
Abstract
ADAMTS13 is an idiosyncratic member of a distinct family of zinc metalloproteases, the ADAMTS family. This 19-member family is related to ADAM proteases and matrix metalloproteinases (MMPs) through sequence homology of their active sites and similar catalytic domain structures. These similarities contrast with the presence of distinct ancillary domains in each family, with those of ADAMTS proteases comprising thrombospondin type 1 repeats and other modules in a characteristic arrangement. Human and animal mutations affecting ADAMTS proteases lead to a variety of inherited human disorders and animal phenotypes, which demonstrate their requirement in crucial biological and disease pathways. ADAMTS proteases are implicated in procollagen maturation (ADAMTS2, ADAMTS3), versican turnover during embryogenesis (ADAMTS1, ADAMTS5, ADAMTS9, ADAMTS15, ADAMTS20) and ovulation (ADAMTS1), cartilage aggrecan destruction in arthritis (ADAMTS4, ADAMTS5), genetic disorders affecting fibrillin microfibrils (ADAMTS10, ADAMTS17), and, recently, VEGF-C processing during lymphangiogenesis (ADAMTS3). Although ADAMTS13 shares many characteristics with the other family members, it has some special attributes and is the only ADAMTS protease presently implicated in hemostasis. Its unique specificity for von Willebrand factor contrasts with that of other family members, which can have shared or multiple substrates.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Thrombotic Thrombocytopenic Purpura
- Chondroitin Sulfate Proteoglycan
- ADAMTS Activity
- Catalytic Module
- ADAMTS Gene
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 What Are ADAMTS Proteases, and How Are They Related to Other Metalloproteases?
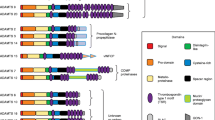
ADAMTS proteases were unknown until 1997, when Kuno et al. [1] identified a novel metalloprotease with a catalytic domain containing a reprolysin (or snake venom-like) active-site sequence motif found in ADAM (a disintegrin and metalloprotease) proteases [2]. Among the predicted features of this new protease that set it apart from the ADAMs were the presence of thrombospondin type 1 repeats (TSRs) and the absence of a membrane-spanning domain, which is present in all ADAMs. Subsequently, sequencing of the human and mouse genomes enabled discovery of additional mammalian gene products that shared the characteristics of ADAMTS1. These 19 gene products resembled each other more closely in domain composition and primary structure than they did other metalloproteases, leading to designation of a new protease family [3, 4]. The protease named as ADAMTS5 [4] was also subsequently named ADAMTS11 [5]; therefore, the designation ADAMTS11 is no longer used. ADAMTS proteases have two functional domains, namely, a protease domain and an adjoining domain comprised of multiple modules, including TSRs, which is termed the ancillary domain (Fig. 2.1) [6]. ADAMTS proteases belong to a superfamily of ADAMTS proteins, which also includes 7 ADAMTS-like (ADAMTSL) proteins in mammals [6]. ADAMTSLs lack a propeptide, catalytic module, and disintegrin-like module, i.e., they lack the regions comprising the protease domain of ADAMTS proteases (Fig. 2.1), and, therefore, are not proteases [7]. ADAMTSLs are encoded by a distinct set of genes and do not result from alternative splicing of, or the use of, alternative promoters within ADAMTS genes. Notably, MMPs and ADAMs do not have such non-protease relatives.

Structure of ADAMTS proteases. The domain backbone shared by each ADAMTS protease is shown at the top, and modules present in every ADAMTS are shown in the box on the left. The modular organization of specialized ADAMTS clades is indicated on the right, and the key to these modules is located at the bottom of the figure. The clades are named according to structural or functional characteristics that best define them. Domain structures are based on reference sequences obtained from GenBank. CUB, complement C1r/C1s, Uegf, Bmp1 domain; PLAC, protease and lacunin domain. Reproduced from Apte, S.S., 2009. A disintegrin-like and metalloprotease (reprolysin type) with thrombospondin type 1 motif (ADAMTS) superfamily: Functions and mechanisms. Journal of Biological Chemistry 284, 31493–31497
Identification of ADAMTS orthologs in invertebrate genomes, such as those of the roundworm C. elegans, fruit fly D. melanogaster, and sea squirt Ciona intestinalis, allowed determination of the evolutionary relationships within the family [8]. This phylogenetic analysis suggested that mammalian ADAMTS proteases arose from duplication and divergence of a small number of related proteases encoded by ancestral genomes [8]. As a consequence of gene duplication, most ADAMTS proteases (ADAMTS13 as exception) have one or more homologous proteases (Fig. 2.1), and thus the mammalian ADAMTS family is divided into several subfamilies [8, 9]. ADAMTS13 is likely to be a relatively recent, chordate innovation, possibly related to evolution of a closed circulation and the need for hemostasis, since none of the invertebrate ADAMTS proteins resemble its primary structure or contain CUB domains. Instead, the most ancient ADAMTS genealogical relationship appears to be between a nematode protein GON-1, Drosophila ADAMTS-A, and two mammalian orthologs, ADAMTS9 and ADAMTS20 [10].
Although ADAMTS genes are dispersed throughout the human and mouse genomes, three pairs of probable tandem duplications are known [11]. Two of the tightly linked pairs involve functionally related ADAMTS proteases, i.e., ADAMTS1/ADAMTS5 and ADAMTS8/ADAMTS15 on human chromosomes 21 and 11, respectively; the corresponding mouse loci are linked on mouse chromosomes 16 and 9, respectively. The third genetic linkage involves ADAMTS13, whose locus is linked to that of ADAMTSL2, on human chromosome 9q34 and mouse chromosome 2. These two genes are 120 and 100 kb apart in the human and mouse genomes, respectively, with CACFD1 intervening in the human genome, along with two additional intervening genes, Slc2a6 and Tmem8c in mouse. There is no evidence for functional interaction between ADAMTSL2 and ADAMTS13, and on the basis of the degenerate synteny in the region, ADAMTS13 and ADAMTSL2 genes probably do not share regulatory regions. ADAMTSL2 is implicated in an inherited connective tissue disorder named geleophysic dysplasia [12, 13], which has no clinical overlap with thrombocytopenic purpura.
Like ADAMs and MMPs, ADAMTS proteases are synthesized as zymogens, which undergo proteolytic excision of the N-terminal propeptide by proprotein convertases such as furin. The similarity of the ADAMTS catalytic domain structure and mechanism to ADAMs and MMPs renders them accessible to the same endogenous inhibitors, namely, tissue inhibitor of metalloproteinases-3 (TIMP3) and α2-macroglobulin [14–16]. These similarities contrast with a number of marked distinctions between the structures and biological roles of ADAMTS proteases and ADAM. In contrast to ADAMs, which are all cell membrane-anchored, as are several membrane-bound MMPs, all ADAMTS proteases are secreted. Several have been shown to bind close to the cell surface through interactions with pericellular matrix components such as proteoglycans [10, 17–19]. Thus, the majority of ADAMTS proteases may be operational cell surface proteases that could have a role in modifying pericellular matrix, modulating signaling molecules, or influencing cell adhesion and migration. ADAMTS1 was shown to shed syndecan-4 [20] and the epidermal growth factor (EGF) receptor ligands heparin-binding EGF and amphiregulin [21]. However, there is little evidence that ADAMTS proteases have a major role in ectodomain shedding, which remains the principal function of ADAMs, a contention supported by strong genetic and biochemical evidence [2]. Paradoxically, while the overall structure and active-site sequences of ADAMTS proteases are closer to ADAMs than MMPs, ADAMTS proteases share with MMPs, but not ADAMs, a propensity for cleavage of extracellular matrix/secreted molecules. This property is attributable to the affinity of the ancillary domain for extracellular matrix.
