules such as inter-cellular adhesion molecule-1 and vascular cell adhesion molecule-1 and chemokines such as monocyte chemoattractant protein-1 by endothelial cells [35–38]. Monocytes migrate into the arterial intima where they differentiate into macrophages. Macrophages express cell surface scavenger receptors which normally function to take up pathogens and apoptotic cells but, in atherosclerosis, are able to take up modified lipoproteins such as oxidized low density lipoprotein (oxLDL) which accumulates in the vessel wall [35, 39, 40]. This uptake and subsequent accumulation of oxLDL transforms macrophages to lipid-laden foam cells which form the fatty streak associated with early stage atherosclerosis. The migration of vascular smooth muscle cells (VSMCs) and T lymphocytes into the artery characterizes the progression of a fatty streak to a more advanced fibrofatty lesion. More complex lesions are formed when VSMCs move into the intima and contribute to foam cell formation through proliferation and lipoprotein uptake. The synthesis of ECM by these cells produces a fibrous cap over the lesion to produce an atherosclerotic plaque that can extend into the arterial lumen. Over time these plaques can become unstable and prone to rupture sometimes leading to thrombosis, myocardial infarction and other acute complications [35–38].
Plaques vulnerable to rupture are characterized by a lipid-rich core, thin fibrous cap, high levels of inflammation, neovascularization and matrix remodelling [35]. Several studies have demonstrated a key role for matrix-degrading proteases in the synthesis and degradation of the ECM in atherosclerosis. Synthesis of MMPs by macrophages and VSMCs is increased in patients with unstable angina and acute myocardial infarction, and their synthesis is increased following macrophage activation by pro-inflammatory cytokines. MMPs have also been demonstrated to contribute to VSMC migration and proliferation [41–43]. Whilst controlled ECM remodelling is essential for migration and proliferation of VSMCs and contributes to plaque stability, matrix degradation can result in the thinning of the fibrous cap of the lesion and decreased collagen content of lesions, rendering them vulnerable to rupture [42].
ADAMTS proteases—links with inflammation and atherosclerosis
Changes in the degradation of ECM by MMPs and other proteases have been associated with a number of pathological inflammatory conditions including atherosclerosis and rheumatoid arthritis. This shift in balance of synthesis and degradation is often driven by inflammation, injury and oxidative stress [30, 44–46]. In atherosclerosis, macrophages and monocytes are known to secrete proteolytic enzymes to influence the development of the lesion and/or the stability of the plaque [45, 47]. Differentiation of monocytes to macrophages is also accompanied by an increase of matrix-degrading proteases [48]. Immunohistochemical analysis has demonstrated that ADAMTS-1, -4, -5 and -8 are present within human carotid lesions and advanced coronary atherosclerotic plaques [45]. Of these, ADAMTS-4, -5 and -8 colocalize with macrophages whilst ADAMTS-1 colocalizes with endothelial and smooth muscle cells [45, 49]. ADAMTS-1 and ADAMTS-4 are the most abundant of the proteases and have consequently been the focus of studies on the link between ADAMTS proteases and atherosclerosis [2]. The potential roles of ADAMTS proteases in atherosclerosis and inflammation are summarized in Fig. 2.

ADAMTS protease role in inflammation and atherosclerosis. A summary of the role of the ADAMTS proteases during atherosclerosis and inflammation
The enhanced expression of ADAMTS-1 in the kidney and heart observed in response to in vivo lipopolysaccharide (LPS) administration first suggested that ADAMTS proteases were associated with inflammatory processes. The observation that the mRNA expression of murine ADAMTS-1 could be upregulated by IL-1 was also consistent with a proposed role for ADAMTS-1 in inflammation [4, 25, 29].