2.2 Biosynthesis, Posttranslational Modification, and Regulation of ADAMTS Activity
As secreted proteins, all ADAMTS proteases have a signal peptide, which directs them to the endoplasmic reticulum (ER), where their folding and posttranslational modification are initiated. There, disulfide bond formation occurs between pairs of cysteine residues brought together by energetically stable states of the newly folded protein, a process that is assisted by chaperones [22]. The ADAMTS9 propeptide provides an intramolecular chaperone required for its secretion, whereas the ADAMTS13 propeptide is not similarly required [23, 24]. Most ADAMTS proteases (ADAMTS4 being an exception) have consensus motifs for N-glycosylation, which occurs co-translationally and is likely to assist folding, thus constituting a potential quality control mechanism. For instance, ADAMTS9 and a nematode ADAMTS named mig-17 are not secreted if they lack N-glycosylation [23, 25, 26].
TSRs undergo two uncommon posttranslational modifications in the ER, namely, protein O-fucosylation [27] and C-mannosylation [28]. O-fucosylation leads to addition of either a mono- or disaccharide to TSRs. The initiating modification is the addition of fucose to a TSR containing the consensus sequence C1XX(S/T)C2XXG by protein O-fucosyltransferase 2 (POFUT2) [27]. Such motifs are present in only 49 proteins encoded by the human genome, of which half belong to the ADAMTS superfamily. This O-linked fucose then acts as the recipient for a glucose residue, a step mediated by β3-glucosyltransferase (B3GLCT), leading to the formation of a glucoseβ1–3fucose disaccharide [29]. O-fucosylation appears to be a quality control step for secretion, since POFUT2 does not modify unfolded peptides containing the consensus sequence, and neither ADAMTS13 nor ADAMTSL2 are secreted in the absence of O-fucosylation [30, 31]. POFUT2, acting via modification of one or more crucial substrates, possibly ADAMTS proteins, is essential for survival, since Pofut2-deficient embryos do not survive past early development [32]. A probable candidate for this severe phenotype is ADAMTS9, since Adamts9-deficient embryos do not survive past gastrulation [33]. Human POFUT2 mutations have not been identified, presumably because they would be lethal. However, B3GLCT mutations lead to a specific disorder named Peters plus syndrome, comprising several ocular and non-ocular manifestations, but not a hemostatic abnormality [34]. From this, it can be surmised that B3GLCT is required for secretion and/or function of some, but not all, ADAMTS proteins and, specifically, not for ADAMTS13 activity.
Protein C-mannosylation occurs on Trp (W0) residues within W0XXW+3 or W0XXC+3 motifs, which lie just upstream of the O-fucosylation consensus sequence in TSRs; this modification has been shown to occur on ADAMTSL1 and is predicted in other superfamily members [35, 36]. Since C-mannosylation occurs on unfolded peptides, it is unlikely to be a quality control mechanism or a prerequisite for folding, and its precise function in ADAMTS proteins is not known. In addition to these three forms of glycosylation, ADAMTS7 and ADAMTS12, which are orthologous proteins, undergo extensive O-glycosylation in regions having the sequence attributes of mucin domains, namely, an abundance of Pro, Ser, and Thr residues [19]. Furthermore, their mucin domains contain sequons for attachment of glycosaminoglycan (GAG) chains. Indeed, ADAMTS7 is modified by attachment of the GAG chondroitin sulfate [19], which makes ADAMTS7 the only protease that is also a proteoglycan, and supports the likelihood of similar modification occurring at sequons present in ADAMTS12 [19].
Most ADAMTS propeptides are over 200 amino acids (aa) in length and contain three Cys residues, whereas the ADAMTS13 propeptide is only 40-residue long and contains two Cys residues [24]. ADAMTS propeptides are excised by proprotein convertases, e.g., furin, in the trans-Golgi or extracellularly, i.e., at the cell surface or in extracellular matrix [10, 14, 18, 19, 37–41]. A typical furin processing site (Arg-Xaa-Arg/Lys-Arg) is present in all ADAMTS proteases but ADAMTS10 at the junction of the propeptide and catalytic module. ADAMTS10 has a suboptimal (Gly-Leu-Lys-Arg) site at the propeptide–catalytic module junction, but contains additional consensus sites within the propeptide [42]. Whereas propeptide excision was not required for activity of ADAMTS9 and ADAMTS13 [23, 24], it is essential for activity of ADAMTS1, ADAMTS4, ADAMTS5, and ADAMTS15 [38–40, 43].
A structure of the ADAMTS13 catalytic domain is presently unavailable. However, three-dimensional structures of the catalytic and disintegrin-like modules of ADAMTS1, ADAMTS4, and ADAMTS5 were obtained by X-ray crystallography and showed a very similar fold [44–46]. These structures demonstrated that the ADAMTS disintegrin-like module did not resemble snake venom disintegrins, but formed a unique conserved fold whose N-terminal half resembled ADAM cysteine-rich domains. These structures also showed that the disintegrin-like module was closely juxtaposed to the catalytic module and formed part of the interacting surface with inhibitors, i.e., it provided a functional extension of the catalytic domain. Indeed, exclusion of the disintegrin-like module from ADAMTS-like proteins [6] further suggests that the ADAMTS protease domain comprises both the catalytic domain and disintegrin-like modules.
ADAMTS protease domains without attached ancillary domains generally lack activity toward native substrates, since the ancillary domains constitute a major substrate-binding region, as shown by several studies employing recombinant ADAMTS proteases, including ADAMTS13 [17, 47–49]. A crystal structure obtained for the ADAMTS13 ancillary domain (excluding the C-terminal TSRs and CUB modules) [50] has been invaluable in understanding the three-dimensional topography and mechanisms of the ADAMTS family and is the only available ancillary domain structure to date. Together with site-directed mutagenesis of the ADAMTS13 ancillary domain and localization of epitopes for ADAMTS13 autoantibodies, the structure demonstrated the importance of exosites that mediate its activity against vWF [51, 52].
There are several examples of posttranslational modification of ADAMTS substrates being crucial determinants of their proteolytic activity. For example, proteolysis of the proteoglycans aggrecan and versican by ADAMTS1, ADAMTS4, and ADAMTS5 requires the chondroitin sulfate side chains on these substrates, since the deglycosylated substrates are poorly cleaved [53, 54]. Indeed, recent work identified two chondroitin sulfate attachment sites on versican V1 that lie in greatest proximity to the cleaved Glu441–Ala442 bond as specific and essential determinants of ADAMTS1 and ADAMTS5 activity [53]. O-linked glycans of vWF have been shown to influence its proteolysis by ADAMTS13 [55]. ADAMTS2 activity against procollagen I requires that this substrate have a triple helical conformation [56]. In the case of ADAMTS13 processing of vWF, stretching of the A1–A3 domains flanking the scissile bond is required, constituting an unusual, shear force-mediated “posttranslational” substrate modification [57, 58]. This requirement may explain why partial vWF denaturation using urea is required for efficient processing by ADAMTS13 in vitro. ADAMTS activity can also be regulated by cofactors or inhibited by interactions with other molecules. For example, ADAMTS13 processing of vWF can be accelerated by factor VIII [52]. Fibulin-1 is a cofactor for ADAMTS1 and ADAMTS5 [59–61], whereas fibronectin inhibits ADAMTS4 [62]. The ADAMTS-like protein papilin was shown to be a noncompetitive inhibitor of ADAMTS2 [63].
2.3 Biological and Disease Pathways Involving ADAMTS Proteases
This chapter highlights ADAMTS proteases that are unequivocally implicated in biological pathways through identification of mutations in Mendelian disorders or via engineered animal mutations.
ADAMTS1 deficiency in mice leads to considerable lethality at birth, together with a high frequency of genitourinary anomalies such as hydronephrosis [64, 65]. Surviving female Adamts1 null mice are infertile because ADAMTS1 is required for versican proteolysis during maturation and rupture of the ovarian follicle [66–68]. ADAMTS1 is also required for versican proteolysis during myocardial compaction, a morphogenetic process in which cardiac myocytes are brought together during the embryonic period to form a functional myocardium [69]. The implication of ADAMTS2 in collagen maturation via a bovine genetic disorder named dermatosparaxis predated the association of ADAMTS13 with von Willebrand factor. Dermatosparaxis results from accumulation of unprocessed cutaneous procollagen having a “hieroglyphic” ultrastructural appearance, leading to extreme skin fragility that is the hallmark of this disorder [70]. The underlying mechanism was identified as the lack of an enzymatic activity essential for the removal of the N-propeptide of procollagen I, the major collagen type in the dermis of the skin [71]. Later, a human connective tissue disorder, Ehlers–Danlos syndrome type VIIc (or dermatosparactic type), having similar skin fragility and collagen fibril anomalies was identified [72] (Table 2.1), and both the bovine and human conditions were attributed to ADAMTS2 mutations in the respective species [73]. ADAMTS3, an ADAMTS2 ortholog, processes procollagen II and procollagen III and is highly expressed in cartilage, where collagen II is a major component [74, 75]. In addition, this enzyme was recently shown to proteolytically activate the pro-angiogenic and pro-lymphangiogenic factor VEGF-C. Proteolysis of VEGF-C is enhanced by the binding of ADAMTS3 to a cofactor, collagen- and calcium-binding epidermal growth factor domain 1 (CCBE1) [76]. The cleavage site in VEGF-C is similar to that in procollagens, and CCBE1 has a C-terminal domain with collagenous repeats, which may provide the basis for its interaction with ADAMTS3.
ADAMTS4 and ADAMTS5 (termed aggrecanase-1 and aggrecanase-2, respectively) are implicated in proteolytic destruction and loss of aggrecan from joint cartilage in osteoarthritis [77]. Aggrecan is a heavily modified chondroitin sulfate proteoglycan that interacts with hyaluronan in cartilage extracellular matrix. The large aggregates thus formed are highly hydrated and endow cartilage with its shock-absorbing properties. Proteolysis of aggrecan is thought to be a major initiating mechanism of arthritis, since it exposes other cartilage components such as collagen II to subsequent destruction by MMPs and other proteases. Aggrecanases are considered to be a major drug target in arthritis, and many small-molecule active-site inhibitors and function-blocking antibodies have been generated and investigated preclinically [77, 78]. ADAMTS5 is strongly expressed in cardiac outflow tract endocardial cushions, where it is required for versican proteolysis during the sculpting of cushions to form thin valve leaflets, and is implicated in TGFβ signaling [79, 80]. Adamts5-deficient mice had reduced sculpting of pulmonic valves during embryogenesis and myxomatous mitral valves in adult hearts [79].
ADAMTS9 is crucial for early mouse development, since embryos lacking this protease do not survive past gastrulation [81]. This highly conserved protease has significant roles in mammalian development gleaned from analysis of single and combinatorial mouse mutants. For example, ADAMTS9 haploinsufficiency leads to cardiac and aortic defects and to a highly penetrant ocular anterior segment dysgenesis [82, 83]. In combination with the Adamts20bt homozygous mutant, Adamts9 haploinsufficiency leads to death at birth from cleft palate [33]. ADAMTS10 mutations lead to Weill–Marchesani syndrome, with short stature, brachydactyly and ectopia lentis (dislocation of the lens) being the major clinical features (Table 2.1) [84]. Since WMS is also caused by fibrillin-1 mutations [85], a functional relationship between ADAMTS10 and fibrillin-1 has emerged and is validated by studies showing that ADAMTS10 binds fibrillin-1 and enhances microfibril biogenesis [42]. ADAMTS10 cleaves fibrillin-1 poorly [42], and ectopia lentis in WMS suggests that ADAMTS10 is primarily required for the formation of the zonule, a microfibril-comprised structure that suspends the lens in the optic path. A WMS-like phenotype in humans, and ectopia lentis in dogs, results from ADAMTS17 mutations [86, 87], suggesting it may function similarly to ADAMTS10 (Table 2.1).
Recently, rats with a targeted mutation of Adamts16 identified its potential role in regulation of blood pressure and male fertility [88, 89], and other works have suggested a connection between Adamts16 and renal development [90]. The related protease ADAMTS18 is implicated in a syndrome comprising microcornea, myopic chorioretinal atrophy, and telecanthus (Table 2.1) [91, 92]. The Adamts20bt mutant has a white spotting phenotype, with the spotting confined to the mid-torso, and results from failure of neural crest-derived melanoblasts to properly colonize hair follicles in that region (hair follicles, but not the intervening skin, are the exclusive domain of melanoblasts in mice) [93, 94]. ADAMTS20 is not required for neural crest cell migration, but for the proliferation and survival of neural crest cells once they reach the hair follicles [94].
As further evidence of cooperativity of ADAMTS proteases, mice with combined Adamts5 and Adamts20 deficiency have soft tissue syndactyly, which results from failure of interdigital web regression in the embryo [59]. Interdigital webs are present not only in aquatic birds and bats but also during embryogenesis in humans, mice, and other mammals, where they participate in the development of digits. They regress by rapid sculpting after digit formation is complete, i.e., by massive apoptosis coupled with matrix proteolysis. ADAMTS proteolysis of versican in the interdigit matrix is required for apoptosis of interdigit mesenchyme, suggesting that these ADAMTS proteases couple matrix proteolysis to cell death during web regression [59]. A similar role for Adamts9 in web regression was elucidated first in combination with Adamts5 or Adamts20 and, more recently, by its limb-specific conditional inactivation [59, 81].
An interesting contrast between ADAMTS13 and other family members relates to substrate specificity. The biology of thrombotic thrombocytopenic purpura suggests an exclusive protease–substrate relationship between ADAMTS13 and von Willebrand factor, whereas other ADAMTS proteases appear not to be as exquisitely specific. For example, numerous substrates have been identified for the prototypic ADAMTS protease, ADAMTS1, including chondroitin sulfate proteoglycans such as aggrecan and versican, collagen I, nidogen-1 and nidogen-2, the matricellular proteins thrombospondin-1 and thrombospondin-2, and the cell-anchored EGFR ligands HB-EGF and amphiregulin [20, 21, 95–99]. Of these, however, a significant biological impact has hitherto been established mostly for proteolysis of versican [67–69], which is also targeted by ADAMTS4, ADAMTS5, ADAMTS9, ADAMTS15, and ADAMTS20 [10, 39, 43, 94, 99, 100].
Genome-wide association studies and transcriptome analysis have identified associations of ADAMTS loci with several common disorders [101–105], but these associations remain suggestive until functionally validated. This is because the single nucleotide polymorphisms used in GWAS map only to the vicinity of the gene locus, i.e., with few exceptions, they are not within the exons of the gene, and do not introduce amino acid changes in the proteins. Furthermore, proximity of the SNP to the intergenic or intronic regions of an ADAMTS gene locus does not imply that it is necessarily in a regulatory region of that gene, since the SNP may actually affect regulation of another locus in the general region or, sometimes, even further away if it lies within an enhancer. Thus, SNPs do not immediately implicate the ADAMTS protease in that disease unless additional conditions are met, for which there are currently few examples. One SNP for which this burden of proof has been partially met is ADAMTS7, which was associated with coronary artery disease [106]. One of the SNPs led to a Ser214Pro substitution in the propeptide. Biochemical analysis following expression of the Ser and Pro variants suggested that the Pro variant quantitatively impaired ADAMTS7 propeptide excision by furin [107], which is thought to be a prerequisite for proteolytic activity. Thus, the Pro variant potentially has lower activity, and individuals with the Pro/Pro ADAMTS7 protein are predicted to have reduced protease activity compared to those with Ser/Ser variants [107].
2.4 Summary and Conclusions
This chapter provides the reader with a concise background on the general molecular aspects of ADAMTS proteases and demonstrates their considerable diversity of structure and function. It is clear that ADAMTS13 is something of an outlier among the 19-member ADAMTS family. ADAMTS13 structural biology, enzyme–substrate interactions, posttranslational modification, biochemical assays, and roles in genetic and acquired disease are better understood than any other ADAMTS protease. It is the only family member presently for which enzymatic replacement via production of recombinant enzyme is sought [108], whereas a specific blockade is sought for ADAMTS4 and ADAMTS5 in osteoarthritis. Although ADAMTS13 has been arguably more extensively studied for longer than most other family members, there is much about it remaining to be investigated. For instance, little is known about its transcriptional regulation, intermolecular interactions, and turnover. Continuing research on ADAMTS13, with the state of the art represented in this volume, as well as other ADAMTS proteases, will continue to elucidate the shared principles and individual distinctions of this remarkable protease family.