In endothelial cells, the expression of ADAMTS-1 can be induced by LPS and the pro-inflammatory cytokine tumour necrosis factor-α (TNF-α). This demonstrates that the upregulation of ADAMTS-1 in response to inflammatory cytokines may promote inflammation [50]. This can be countered by high density lipoprotein (HDL), possibly due to its protective role in endothelial cell function and atherosclerosis. It is possible that HDL may inhibit the expression of genes coding for proteolytic enzymes by endothelial cells similar to what has been observed for several adhesion molecules [50]. ADAMTS-1 is a potent inhibitor of angiogenesis and has been shown to inhibit the proliferation of endothelial cells by sequestration of vascular endothelial growth factor (VEGF). This is achieved through direct binding of the protease to VEGF, thereby preventing the interaction of VEGF with its receptor [50, 51]. Conversely, the expression of ADAMTS-1 can be upregulated by VEGF, and this is protein kinase C-dependent, suggesting a feedback mechanism for the regulation of angiogenesis [52]. The expression of both ADAMTS-1 and VEGF in endothelial cells can be induced by hypoxia [53].
ADAMTS-1 has been linked with atherosclerosis development through a role in promoting inflammation and VSMC migration. Immunohistochemistry has shown that ADAMTS-1 is expressed at high levels in the aorta and colocalizes with endothelial cells and VSMCs in atherosclerotic lesions [45, 49]. Expression of ADAMTS-1 is higher in migrating and proliferating VSMCs. Mice overexpressing ADAMTS-1 crossed with ApoE-deficient mice (a mouse model system for atherosclerosis) show an increased thickening of the arterial intima suggesting that ADAMTS-1 could be involved with expansion of lesions [49]. ADAMTS-1 expression is also upregulated by shear stress in human endothelial cells suggesting a role for the protease in the adaptations in the vessel structure during vascular events [54].
In contrast with ADAMTS-1, ADAMTS-4 colocalizes with macrophages of atherosclerotic lesions, and its expression can be upregulated by the pro-inflammatory cytokines IFN-γ and TNF-α [45]. Studies using the LDL-receptor and ApoB 100/100-deficient, atherosclerotic mouse model have shown that the levels of ADAMTS-4 mRNA are induced almost threefold during lesion development [45]. In vitro experiments in THP-1 cells and primary monocytes have shown that the expression of ADAMTS-4 and -8 are increased following the differentiation of monocytes to macrophages whilst the expression of ADAMTS-1, -2, -5, -7 and -10 remains unchanged [1, 45, 47]. The induction of ADAMTS-4 and -8 expression following monocyte–macrophage differentiation is thought to be mediated through a secondary signal. IL-1β is a suggested candidate for this as its expression in monocytes is induced by PMA, which is used for differentiation of THP-1 cells into macrophages, and it has also been demonstrated to induce ADAMTS-4 expression in human chondrocytes [47] and decrease versican synthesis in arterial smooth muscle cells [55]. However, studies have shown that IL-1β does not induce ADAMTS-4 expression in differentiating THP-1 cells [45] suggesting that such regulation may be differentiation specific.
The ability of both ADAMTS-1 and -4 to cleave the proteoglycan versican is likely to be central to the hypothesized roles for the proteases in atherosclerosis [47]. Versican fragments are present in both arterial samples and in nonhuman primate vascular graft models, indicating that versican undergoes processing, and this processing is likely to be carried out by the ADAMTS proteases [56].
Versican
Versican is a proteoglycan belonging to the same ‘hyaluronan-binding’ family as aggrecan and brevican and is similar in structure to aggrecan in particular in the N- and C-terminal domains [57]. Versican is a key component of the ECM where it can interact with hyaluronan and link proteins to form large aggregates that can regulate cellular processes including adhesion, proliferation and migration, as previously reviewed [58].
Four isoforms of versican (V0, V1, V2 and V3) arise from alternative splicing of the same gene, located on chromosome 5q in humans [58, 59]. The four isoforms differ by the presence or absence of two glycosaminoglycan binding domains designated α-GAG and β-GAG. All versican isoforms are made up of an N-terminal globular domain, which contains binding elements for interaction with ECM components such as hyaluronan, and a C-terminal globular domain containing two EGF-like repeats, a complement-regulatory protein-like repeat and a C-type lectin domain [13, 59]. The glycosaminoglycan binding domains contain between 5 and 23 chondroitin sulphate attachment regions depending on versican isoform and tissue type and location [59]. V0 contains both α-GAG and β-GAG binding domains, V1 contains α-GAG, V2 contains β-GAG and V3 has no glycosaminoglycan binding domains [58, 60].