References
Kuno K, Kanada N, Nakashima E, Fujiki F, Ichimura F, Matsushima K. Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J Biol Chem. 1997;272(1):556–62.
Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6(1):32–43.
Tang BL, Hong W. ADAMTS: a novel family of proteases with an ADAM protease domain and thrombospondin 1 repeats. FEBS Lett. 1999;445(2-3):223–5.
Hurskainen TL, Hirohata S, Seldin MF, Apte SS. ADAM-TS5, ADAM-TS6, and ADAM-TS7, novel members of a new family of zinc metalloproteases. General features and genomic distribution of the ADAM-TS family. J Biol Chem. 1999;274(36):25555–63.
Abbaszade I, Liu RQ, Yang F, Rosenfeld SA, Ross OH, Link JR, Ellis DM, Tortorella MD, Pratta MA, Hollis JM, Wynn R, Duke JL, George HJ, Hillman Jr MC, Murphy K, Wiswall BH, Copeland RA, Decicco CP, Bruckner R, Nagase H, Itoh Y, Newton RC, Magolda RL, Trzaskos JM, Burn TC, et al. Cloning and characterization of ADAMTS11, an aggrecanase from the ADAMTS family. J Biol Chem. 1999;274(33):23443–50.
Apte SS. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009;284(46):31493–7. pii: R109.052340. doi:10.1074/jbc.R109.052340. Epub 2009/09/08.
Hirohata S, Wang LW, Miyagi M, Yan L, Seldin MF, Keene DR, Crabb JW, Apte SS. Punctin, a novel ADAMTS-like molecule (ADAMTSL-1) in extracellular matrix. J Biol Chem. 2002;22:22.
Huxley-Jones J, Apte SS, Robertson DL, Boot-Handford RP. The characterisation of six ADAMTS proteases in the basal chordate Ciona intestinalis provides new insights into the vertebrate ADAMTS family. Int J Biochem Cell Biol. 2005;37(9):1838–45.
Apte SS. A disintegrin-like and metalloprotease (reprolysin type) with thrombospondin type 1 motifs: the ADAMTS family. Int J Biochem Cell Biol. 2004;36(6):981–5.
Somerville RP, Longpre JM, Jungers KA, Engle JM, Ross M, Evanko S, Wight TN, Leduc R, Apte SS. Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J Biol Chem. 2003;278(11):9503–13. doi:10.1074/jbc.M211009200.
Koo BH, Le Goff C, Jungers KA, Vasanji A, O’Flaherty J, Weyman CM, Apte SS. ADAMTS-like 2 (ADAMTSL2) is a secreted glycoprotein that is widely expressed during mouse embryogenesis and is regulated during skeletal myogenesis. Matrix Biol. 2007;26(6):431–41. pii: S0945-053X(07)00042-X. doi:10.1016/j.matbio.2007.03.003. Epub 2007/05/19.
Hubmacher D, Apte SS. Genetic and functional linkage between ADAMTS superfamily proteins and fibrillin-1: a novel mechanism influencing microfibril assembly and function. Cell Mol Life Sci. 2011;68(19):3137–48. doi:10.1007/s00018-011-0780-9. Epub 2011/08/23.
Le Goff C, Morice-Picard F, Dagoneau N, Wang LW, Perrot C, Crow YJ, Bauer F, Flori E, Prost-Squarcioni C, Krakow D, Ge G, Greenspan DS, Bonnet D, Le Merrer M, Munnich A, Apte SS, Cormier-Daire V. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat Genet. 2008;40:1119–23.
Kuno K, Terashima Y, Matsushima K. ADAMTS-1 is an active metalloproteinase associated with the extracellular matrix. J Biol Chem. 1999;274(26):18821–6.
Tortorella MD, Arner EC, Hills R, Easton A, Korte-Sarfaty J, Fok K, Wittwer AJ, Liu RQ, Malfait AM. Alpha2-macroglobulin is a novel substrate for ADAMTS-4 and ADAMTS-5 and represents an endogenous inhibitor of these enzymes. J Biol Chem. 2004;279(17):17554–61.
Kashiwagi M, Tortorella M, Nagase H, Brew K. TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J Biol Chem. 2001;276(16):12501–4.
Kashiwagi M, Enghild JJ, Gendron C, Hughes C, Caterson B, Itoh Y, Nagase H. Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing. J Biol Chem. 2004;279(11):10109–19.
Somerville RP, Jungers KA, Apte SS. ADAMTS10: discovery and characterization of a novel, widely expressed metalloprotease and its proteolytic activation. J Biol Chem. 2004;279:51208–17.
Somerville RP, Longpre JM, Apel ED, Lewis RM, Wang LW, Sanes JR, Leduc R, Apte SS. ADAMTS7B, the full-length product of the ADAMTS7 gene, is a chondroitin sulfate proteoglycan containing a mucin domain. J Biol Chem. 2004;279(34):35159–75.
Rodriguez-Manzaneque JC, Carpizo D, Plaza-Calonge Mdel C, Torres-Collado AX, Thai SN, Simons M, Horowitz A, Iruela-Arispe ML. Cleavage of syndecan-4 by ADAMTS1 provokes defects in adhesion. Int J Biochem Cell Biol. 2009;41(4):800–10. pii: S1357-2725(08)00334-8. doi:10.1016/j.biocel.2008.08.014. Epub 2008/09/09.
Liu YJ, Xu Y, Yu Q. Full-length ADAMTS-1 and the ADAMTS-1 fragments display pro- and antimetastatic activity, respectively. Oncogene. 2006;25(17):2452–67.
Koo BH, Apte SS. Cell-surface processing of the metalloprotease pro-ADAMTS9 is influenced by the chaperone GRP94/gp96. J Biol Chem. 2010;285(1):197–205. pii: M109.039677. doi:10.1074/jbc.M109.039677. Epub 2009/10/31.
Koo BH, Longpre JM, Somerville RP, Alexander JP, Leduc R, Apte SS. Regulation of ADAMST9 secretion and enzymatic activity by its propeptide. J Biol Chem. 2007;282:16146–54.
Majerus EM, Zheng X, Tuley EA, Sadler JE. Cleavage of the ADAMTS13 propeptide is not required for protease activity. J Biol Chem. 2003;278(47):46643–8.
Kubota Y, Sano M, Goda S, Suzuki N, Nishiwaki K. The conserved oligomeric Golgi complex acts in organ morphogenesis via glycosylation of an ADAM protease in C. elegans. Development. 2006;133(2):263–73.
Nishiwaki K, Kubota Y, Chigira Y, Roy SK, Suzuki M, Schvarzstein M, Jigami Y, Hisamoto N, Matsumoto K. An NDPase links ADAM protease glycosylation with organ morphogenesis in C. elegans. Nat Cell Biol. 2004;6(1):31–7.
Luo Y, Koles K, Vorndam W, Haltiwanger RS, Panin VM. Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats. J Biol Chem. 2006;281(14):9393–9.
Gonzalez de Peredo A, Klein D, Macek B, Hess D, Peter-Katalinic J, Hofsteenge J. C-mannosylation and o-fucosylation of thrombospondin type 1 repeats. Mol Cell Proteomics. 2002;1(1):11–8.
Kozma K, Keusch JJ, Hegemann B, Luther KB, Klein D, Hess D, Haltiwanger RS, Hofsteenge J. Identification and characterization of abeta1,3-glucosyltransferase that synthesizes the Glc-beta1,3-Fuc disaccharide on thrombospondin type 1 repeats. J Biol Chem. 2006;281(48):36742–51.
Ricketts LM, Dlugosz M, Luther KB, Haltiwanger RS, Majerus EM. O-fucosylation is required for ADAMTS13 secretion. J Biol Chem. 2007;282(23):17014–23.
Wang LW, Dlugosz M, Somerville RP, Raed M, Haltiwanger RS, Apte SS. O-fucosylation of thrombospondin type 1 repeats in ADAMTS-like-1/punctin-1 regulates secretion: implications for the ADAMTS superfamily. J Biol Chem. 2007;282(23):17024–31.
Du J, Takeuchi H, Leonhard-Melief C, Shroyer KR, Dlugosz M, Haltiwanger RS, Holdener BC. O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Dev Biol. 2010;346(1):25–38. doi:10.1016/j.ydbio.2010.07.008.
Enomoto H, Nelson CM, Somerville RP, Mielke K, Dixon LJ, Powell K, Apte SS. Cooperation of two ADAMTS metalloproteases in closure of the mouse palate identifies a requirement for versican proteolysis in regulating palatal mesenchyme proliferation. Development. 2010;137(23):4029–38. doi:10.1242/dev.050591.
Lesnik Oberstein SA, Kriek M, White SJ, Kalf ME, Szuhai K, den Dunnen JT, Breuning MH, Hennekam RC. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am J Hum Genet. 2006;79(3):562–6. doi:10.1086/507567.
Hofsteenge J, Huwiler KG, Macek B, Hess D, Lawler J, Mosher DF, Peter-Katalinic J. C-mannosylation and O-fucosylation of the thrombospondin type 1 module. J Biol Chem. 2001;276(9):6485–98.
Wang LW, Leonhard-Melief C, Haltiwanger RS, Apte SS. Post-translational modification of thrombospondin type-1 repeats in ADAMTS-like 1/punctin-1 by C-mannosylation of tryptophan. J Biol Chem. 2009;284(44):30004–15. pii: M109.038059. doi:10.1074/jbc.M109.038059. Epub 2009/08/13.
Koo BH, Longpre JM, Somerville RP, Alexander JP, Leduc R, Apte SS. Cell-surface processing of pro-ADAMTS9 by furin. J Biol Chem. 2006;281(18):12485–94.
Longpre JM, Leduc R. Identification of prodomain determinants involved in ADAMTS-1 biosynthesis. J Biol Chem. 2004;279(32):33237–45.
Longpre JM, McCulloch DR, Koo BH, Alexander JP, Apte SS, Leduc R. Characterization of proADAMTS5 processing by proprotein convertases. Int J Biochem Cell Biol. 2009;41(5):1116–26. pii: S1357-2725(08)00414-7. doi:10.1016/j.biocel.2008.10.008. Epub 2008/11/11.
Tortorella MD, Arner EC, Hills R, Gormley J, Fok K, Pegg L, Munie G, Malfait AM. ADAMTS-4 (aggrecanase-1): N-terminal activation mechanisms. Arch Biochem Biophys. 2005;444(1):34–44.
Wylie JD, Ho JC, Singh S, McCulloch DR, Apte SS. Adamts5 (aggrecanase-2) is widely expressed in the mouse musculoskeletal system and is induced in specific regions of knee joint explants by inflammatory cytokines. J Orthop Res. 2012;30(2):226–33. doi:10.1002/jor.21508. Epub 2011/07/30.
Kutz WE, Wang LW, Bader HL, Majors AK, Iwata K, Traboulsi EI, Sakai LY, Keene DR, Apte SS. ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts. J Biol Chem. 2011;286(19):17156–67. pii: M111.231571. doi:10.1074/jbc.M111.231571. Epub 2011/03/16.
Stupka N, Kintakas C, White JD, Fraser FW, Hanciu M, Aramaki-Hattori N, Martin S, Coles C, Collier F, Ward AC, Apte SS, McCulloch DR. Versican processing by a disintegrin-like and metalloproteinase domain with thrombospondin-1 repeats proteinases-5 and -15 facilitates myoblast fusion. J Biol Chem. 2013;288(3):1907–17. doi:10.1074/jbc.M112.429647. Epub 2012/12/13.
Gerhardt S, Hassall G, Hawtin P, McCall E, Flavell L, Minshull C, Hargreaves D, Ting A, Pauptit RA, Parker AE, Abbott WM. Crystal structures of human ADAMTS-1 reveal a conserved catalytic domain and a disintegrin-like domain with a fold homologous to cysteine-rich domains. J Mol Biol. 2007;373(4):891–902.
Mosyak L, Georgiadis K, Shane T, Svenson K, Hebert T, McDonagh T, Mackie S, Olland S, Lin L, Zhong X, Kriz R, Reifenberg EL, Collins-Racie LA, Corcoran C, Freeman B, Zollner R, Marvell T, Vera M, Sum PE, Lavallie ER, Stahl M, Somers W. Crystal structures of the two major aggrecan degrading enzymes, ADAMTS4 and ADAMTS5. Protein Sci. 2008;17(1):16–21.
Shieh HS, Mathis KJ, Williams JM, Hills RL, Wiese JF, Benson TE, Kiefer JR, Marino MH, Carroll JN, Leone JW, Malfait AM, Arner EC, Tortorella MD, Tomasselli A. High resolution crystal structure of the catalytic domain of ADAMTS-5 (aggrecanase-2). J Biol Chem. 2008;283(3):1501–7.
Colige A, Ruggiero F, Vandenberghe I, Dubail J, Kesteloot F, Van Beeumen J, Beschin A, Brys L, Lapiere CM, Nusgens B. Domains and maturation processes that regulate the activity of ADAMTS-2, a metalloproteinase cleaving the aminopropeptide of fibrillar procollagens type I, II, III and V. J Biol Chem. 2005;280:34397–408.
Kuno K, Matsushima K. ADAMTS-1 protein anchors at the extracellular matrix through the thrombospondin type I motifs and its spacing region. J Biol Chem. 1998;273(22):13912–7.
Zheng X, Nishio K, Majerus EM, Sadler JE. Cleavage of von Willebrand factor requires the spacer domain of the metalloprotease ADAMTS13. J Biol Chem. 2003;278(32):30136–41. pii: M305331200. doi:10.1074/jbc.M305331200. Epub 2003/06/07.
Akiyama M, Takeda S, Kokame K, Takagi J, Miyata T. Crystal structures of the noncatalytic domains of ADAMTS13 reveal multiple discontinuous exosites for von Willebrand factor. Proc Natl Acad Sci U S A. 2009;106(46):19274–9. pii: 0909755106. doi:10.1073/pnas.0909755106. Epub 2009/11/03.
Wu JJ, Fujikawa K, McMullen BA, Chung DW. Characterization of a core binding site for ADAMTS-13 in the A2 domain of von Willebrand factor. Proc Natl Acad Sci U S A. 2006;103(49):18470–4. doi:10.1073/pnas.0609190103.
Zheng XL. Structure–function and regulation of ADAMTS-13 protease. J Thromb Haemost. 2013;11 Suppl 1:11–23. doi:10.1111/jth.12221.
Foulcer SJ, Nelson CM, Quintero MV, Kuberan B, Larkin J, Dours-Zimmermann MT, Zimmermann DR, Apte SS. Determinants of versican-V1 proteoglycan processing by the metalloproteinase ADAMTS5. J Biol Chem. 2014;289(40):27859–73. doi:10.1074/jbc.M114.573287.
Tortorella M, Pratta M, Liu RQ, Abbaszade I, Ross H, Burn T, Arner E. The thrombospondin motif of aggrecanase-1 (ADAMTS-4) is critical for aggrecan substrate recognition and cleavage. J Biol Chem. 2000;275(33):25791–7.
Nowak AA, McKinnon TA, Hughes JM, Chion AC, Laffan MA. The O-linked glycans of human von Willebrand factor modulate its interaction with ADAMTS-13. J Thromb Haemost. 2014;12(1):54–61. doi:10.1111/jth.12451.
Tuderman L, Kivirikko KI, Prockop DJ. Partial purification and characterization of a neutral protease which cleaves the N-terminal propeptides from procollagen. Biochemistry. 1978;17(15):2948–54.
Dong JF, Moake JL, Bernardo A, Fujikawa K, Ball C, Nolasco L, Lopez JA, Cruz MA. ADAMTS-13 metalloprotease interacts with the endothelial cell-derived ultra-large von Willebrand factor. J Biol Chem. 2003;278(32):29633–9.
Wu T, Lin J, Cruz MA, Dong JF, Zhu C. Force-induced cleavage of single VWFA1A2A3 tridomains by ADAMTS-13. Blood. 2010;115(2):370–8. pii: blood-2009-03-210369. doi:10.1182/blood-2009-03-210369. Epub 2009/11/10.