Versican and atherosclerosis
The contribution of versican to atherosclerosis is not straightforward. In addition to being present within developing blood vessels and within all three layers of the arterial wall, its expression is upregulated in all forms of vascular disease and has also been found to accumulate in different arterial lesions including restenotic lesions and atherosclerotic plaques [47, 55]. Of the different isoforms, the V0, V1 and V3 forms are detectable in the human aorta and expressed by VSMCs [57]. Versican is synthesized by arterial smooth muscle cells and is able to influence the phenotypes of arterial smooth muscle cells and endothelial cells [55]. In addition, versican expression has been detected at the plaque thrombus interface where it is thought to promote platelet adhesion and aggregation [61].
In non-disease states, versican is likely to provide structure to the vasculature through its interaction with hyaluronan [13]. Formation of complexes between hyaluronan and versican are necessary for the migration and proliferation of VSMCs following wounding. This complex formation is rapid and occurs during the detachment stage of VSMC migration [62] and can be upregulated by platelet derived growth factor (PDGF) in arterial smooth muscle cells [63]. In addition to binding to versican, hyaluronan can serve as an attachment ligand for macrophages and lymphocytes suggesting that hyaluronan–versican complexes may also influence the retention and adhesion of inflammatory cells [13]. The chondroitin sulphate (CS) chains of versican can interact with a number of adhesion molecules and chemokines such as L-selectin and P-selectin that may influence the migration and recruitment of vascular cells including macrophages [64, 65].
In advanced atherosclerosis, the location of versican close to accumulated lipoproteins at the edge of lesions suggests that versican and versican complexes may have a role in the retention of lipoproteins in addition to inflammatory cells [13]. Multiple LDL particles are able to bind to the CS chains of versican and other proteoglycans. Elongation of CS chains is observed in vascular injury and promotes binding to LDL [13]. Versican–LDL complex formation can increase lipoprotein uptake in both VSMCs and macrophages [66, 67]. Versican–LDL complexes are rapidly taken up by macrophages and can enter through the LDL receptor pathway in smooth muscle cells [68, 69]. Formation of these complexes is enhanced by lipoprotein lipase [70].
Cleavage of versican by ADAMTS proteases–relevance to atherosclerosis
Sandy et al. [57] first showed that in the human aorta, versican fragments could be generated by ADAMTS-1 and ADAMTS-4 digestion of human intact versican. The production of a 70-kDa fragment demonstrated that consensus motifs (Glu–X bonds) for ADAMTS cleavage were present within versican and other aggregating proteoglycans. The inability of other MMP enzymes to cleave at this site (despite their ability to degrade the protein) further supported the cleavage of versican by ADAMTS proteases [57]. It is now known that the ADAMTS-1 and -4 proteases cleave versican (isoformsV0 and V1) at the Glu441–Ala442/Glu1428–Ala1429 bond [49, 57]. In the case of ADAMTS-1, the degradation of the primary proteoglycan component of the vascular ECM could facilitate the migration of VSMCs [49]. Interestingly, ADAMTS-7 has been demonstrated to facilitate the migration of VSMCs and intimal thickening in a rat vascular balloon-injury model, and this is thought to be mediated by the breakdown of cartilage oligomatrix protein by the protease [71].
In addition to increased proliferation and migration of VSMCs, versican fragments have also been linked with changes in ECM volume of vascular lesions and therefore with intimal growth and regression [56, 57]. The ECM plays a large role in neointimal thickening, contributing between 60% and 80% to the mass in vascular lesions. The high number of glycosaminoglycan chains present in versican contributes to the water content and volume of the intima making the removal/deposition of proteoglycans a highly effective way to control ECM volume [56]. Using a graft repair model, it is possible to show that generation of versican fragments can be increased by high blood flow, indicating that the activity of ADAMTS proteases in blood vessels may be mediated by shear stress [56]. Intimal tissue regression following balloon- or stent-mediated injury has been shown to be associated with increased blood flow and loss of versican. In a baboon vascular graft model, high blood flow causes cell death and loss of ECM. This is accompanied by an increase of a versican cleavage product that can be generated by the ADAMTS proteases-1, -4, -5 and -9. Kenagy and colleagues measured the mRNA levels of ADAMTS proteases in this graft model and found that levels of ADAMTS-4 were significantly increased following a switch to high blood flow and that this was linked with tissue atrophy [56, 72].