McCulloch DR, Nelson CM, Dixon LJ, Silver DL, Wylie JD, Lindner V, Sasaki T, Cooley MA, Argraves WS, Apte SS. ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression. Dev Cell. 2009;17(5):687–98. doi:10.1016/j.devcel.2009.09.008.
Cooley MA, Fresco VM, Dorlon ME, Twal WO, Lee NV, Barth JL, Kern CB, Iruela-Arispe ML, Argraves WS. Fibulin-1 is required during cardiac ventricular morphogenesis for versican cleavage, suppression of ErbB2 and Erk1/2 activation, and to attenuate trabecular cardiomyocyte proliferation. Dev Dyn. 2012;241(2):303–14. doi:10.1002/dvdy.23716.
Lee NV, Rodriguez-Manzaneque JC, Thai SN, Twal WO, Luque A, Lyons KM, Argraves WS, Iruela-Arispe ML. Fibulin-1 acts as a cofactor for the matrix metalloprotease ADAMTS-1. J Biol Chem. 2005;280:34796–804.
Hashimoto G, Shimoda M, Okada Y. ADAMTS4 (aggrecanase-1) interaction with the C-terminal domain of fibronectin inhibits proteolysis of aggrecan. J Biol Chem. 2004;279(31):32483–91.
Kramerova IA, Kawaguchi N, Fessler LI, Nelson RE, Chen Y, Kramerov AA, Kusche-Gullberg M, Kramer JM, Ackley BD, Sieron AL, Prockop DJ, Fessler JH. Papilin in development; a pericellular protein with a homology to the ADAMTS metalloproteinases. Development. 2000;127(24):5475–85.
Shindo T, Kurihara H, Kuno K, Yokoyama H, Wada T, Kurihara Y, Imai T, Wang Y, Ogata M, Nishimatsu H, Moriyama N, Oh-hashi Y, Morita H, Ishikawa T, Nagai R, Yazaki Y, Matsushima K. ADAMTS-1: a metalloproteinase-disintegrin essential for normal growth, fertility, and organ morphology and function. J Clin Invest. 2000;105(10):1345–52.
Mittaz L, Russell DL, Wilson T, Brasted M, Tkalcevic J, Salamonsen LA, Hertzog PJ, Pritchard MA. Adamts-1 is essential for the development and function of the urogenital system. Biol Reprod. 2004;70(4):1096–105.
Brown HM, Dunning KR, Robker RL, Boerboom D, Pritchard M, Lane M, Russell DL. ADAMTS1 cleavage of versican mediates essential structural remodeling of the ovarian follicle and cumulus-oocyte matrix during ovulation in mice. Biol Reprod. 2010;83(4):549–57. pii: biolreprod.110.084434. doi:10.1095/biolreprod.110.084434. Epub 2010/07/02.
Brown HM, Dunning KR, Robker RL, Pritchard M, Russell DL. Requirement for ADAMTS-1 in extracellular matrix remodeling during ovarian folliculogenesis and lymphangiogenesis. Dev Biol. 2006;300(2):699–709.
Russell DL, Doyle KM, Ochsner SA, Sandy JD, Richards JS. Processing and localization of ADAMTS-1 and proteolytic cleavage of versican during cumulus matrix expansion and ovulation. J Biol Chem. 2003;278(43):42330–9.
Stankunas K, Hang CT, Tsun ZY, Chen H, Lee NV, Wu JI, Shang C, Bayle JH, Shou W, Iruela-Arispe ML, Chang CP. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev Cell. 2008;14(2):298–311.
Hanset R, Lapiere CM. Inheritance of dermatosparaxis in the calf. A genetic defect of connective tissues. J Hered. 1974;65(6):356–8.
Lapiere CM, Lenaers A, Kohn LD. Procollagen peptidase: an enzyme excising the coordination peptides of procollagen. Proc Natl Acad Sci U S A. 1971;68(12):3054–8.
Nusgens BV, Verellen-Dumoulin C, Hermanns-Le T, De Paepe A, Nuytinck L, Pierard GE, Lapiere CM. Evidence for a relationship between Ehlers-Danlos type VII C in humans and bovine dermatosparaxis. Nat Genet. 1992;1(3):214–7.
Colige A, Sieron AL, Li SW, Schwarze U, Petty E, Wertelecki W, Wilcox W, Krakow D, Cohn DH, Reardon W, Byers PH, Lapiere CM, Prockop DJ, Nusgens BV. Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am J Hum Genet. 1999;65(2):308–17.
Fernandes RJ, Hirohata S, Engle JM, Colige A, Cohn DH, Eyre DR, Apte SS. Procollagen II amino propeptide processing by ADAMTS-3. Insights on dermatosparaxis. J Biol Chem. 2001;276(34):31502–9.
Le Goff C, Somerville RP, Kesteloot F, Powell K, Birk DE, Colige AC, Apte SS. Regulation of procollagen amino-propeptide processing during mouse embryogenesis by specialization of homologous ADAMTS proteases: insights on collagen biosynthesis and dermatosparaxis. Development. 2006;133(8):1587–96.
Jeltsch M, Jha SK, Tvorogov D, Anisimov A, Leppanen VM, Holopainen T, Kivela R, Ortega S, Karpanen T, Alitalo K. CCBE1 enhances lymphangiogenesis via a disintegrin and metalloprotease with thrombospondin motifs-3-mediated vascular endothelial growth factor-C activation. Circulation. 2014;129(19):1962–71. doi:10.1161/CIRCULATIONAHA.113.002779.
Fosang AJ, Little CB. Drug insight: aggrecanases as therapeutic targets for osteoarthritis. Nat Clin Pract Rheumatol. 2008;4(8):420–7. pii: ncprheum0841. doi:10.1038/ncprheum0841. Epub 2008/06/26.
Gilbert AM, Bursavich MG, Lombardi S, Georgiadis KE, Reifenberg E, Flannery CR, Morris EA. 5-((1H-pyrazol-4-yl)methylene)-2-thioxothiazolidin-4-one inhibitors of ADAMTS-5. Bioorg Med Chem Lett. 2007;17(5):1189–92.
Dupuis LE, McCulloch DR, McGarity JD, Bahan A, Wessels A, Weber D, Diminich AM, Nelson CM, Apte SS, Kern CB. Altered versican cleavage in ADAMTS5 deficient mice; a novel etiology of myxomatous valve disease. Dev Biol. 2011;357(1):152–64. pii: S0012-1606(11)01101-8. doi:10.1016/j.ydbio.2011.06.041. Epub 2011/07/14.
Dupuis LE, Osinska H, Weinstein MB, Hinton RB, Kern CB. Insufficient versican cleavage and Smad2 phosphorylation results in bicuspid aortic and pulmonary valves. J Mol Cell Cardiol. 2013;60:50–9. doi:10.1016/j.yjmcc.2013.03.010.
Dubail J, Aramaki-Hattori N, Bader HL, Nelson CM, Katebi N, Matuska B, Olsen BR, Apte SS. A new Adamts9 conditional mouse allele identifies its non-redundant role in interdigital web regression. Genesis. 2014;52(7):702–12. doi:10.1002/dvg.22784.
Kern CB, Wessels A, McGarity J, Dixon LJ, Alston E, Argraves WS, Geeting D, Nelson CM, Menick DR, Apte SS. Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biol. 2010;29(4):304–16. pii: S0945-053X(10)00009-0. doi:10.1016/j.matbio.2010.01.005. Epub 2010/01/26.
Koo BH, Coe DM, Dixon LJ, Somerville RP, Nelson CM, Wang LW, Young ME, Lindner DJ, Apte SS. ADAMTS9 is a cell-autonomously acting, anti-angiogenic metalloprotease expressed by microvascular endothelial cells. Am J Pathol. 2010. pii: ajpath.2010.090655. doi:10.2353/ajpath.2010.090655. Epub 2010/01/23.
Dagoneau N, Benoist-Lasselin C, Huber C, Faivre L, Megarbane A, Alswaid A, Dollfus H, Alembik Y, Munnich A, Legeai-Mallet L, Cormier-Daire V. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet. 2004;75(5):801–6. doi:10.1086/425231. Epub 2004/09/16.
Faivre L, Gorlin RJ, Wirtz MK, Godfrey M, Dagoneau N, Samples JR, Le Merrer M, Collod-Beroud G, Boileau C, Munnich A, Cormier-Daire V. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J Med Genet. 2003;40(1):34–6.
Farias FH, Johnson GS, Taylor JF, Giuliano E, Katz ML, Sanders DN, Schnabel RD, McKay SD, Khan S, Gharahkhani P, O’Leary CA, Pettitt L, Forman OP, Boursnell M, McLaughlin B, Ahonen S, Lohi H, Hernandez-Merino E, Gould DJ, Sargan D, Mellersh CS. An ADAMTS17 splice donor site mutation in dogs with primary lens luxation. Invest Ophthalmol Vis Sci. 2010;51:4716–21. pii: iovs.09-5142. doi:10.1167/iovs.09-5142. Epub 2010/04/09.
Morales J, Al-Sharif L, Khalil DS, Shinwari JM, Bavi P, Al-Mahrouqi RA, Al-Rajhi A, Alkuraya FS, Meyer BF, Al Tassan N. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am J Hum Genet. 2009;85(5):558–68. pii: S0002-9297(09)00407-8. doi:10.1016/j.ajhg.2009.09.011. Epub 2009/10/20.
Abdul-Majeed S, Mell B, Nauli SM, Joe B. Cryptorchidism and infertility in rats with targeted disruption of the Adamts16 locus. PLoS One. 2014;9(7), e100967. doi:10.1371/journal.pone.0100967.
Gopalakrishnan K, Kumarasamy S, Abdul-Majeed S, Kalinoski AL, Morgan EE, Gohara AF, Nauli SM, Filipiak WE, Saunders TL, Joe B. Targeted disruption of Adamts16 gene in a rat genetic model of hypertension. Proc Natl Acad Sci U S A. 2012;109(50):20555–9. doi:10.1073/pnas.1211290109.
Jacobi CL, Rudigier LJ, Scholz H, Kirschner KM. Transcriptional regulation by the Wilms tumor protein, Wt1, suggests a role of the metalloproteinase Adamts16 in murine genitourinary development. J Biol Chem. 2013;288(26):18811–24. doi:10.1074/jbc.M113.464644.
Aldahmesh MA, Alshammari MJ, Khan AO, Mohamed JY, Alhabib FA, Alkuraya FS. The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) is caused by mutations in ADAMTS18. Hum Mutat. 2013;34(9):1195–9. doi:10.1002/humu.22374.
Peluso I, Conte I, Testa F, Dharmalingam G, Pizzo M, Collin RW, Meola N, Barbato S, Mutarelli M, Ziviello C, Barbarulo AM, Nigro V, Melone MA, European Retinal Disease C, Simonelli F, Banfi S. The ADAMTS18 gene is responsible for autosomal recessive early onset severe retinal dystrophy. Orphanet J Rare Dis. 2013;8:16. doi:10.1186/1750-1172-8-16.
Rao C, Foernzler D, Loftus SK, Liu S, McPherson JD, Jungers KA, Apte SS, Pavan WJ, Beier DR. A defect in a novel ADAMTS family member is the cause of the belted white-spotting mutation. Development. 2003;130(19):4665–72.
Silver DL, Hou L, Somerville R, Young ME, Apte SS, Pavan WJ. The secreted metalloprotease ADAMTS20 is required for melanoblast survival. PLoS Genet. 2008;4(2):1–15.
Rodriguez-Manzaneque JC, Westling J, Thai SN, Luque A, Knauper V, Murphy G, Sandy JD, Iruela-Arispe ML. ADAMTS1 cleaves aggrecan at multiple sites and is differentially inhibited by metalloproteinase inhibitors. Biochem Biophys Res Commun. 2002;293(1):501–8.
Torres-Collado AX, Kisiel W, Iruela-Arispe ML, Rodriguez-Manzaneque JC. ADAMTS1 interacts with, cleaves, and modifies the extracellular location of the matrix inhibitor tissue factor pathway inhibitor-2. J Biol Chem. 2006;281(26):17827–37.
Canals F, Colome N, Ferrer C, Plaza-Calonge Mdel C, Rodriguez-Manzaneque JC. Identification of substrates of the extracellular protease ADAMTS1 by DIGE proteomic analysis. Proteomics. 2006;6 Suppl 1:S28–35.
Lee NV, Sato M, Annis DS, Loo JA, Wu L, Mosher DF, Iruela-Arispe ML. ADAMTS1 mediates the release of antiangiogenic polypeptides from TSP1 and 2. EMBO J. 2006;25(22):5270–83.
Lind T, Birch MA, McKie N. Purification of an insect derived recombinant human ADAMTS-1 reveals novel gelatin (type I collagen) degrading activities. Mol Cell Biochem. 2006;281(1-2):95–102. doi:10.1007/s11010-006-0637-y.
Sandy JD, Westling J, Kenagy RD, Iruela-Arispe ML, Verscharen C, Rodriguez-Mazaneque JC, Zimmermann DR, Lemire JM, Fischer JW, Wight TN, Clowes AW. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441–Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J Biol Chem. 2001;276(16):13372–8.
Lo PH, Lung HL, Cheung AK, Apte SS, Chan KW, Kwong FM, Ko JM, Cheng Y, Law S, Srivastava G, Zabarovsky ER, Tsao SW, Tang JC, Stanbridge EJ, Lung ML. Extracellular protease ADAMTS9 suppresses esophageal and nasopharyngeal carcinoma tumor formation by inhibiting angiogenesis. Cancer Res. 2010. pii: 0008-5472.CAN-09-4510. doi:10.1158/0008-5472.CAN-09-4510. Epub 2010/06/17.
Viloria CG, Obaya AJ, Moncada-Pazos A, Llamazares M, Astudillo A, Capella G, Cal S, Lopez-Otin C. Genetic inactivation of ADAMTS15 metalloprotease in human colorectal cancer. Cancer Res. 2009;69(11):4926–34. pii: 0008-5472.CAN-08-4155. doi:10.1158/0008-5472.CAN-08-4155. Epub 2009/05/22.
Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, Igo RP, Jr., Buitendijk GH, Sim X, Weeks DE, Guymer RH, Merriam JE, Francis PJ, Hannum G, Agarwal A, Armbrecht AM, Audo I, Aung T, Barile GR, Benchaboune M, Bird AC, Bishop PN, Branham KE, Brooks M, Brucker AJ, Cade WH, Cain MS, Campochiaro PA, Chan CC, Cheng CY, Chew EY, Chin KA, Chowers I, Clayton DG, Cojocaru R, Conley YP, Cornes BK, Daly MJ, Dhillon B, Edwards AO, Evangelou E, Fagerness J, Ferreyra HA, Friedman JS, Geirsdottir A, George RJ, Gieger C, Gupta N, Hagstrom SA, Harding SP, Haritoglou C, Heckenlively JR, Holz FG, Hughes G, Ioannidis JP, Ishibashi T, Joseph P, Jun G, Kamatani Y, Katsanis N, C NK, Khan JC, Kim IK, Kiyohara Y, Klein BE, Klein R, Kovach JL, Kozak I, Lee CJ, Lee KE, Lichtner P, Lotery AJ, Meitinger T, Mitchell P, Mohand-Said S, Moore AT, Morgan DJ, Morrison MA, Myers CE, Naj AC, Nakamura Y, Okada Y, Orlin A, Ortube MC, Othman MI, Pappas C, Park KH, Pauer GJ, Peachey NS, Poch O, Priya RR, Reynolds R, Richardson AJ, Ripp R, Rudolph G, Ryu E, Sahel JA, Schaumberg DA, Scholl HP, Schwartz SG, Scott WK, Shahid H, Sigurdsson H, Silvestri G, Sivakumaran TA, Smith RT, Sobrin L, Souied EH, Stambolian DE, Stefansson H, Sturgill-Short GM, Takahashi A, Tosakulwong N, Truitt BJ, Tsironi EE, Uitterlinden AG, van Duijn CM, Vijaya L, Vingerling JR, Vithana EN, Webster AR, Wichmann HE, Winkler TW, Wong TY, Wright AF, Zelenika D, Zhang M, Zhao L, Zhang K, Klein ML, Hageman GS, Lathrop GM, Stefansson K, Allikmets R, Baird PN, Gorin MB, Wang JJ, Klaver CC, Seddon JM, Pericak-Vance MA, Iyengar SK, Yates JR, Swaroop A, Weber BH, Kubo M, Deangelis MM, Leveillard T, Thorsteinsdottir U, Haines JL, Farrer LA, Heid IM, Abecasis GR, Consortium AMDG. Seven new loci associated with age-related macular degeneration. Nat Genet. 2013;45(4):433–9, 9e1–2. doi:10.1038/ng.2578.