Cytokine regulation of versican expression and degradation
All stages of atherosclerosis are regulated by pro- and anti-inflammatory cytokines. It is therefore possible that the expression and activity of versican and of ADAMTS proteases in the vascular system will be regulated by cytokines. Little is currently known about cytokine regulation of ADAMTS proteases in relation to atherosclerosis although the expression of ADAMTS-1 and ADAMTS-4 is upregulated by pro-inflammatory cytokines [45]. In contrast, versican expression is known to be regulated by a number of cytokines including PDGF, TGF-β and IL-1β [55, 73]. Studies in monkey arterial smooth muscle cells showed that PDGF and TGF-β upregulate the expression of versican mRNA [73]. In addition, immunostaining of atherosclerotic lesions from hypercholesterolemic nonhuman primates has shown that versican–hyaluronan complexes colocalize in the ECM with TGF-β and PDGF positive cells [74]. Further studies in arterial smooth muscle cells have shown that IL-1β reduces the mRNA stability of versican leading to decreased levels of synthesis [55], and interestingly, the cytokine is also able to upregulate decorin expression and induce aggrecan degradation through the upregulation of ADAMTS aggrecanase activity [55]. This is an area for further study.
Concluding remarks
In summary, proteases play a critical role in atherosclerosis. Inflammation drives a shift in the balance between ECM synthesis and its degradation by proteases. Degradation of the ECM contributes to plaque instability and the subsequent rupture and thrombosis that are the culmination of atherosclerosis. A large number of studies have demonstrated a crucial role for macrophage-produced proteases, particularly MMP-2 and MMP-9 in the degradation of ECM in atherosclerotic plaques [30]. Recent studies have revealed potentially important roles for ADAMTS proteases in atherosclerosis and inflammation (summarized in Fig. 2). The expression of the ADAMTS proteases, particularly ADAMTS-1 and ADAMTS-4, in atherosclerotic lesions and their ability to breakdown versican, a key component of the vascular ECM, implicates them in the disease. ADAMTS-1 and -4 have been shown to colocalize with smooth muscle cells and macrophages, respectively, in lesions, and fragments of versican present in human aorta and atherosclerotic lesions can be generated by the proteases. In vivo, ADAMTS-1 knockout mice show accumulation of collagen and defective ECM degradation suggesting that the ADAMTS proteases may play a role similar to that of the MMPs [49]. Additionally, PPARγ agonists with anti-inflammatory properties such as the thiazolidinediones, used in the treatment of type II diabetes mellitus, have also been demonstrated to inhibit macrophage-produced metalloproteases including MMP-9 and ADAMTS-4 which may contribute to plaque stability [47].
Involvement of the ADAMTS proteases in inflammatory disease is suggested by the role of ADAMTS-4 and -5 in osteoarthritis and RA where they induce cartilage breakdown through the degradation of the key proteoglycan component of cartilage, aggrecan, a molecule that is highly structurally similar to versican [75]. RA has been linked with accelerated atherosclerosis, with cardiovascular disease as the highest cause of mortality in RA patients [76]. More research will be required to further the understanding of the role of ADAMTS proteases in both inflammation and atherosclerosis, and in vivo, functional data will be particularly important for this. Many questions remain to be answered regarding the molecular activity, regulation and function of ADAMTS proteases in the vasculature. Are the ADAMTS proteases pro- or anti-atherogenic in nature? How are they regulated by cytokines and other inflammatory mediators? Are versican fragments generated by ADAMTS cleavage biologically active? In particular, the role of ADAMTS proteases may be much more complex than cleavage of versican and ECM degradation.