Heid IM, Jackson AU, Randall JC, Winkler TW, Qi L, Steinthorsdottir V, Thorleifsson G, Zillikens MC, Speliotes EK, Magi R, Workalemahu T, White CC, Bouatia-Naji N, Harris TB, Berndt SI, Ingelsson E, Willer CJ, Weedon MN, Luan J, Vedantam S, Esko T, Kilpelainen TO, Kutalik Z, Li S, Monda KL, Dixon AL, Holmes CC, Kaplan LM, Liang L, Min JL, Moffatt MF, Molony C, Nicholson G, Schadt EE, Zondervan KT, Feitosa MF, Ferreira T, Allen HL, Weyant RJ, Wheeler E, Wood AR, Estrada K, Goddard ME, Lettre G, Mangino M, Nyholt DR, Purcell S, Smith AV, Visscher PM, Yang J, McCarroll SA, Nemesh J, Voight BF, Absher D, Amin N, Aspelund T, Coin L, Glazer NL, Hayward C, Heard-Costa NL, Hottenga JJ, Johansson A, Johnson T, Kaakinen M, Kapur K, Ketkar S, Knowles JW, Kraft P, Kraja AT, Lamina C, Leitzmann MF, McKnight B, Morris AP, Ong KK, Perry JR, Peters MJ, Polasek O, Prokopenko I, Rayner NW, Ripatti S, Rivadeneira F, Robertson NR, Sanna S, Sovio U, Surakka I, Teumer A, van Wingerden S, Vitart V, Zhao JH, Cavalcanti-Proenca C, Chines PS, Fisher E, Kulzer JR, Lecoeur C, Narisu N, Sandholt C, Scott LJ, Silander K, Stark K, Tammesoo ML, Teslovich TM, Timpson NJ, Watanabe RM, Welch R, Chasman DI, Cooper MN, Jansson JO, Kettunen J, Lawrence RW, Pellikka N, Perola M, Vandenput L, Alavere H, Almgren P, Atwood LD, Bennett AJ, Biffar R, Bonnycastle LL, Bornstein SR, Buchanan TA, Campbell H, Day IN, Dei M, Dorr M, Elliott P, Erdos MR, Eriksson JG, Freimer NB, Fu M, Gaget S, Geus EJ, Gjesing AP, Grallert H, Grassler J, Groves CJ, Guiducci C, Hartikainen AL, Hassanali N, Havulinna AS, Herzig KH, Hicks AA, Hui J, Igl W, Jousilahti P, Jula A, Kajantie E, Kinnunen L, Kolcic I, Koskinen S, Kovacs P, Kroemer HK, Krzelj V, Kuusisto J, Kvaloy K, Laitinen J, Lantieri O, Lathrop GM, Lokki ML, Luben RN, Ludwig B, McArdle WL, McCarthy A, Morken MA, Nelis M, Neville MJ, Pare G, Parker AN, Peden JF, Pichler I, Pietilainen KH, Platou CG, Pouta A, Ridderstrale M, Samani NJ, Saramies J, Sinisalo J, Smit JH, Strawbridge RJ, Stringham HM, Swift AJ, Teder-Laving M, Thomson B, Usala G, van Meurs JB, van Ommen GJ, Vatin V, Volpato CB, Wallaschofski H, Walters GB, Widen E, Wild SH, Willemsen G, Witte DR, Zgaga L, Zitting P, Beilby JP, James AL, Kahonen M, Lehtimaki T, Nieminen MS, Ohlsson C, Palmer LJ, Raitakari O, Ridker PM, Stumvoll M, Tonjes A, Viikari J, Balkau B, Ben-Shlomo Y, Bergman RN, Boeing H, Smith GD, Ebrahim S, Froguel P, Hansen T, Hengstenberg C, Hveem K, Isomaa B, Jorgensen T, Karpe F, Khaw KT, Laakso M, Lawlor DA, Marre M, Meitinger T, Metspalu A, Midthjell K, Pedersen O, Salomaa V, Schwarz PE, Tuomi T, Tuomilehto J, Valle TT, Wareham NJ, Arnold AM, Beckmann JS, Bergmann S, Boerwinkle E, Boomsma DI, Caulfield MJ, Collins FS, Eiriksdottir G, Gudnason V, Gyllensten U, Hamsten A, Hattersley AT, Hofman A, Hu FB, Illig T, Iribarren C, Jarvelin MR, Kao WH, Kaprio J, Launer LJ, Munroe PB, Oostra B, Penninx BW, Pramstaller PP, Psaty BM, Quertermous T, Rissanen A, Rudan I, Shuldiner AR, Soranzo N, Spector TD, Syvanen AC, Uda M, Uitterlinden A, Volzke H, Vollenweider P, Wilson JF, Witteman JC, Wright AF, Abecasis GR, Boehnke M, Borecki IB, Deloukas P, Frayling TM, Groop LC, Haritunians T, Hunter DJ, Kaplan RC, North KE, O’Connell JR, Peltonen L, Schlessinger D, Strachan DP, Hirschhorn JN, Assimes TL, Wichmann HE, Thorsteinsdottir U, van Duijn CM, Stefansson K, Cupples LA, Loos RJ, Barroso I, McCarthy MI, Fox CS, Mohlke KL, Lindgren CM. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010. pii: ng.685. doi:10.1038/ng.685. Epub 2010/10/12.
Helisalmi S, Immonen I, Losonczy G, Resch MD, Benedek S, Balogh I, Papp A, Berta A, Uusitupa M, Hiltunen M, Kaarniranta K. ADAMTS9 locus associates with increased risk of wet AMD. Acta Ophthalmol. 2014. doi:10.1111/aos.12341.
Reilly MP, Li M, He J, Ferguson JF, Stylianou IM, Mehta NN, Burnett MS, Devaney JM, Knouff CW, Thompson JR, Horne BD, Stewart AF, Assimes TL, Wild PS, Allayee H, Nitschke PL, Patel RS, Martinelli N, Girelli D, Quyyumi AA, Anderson JL, Erdmann J, Hall AS, Schunkert H, Quertermous T, Blankenberg S, Hazen SL, Roberts R, Kathiresan S, Samani NJ, Epstein SE, Rader DJ. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 2011;377(9763):383–92. pii: S0140-6736(10)61996-4. doi:10.1016/S0140-6736(10)61996-4. Epub 2011/01/18.
Pu X, Xiao Q, Kiechl S, Chan K, Ng FL, Gor S, Poston RN, Fang C, Patel A, Senver EC, Shaw-Hawkins S, Willeit J, Liu C, Zhu J, Tucker AT, Xu Q, Caulfield MJ, Ye S. ADAMTS7 cleavage and vascular smooth muscle cell migration is affected by a coronary-artery-disease-associated variant. Am J Hum Genet. 2013;92(3):366–74. doi:10.1016/j.ajhg.2013.01.012.
Plaimauer B, Scheiflinger F. Expression and characterization of recombinant human ADAMTS-13. Semin Hematol. 2004;41(1):24–33.
Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw Jr JD, Ginsburg D, Tsai HM. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488–94.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Apte, S.S. (2015). Overview of the ADAMTS Superfamily. In: Rodgers, G. (eds) ADAMTS13. Springer, Cham. https://doi.org/10.1007/978-3-319-08717-7_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-08717-7_2
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-08716-0
Online ISBN: 978-3-319-08717-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)