References
Porter S, Clark I, Keveorkian L, Edwards D (2005) The ADAMTS metalloproteinases. Biochem J 386:15–27
Jones GC, Riley GP (2005) ADAMTS proteinases: a multi-domain, multi-functional family with roles in extracellular matrix turnover and arthritis. Arthritis Res Ther 7:160–169
Wang W, Lee S, Steiglitz B, Scott I, Lebares C, Allen M, Brenner M, Takahara K, Greenspan D (2003) Transforming growth factor-beta induces secretion of activated ADAMTS-2. A procollagen III N-proteinase. J Biol Chem 278:19549–19557
Apte S (2009) A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem 284:31493–31497
Shindo T, Kurihara H, Kuno K, Yokoyama H, Wada T, Kurihara Y, Imai T, Wang Y, Ogata M, Nishimatsu H, Moriyama N, Oh-hashi Y, Morita H, Ishikawa T, Nagai R, Yazaki Y, Matsushima K (2000) ADAMTS-1: a metalloproteinase-disintegrin essential for normal growth, fertility, and organ morphology and function. J Clin Invest 105:1345–1352
Kuno K, Kanada N, Nakashima E, Fujiki F, Ichimura F, Matsushima K (1997) Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J Biol Chem 272:556–562
Vázquez F, Hastings G, Ortega M, Lane T, Oikemus S, Lombardo M, Iruela-Arispe M (1999) METH-1, a human ortholog of ADAMTS-1, and METH-2 are members of a new family of proteins with angio-inhibitory activity. J Biol Chem 274:23349–23357
Kuno K, Okada Y, Kawashima H, Nakamura H, Miyasaka M, Ohno H, Matsushima K (2000) ADAMTS-1 cleaves a cartilage proteoglycan, aggrecan. FEBS Lett 478:241–245
Naito S, Shiomi T, Okada A, Kimura T, Chijiiwa M, Fujita Y, Yatabe T, Komiya K, Enomoto H, Fujikawa K, Okada Y (2007) Expression of ADAMTS-4 (aggrecanase-1) in human osteoarthritic cartilage. Pathol Int 57:703–711
Miwa H, Gerken T, Huynh T, Duesler L, Cotter M, Hering T (2009) Conserved sequence in the aggrecan interglobular domain modulates cleavage by ADAMTS-4 and ADAMTS-5. Biochim Biophys Acta 1790:161–172
Huang K, Wu L (2008) Aggrecanase and aggrecan degradation in osteoarthritis: a review. J Int Med Res 36:1149–1160
Bondeson J, Wainwright S, Hughes C, Caterson B (2008) The regulation of the ADAMTS4 and ADAMTS5 aggrecanases in osteoarthritis: a review. Clin Exp Rheumatol 26:139–145
Wight T, Merrilees M (2004) Proteoglycans in atherosclerosis and restenosis: key roles for versican. Circ Res 94:1158–1167
Haddock G, Cross A, Allan S, Sharrack B, Callaghan J, Bunning R, Buttle D, Woodroofe M (2007) Brevican and phosphacan expression and localization following transient middle cerebral artery occlusion in the rat. Biochem Soc Trans 35:692–694
Nakamura H, Fujii Y, Inoki I, Sukimoto K, Tanzawa K, Matsuki H, Miura R, Yamaguchi Y, Okada Y (2000) Brevican is degraded by matrix metalloproteinases and aggrecanase-1 (ADAMTS4) at different sites. J Biol Chem 275:38885–38890
Viapiano M, Hockfield S, Matthews R (2008) BEHAB/brevican requires ADAMTS-mediated proteolytic cleavage to promote glioma invasion. J Neurooncol 88:261–272
Hofer T, Frankenburger M, Mages J, Lang R, Hoffmann R, Colige A, Ziegler-Heitbrock L (2008) Tissue-specific induction of ADAMTS2 in monocytes and macrophages by glucocorticoids. J Mol Med 86:323–332
Fujikawa K, Suzuki H, McMullen B, Chung D (2001) Purification of human von Willebrand factor-cleaving protease and its identification as a new member of the metalloproteinase family. Blood 98:1662–1666
Moriguchi-Goto S, Yamashita A, Tamura N, Soejima K, Takahashi M, Nakagaki T, Goto S, Asada Y (2009) ADAMTS-13 attenuates thrombus formation on type I collagen surface and disrupted plaques under flow conditions. Atherosclerosis 203:409–416
Long Zheng X (2010) ADAMTS13 testing: why bother? Blood 115:1475–1476
Kuno K, Terashima Y, Matsushima K (1999) ADAMTS-1 is an active metalloproteinase associated with the extracellular matrix. J Biol Chem 274:18821–18826
Wang P, Tortorella M, England K, Malfait A, Thomas G, Arner E, Pei D (2004) Proprotein convertase furin interacts with and cleaves pro-ADAMTS4 (Aggrecanase-1) in the trans-Golgi network. J Biol Chem 279:15434–15440
Gerhardt S, Hassall G, Hawtin P, McCall E, Flavell L, Minshull C, Hargreaves D, Ting A, Pauptit R, Parker A, Abbott W (2007) Crystal structures of human ADAMTS-1 reveal a conserved catalytic domain and a disintegrin-like domain with a fold homologous to cysteine-rich domains. J Mol Biol 373:891–902
Gao G, Westling J, Thompson V, Howell T, Gottschall P, Sandy J (2002) Activation of the proteolytic activity of ADAMTS4 (aggrecanase-1) by C-terminal truncation. J Biol Chem 277:11034–11041
Kuno K, Matsushima K (1998) ADAMTS-1 protein anchors at the extracellular matrix through the thrombospondin type 1 motifs and its spacing region. J Biol Chem 273:13912–13917
Flannery C, Zeng W, Corcoran C, Collins-Racie L, Chockalingam P, Hebert T, Mackie S, McDonagh T, Crawford T, Tomkinson K, LaVallie E, Morris E (2002) Autocatalytic cleavage of ADAMTS-4 (aggrecanase-1) reveals multiple glycosaminoglycan-binding sites. J Biol Chem 277:42775–42780
Tortorella M, Pratta M, Liu R, Abbaszade I, Ross H, Burn T, Arner E (2000) The thrombospondin motif of aggrecanase-1 (ADAMTS-4) is critical for aggrecan substrate recognition and cleavage. J Biol Chem 275:25791–25797
Hashimoto G, Shimoda M, Okada Y (2004) ADAMTS-4 (aggrecanase-1) interaction with the C-terminal domain of fibronectin inhibits proteolysis of aggrecan. J Biol Chem 279:32483–32491
Wight TN (2005) The ADAMTS proteases, extracellular matrix, and vascular disease: waking the sleeping giant(s)! Arterioscler Thromb Vasc Biol 25:12–14
Galis Z, Khatri J (2002) Matrix metalloproteinases in vascular remodelling and atherogenesis: the good, the bad and the ugly. Circ Res 90:251–262
Rodríguez-Manzaneque J, Westling J, Thai S, Luque A, Knauper V, Murphy G, Sandy J, Iruela-Arispe M (2002) ADAMTS1 cleaves aggrecan at multiple sites and is differentially inhibited by metalloproteinase inhibitors. Biochem Biophys Res Commun 293:501–508
Hashimoto G, Aoki H, Nakamura K, Tanzawa Y, Okada Y (2001) Inhibition of ADAMTS4 (aggrecanase-1) by tissue inhibitors of metalloproteinases (TIMP-1, 2, 3 and 4). FEBS Lett 494:192–195
Kashiwagi M, Tortorella M, Nagase H, Brew K (2001) TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J Biol Chem 276:12501–12504
Troeberg L, Fushimi K, Scilabra S, Nakamura H, Dive V, Thøgersen I, Enghild J, Nagase H (2009) The C-terminal domains of ADAMTS-4 and ADAMTS-5 promote association with N-TIMP-3. Matrix Biol 28:463–469
Bui Q, Prempeh M, Wilensky R (2009) Atherosclerotic plaque development. Int J Biochem Cell Biol 41:2109–2113
Lusis A, Mar R, Pajukanta P (2004) Genetics of atherosclerosis. Annu Rev Genomics Hum Genet 5:189–218
Glass C, Witztum J (2001) Atherosclerosis: the road ahead. Cell 104:503–516
Weber C, Zernecke A, Libby P (2008) The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol 8:802–815
Li A, Glass C (2002) The macrophage foam cell as a target for therapeutic intervention. Nat Med 8:1235–1242
Shashkin P, Dragulev B, Ley K (2005) Macrophage differentiation to foam cells. Curr Pharm Des 11:3061–3072
Halvorsen B, Otterdal K, Dahl T, Skjelland M, Gullestad L, Øie E, Aukrust P (2008) Atherosclerotic plaque stability—what determines the fate of a plaque? Prog Cardiovasc Dis 51:183–194
Newby A (2007) Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc Med 17:253–258
Raffetto J, Khalil R (2008) Matrix metalloproteases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol 75:346–359
Galis Z, Sukhova G, Lark M, Libby P (1994) Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest 94:2493–2503
Wågsäter D, Björk H, Zhu C, Björkegren J, Valen G, Hamsten A, Eriksson P (2008) ADAMTS-4 and -8 are inflammatory regulated enzymes expressed in macrophage-rich areas of human atherosclerotic plaques. Atherosclerosis 196:514–522
Malemud C (2006) Matrix metalloproteinases (MMPs) in health and disease: an overview. Front Biosci 11:1696–1701
Worley J, Baugh M, Hughes D, Edwards D, Hogan A, Sampson M, Gavrilovic J (2003) Metalloproteinase expression in PMA-stimulated THP-1 cells. Effects of peroxisome proliferator-activated receptor-gamma (PPAR gamma) agonists and 9-cis-retinoic acid. J Biol Chem 278:51340–51346
Whatling C, Björk H, Gredmark S, Hamsten A, Eriksson P (2004) Effect of macrophage differentiation and exposure to mildly oxidised LDL on the proteolytic repertoire of THP-1 monocytes. J Lipid Res 45:1768–1776
Jönsson-Rylander A, Nilsson T, Fritsche-Danielson R, Hammarström A, Behrendt M, Andersson J, Lindgren K, Andersson A, Wallbrandt P, Rosengren B, Brodin P, Thelin A, Westin A, Hurt-Camejo E, Lee-Søgaard C (2005) Role of ADAMTS-1 in atherosclerosis: Remodeling of carotid artery, immunohistochemistry, and proteolysis of Versican. Arterioscler Thromb Vasc Biol 25:180–185
Norata G, Björk H, Hamsten A, Catapano A, Eriksson P (2004) High-density lipoprotein subfraction 3 decreases ADAMTS-1 expression induced by lipopolysaccharide and tumor necrosis factor-alpha in human endothelial cells. Matrix Biol 22:557–560
Luque A, Carpizo D, Iruela-Arispe M (2003) ADAMTS-1/METH-1 inhibits endothelial cell proliferation by direct binding and sequestration of VEGF 165. J Biol Chem 278:23656–23665
Xu Z, Yu Y, Duh E (2006) Vascular endothelial growth factor upregulates expression of ADAMTS1 in endothelial cells through protein kinase C signaling. Invest Opthalmol Vis Sci 47:4059–4066
Hatipoglu O, Hirohata S, Cilek M, Ogawa H, Miyoshi T, Obika M, Demircan K, Shinohata R, Kusachi S, Ninomiya Y (2009) ADAMTS1 is a unique hypoxic early response gene expressed by endothelial cells. J Biol Chem 284:16325–16333
Bongrazio M, Baumann C, Zakrzewicz A, Pries A, Gaehtgens P (2000) Evidence for modulation of genes involved in vascular adaptation by prolonged exposure of endothelial cells to shear stress. Cardiovasc Res 47:384–393
Lemire J, Chan C, Bressler S, Miller J, LeBaron R, Wight T (2007) Interleukin-1beta selectively decreases the synthesis of versican by arterial smooth muscle cells. J Cell Biochem 101:753–766
Kenagy R, Fischer J, Lara S, Sandy J, Clowes A, Wight T (2005) Accumulation and loss of extracellular matrix during shear stress-mediated intimal growth and regression in baboon vascular grafts. J Histochem Cytochem 53:131–140
Sandy J, Westling J, Kenagy R, Iruela-Arispe M, Verscharen C, Rodriguez-Mazaneque J, Zimmermann D, Lemire J, Fischer J, Wight T, Clowes A (2001) Versican V1 proteolysis in human aorta in vivo occurs at the Glu441–Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J Biol Chem 276:13372–13378
Wight T (2002) Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol 14:617–623
Rahmani M, Wong B, Ang L, Cheung C, Carthy J, Walinski H, McManus B (2006) Versican: signaling to transcriptional control pathways. Can J Physiol Pharmacol 84:77–92
Kenagy R, Plaas A, Wight T (2006) Versican degradation and vascular disease. Trends Cardiovasc Med 16:209–215
Mazzucato M, Cozzi M, Pradella P, Perissinotto D, Malmstrom A, Morgelin M, Spessotto P, Colombatti A, De Marco L, Perris R (2002) Vascular PG-M/versican variants promote platelet adhesion at low shear rates and cooperate with collagens to induce aggregation. FASEB J 16:1903–1916
Evanko S, Angello J, Wight T (1999) Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 19:1004–1013
Evanko S, Johnson P, Braun K, Underhill C, Dudhia J, Wight T (2001) Platelet-derived growth factor stimulates the formation of versican–hyaluronan aggregates and pericellular matrix expansion in arterial smooth muscle cells. Arch Biochem Biophys 394:29–38
Hirose J, Kawashima H, Yoshie O, Tashiro K, Miyasaka M (2001) Versican interacts with chemokines and modulates cellular responses. J Biol Chem 276:5228–5234
Kawashima H, Hirose M, Hirose J, Nagakubo D, Plaas A, Miyasaka M (2000) Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan, v, to L-selectin, P-selectin, and CD44. J Biol Chem 275:35448–35456
Ismail N, Alavi M, Moore S (1994) Lipoprotein–proteoglycan complexes from injured rabbit aortas accelerate lipoprotein uptake by arterial smooth muscle cells. Atherosclerosis 105:79–87
Srinivasan S, Xu J, Vijayagopal P, Radhakrishnamurthy B, Berenson G (1995) Low-density lipoprotein binding affinity of arterial chondroitin sulfate proteoglycan variants modulates cholesteryl ester accumulation in macrophages. Biochim Biophys Acta 1272:61–67
Hurt-Camejo E, Camejo G, Rosengren B, López F, Ahlström C, Fager G, Bondjers G (1992) Effect of arterial proteoglycans and glycosaminoglycans on low density lipoprotein oxidation and its uptake by human macrophages and arterial smooth muscle cells. Arterioscler Thromb Vasc Biol 12:569–583
Llorente-Cortés V, Otero-Viñas M, Hurt-Camejo E, Martínez-González J, Badimon L (2002) Human coronary smooth muscle cells internalize versican-modified LDL through LDL receptor-related protein and LDL receptors. Arterioscler Thromb Vasc Biol 22:387–393
Olin K, Potter-Perigo S, Barrett P, Wight T, Chait A (1999) Lipoprotein lipase enhances the binding of native and oxidized low density lipoproteins to versican and biglycan synthesized by cultured arterial smooth muscle cells. J Biol Chem 274:34629–34636
Wang L, Zheng J, Bai X, Liu B, Liu C, Xu Q, Zhu Y (2009) ADAMTS-7 mediates vascular smooth muscle cell migration and neointima formation in balloon-injured rat arteries. Circ Res 104:688–698
Kenagy R, Min S, Clowes A, Sandy J (2009) Cell death-associated ADAMTS4 and versican degradation in vascular tissue. J Histochem Cytochem 57:889–897
Schönherr E, Järveläinen H, Sandell L, Wight T (1991) Effects of platelet-derived growth factor and transforming growth factor-beta 1 on the synthesis of a large versican-like chondroitin sulfate proteoglycan by arterial smooth muscle cells. J Biol Chem 266:17640–17647
Evanko S, Raines E, Ross R, Gold L, Wight T (1998) Proteoglycan distribution in lesions of atherosclerosis depends on lesion severity, structural characteristics, and the proximity of platelet-derived growth factor and transforming growth factor-beta. Am J Pathol 152:533–546
Malfait A, Liu R, Ijiri K, Komiya S, Tortorella M (2002) Inhibition of ADAMTS-4 and ADAMTS-5 prevents aggrecan degradation in osteoarthritic cartilage. J Biol Chem 277:22201–22208
Ozbalkan Z, Efe C, Cesur M, Ertek S, Nairoglu N, Berneis K, Rizzo M (2010) An update on the relationships between rheumatoid arthritis and atherosclerosis. Atherosclerosis. doi:10.1016/j.atherosclerosis.2010.03.035
Acknowledgements
Rebecca C. Salter and Tim G. Ashlin were recipients of BBSRC studentships.
Disclosure of potential conflict of interests
The authors declare no conflict of interests related to this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Salter, R.C., Ashlin, T.G., Kwan, A.P.L. et al. ADAMTS proteases: key roles in atherosclerosis?. J Mol Med 88, 1203–1211 (2010). https://doi.org/10.1007/s00109-010-0654-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-010-0654-x