
Abstract
Dynamin 2 (DNM2) mutations cause autosomal dominant centronuclear myopathy, a rare form of congenital myopathy, and intermediate and axonal forms of Charcot–Marie-Tooth disease, a peripheral neuropathy. DNM2 is a large GTPase mainly involved in membrane trafficking through its function in the formation and release of nascent vesicles from biological membranes. DNM2 participates in clathrin-dependent and clathrin-independent endocytosis and intracellular membrane trafficking (from endosomes and Golgi apparatus). Recent studies have also implicated DNM2 in exocytosis. DNM2 belongs to the machinery responsible for the formation of vesicles and regulates the cytoskeleton providing intracellular vesicle transport. In addition, DNM2 tightly interacts with and is involved in the regulation of actin and microtubule networks, independent from membrane trafficking processes. We summarize here the molecular, biochemical, and functional data on DNM2 and discuss the possible pathophysiological mechanisms via which DNM2 mutations can lead to two distinct neuromuscular disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Dynamin 2 (DNM2) belongs to a superfamily of large GTPases, including three classical dynamins and several dynamin-like proteins, which are involved in a wide range of cell functions [1]. The importance of DNM2 was emphasized in 2005 with the demonstration of DNM2 gene mutations causing two distinct human diseases [2, 3]. Our purpose is to review the molecular and functional data on DNM2 to highlight the pathophysiological hypotheses in DNM2-related diseases. Knowledge of the dynamins mainly comes from studies of the neuronal dynamin 1 (DNM1). However, we have focused this review on DNM2 since several studies have demonstrated notable differences between DNM1 and DNM2 [4–7].
DNM2 gene organization and isoforms
DNM2, one of three classical dynamins, was identified in rat liver and brain cDNA libraries [4, 8]. A human homologue was thereafter identified by screening of a fibroblast library [9]. The human transcript (3.6 kilobases) is ubiquitously expressed, with higher abundance in heart and skeletal muscle [9]. Human DNM2 is encoded by the DNM2 gene located on the short arm of chromosome 19 (19p13.2). The gene is composed of 22 exons in a 114-kilobase region. Four major isoforms are expressed by the DNM2 gene using a combination of two alternative splice sites (Fig. 1a). Exons 10 and 10bis have the same length (139 base pairs encoding the amino acids 399–445 in the middle domain) and are alternatively spliced. In addition, the exon 13bis (12-base pair length) can be spliced leading to the translation of proteins of 866 or 870 amino acids (Fig. 1) without or with the GEIL sequence at position 516–519 in the C-terminal part of the middle domain (MD). The four major isoforms have been shown to be expressed in a panel of rat tissues including brain, heart, kidney, liver, lung, pancreas, and testis [10]. The human tissue expression pattern is unknown, but we have shown expression of the four isoforms in skeletal muscle and peripheral nerve [11]. Specific functions of these isoforms will be discussed below.

DNM2 gene organization and mutations. a Schematic organization of the human DNM2 gene showing alternative splicing. Asterisks indicate the seven exons in which disease-associated mutations have been identified. Exons were colored relative to the encoded protein domain illustrated in (b). The combination of the two alternative splice sites leads to the translation of four DNM2 isoforms. Isoforms 1, 2, 3, and 4 are also known as isoforms aa, ba, ab, and bb, respectively. b Schematic representation of DNM2 showing the five protein domains and the position of the 19 disease-associated mutations. CMT-mutations are indicated in green and CNM-mutations in red. The two regions of variation (at positions 399-445 and 516-519) between the four isoforms were indicated in the MD by black lines. In black are indicated the sites of post-translational modifications (phosphorylation, nitrosylation, and cathepsin L cleavage). The CMT-mutation G358R is located in the cathepsin L cleavage site. In blue are indicated the DNM2 constructs with point mutations or small deletions overexpressed in vitro [60]. Insert inb: Position of the CNM- and CMT-mutations on the 3D structure of the PH domain (accession number 63660 in the NCBI 3D structure database). The N-terminal part of the domain, bearing CMT-mutations and only one CNM mutation, is composed of β-sheets involved in the interaction with membrane phosphoinositides
DNM2 structure and regulation
The 98 kDa DNM2 is a large GTPase composed of a N-terminal GTPase domain, an MD, a pleckstrin homology domain (PH), a GTPase effector domain (GED), and a C-terminal proline rich domain (PRD; Fig. 1b). The catalytic GTPase domain is responsible for GTP binding and hydrolysis, whereas the MD is involved in DNM2 self-assembly [12] and in GTP hydrolysis-induced conformational change of the protein [13]. The PH domain interacts with membrane phosphoinositides and therefore is involved in the targeting of dynamin to membranes [14]. The DNM2-PH domain displays phosphoinositide binding affinity following the order: PI4,5P2 ≈ PI3,4,5P3 ≈ PI3,4P2 > PI4P ≈ PI3P, and DNM2 oligomerization appears crucial for high affinity [15]. The GED probably participates in the self-assembly of DNM2 and acts as a GTPase-activating protein [16]. The PRD contains multiple Src homology 3 (SH3) binding motifs and mediates multiple protein–protein interactions (Table 1). A general model of dynamin intramolecular interaction was proposed, in which the GTPase domain, MD and GED interact to drive self-assembly and the PH domain mediates interaction with membrane lipids [1].
In vitro at high ionic strength, DNM2 is in monomer–tetramer equilibrium. At low ionic strength, DNM2 self-assembles into higher-order aggregates leading to a drastic increase in GTPase activity [17, 18]. Microtubules or phospholipid vesicles, especially those containing PI4,5P2, also induce self-assembly and increase DNM2 GTPase activity [5, 17, 19]. Purified from baculovirus, GTP-bound and GDP-bound monomer DNM2 has Kd values of 13.2 and 7.1 µM, respectively, with GTPase activity of 37 nmol/mg/min. When in an oligomeric state, the GTPase activity of DNM2 markedly increased and Kd values decreased [20]. When compared with small GTPases, DNM2 exhibits a relatively low affinity for GTP (Km = 12 µM) but high intrinsic rates of GTP hydrolysis.
DNM2 activity is regulated by post-translational modifications. DNM2 becomes phosphorylated on Tyr231 (MD) and Tyr597 (PH domain) through Src-mediated phosphorylation, leading to albumin endocytosis [21]. In contrast, dopamine leads to the dephosphorylation of DNM2 by increasing protein phosphatase 2A activity, necessary for dopamine-induced Na+K+-ATPase endocytosis [22]. S-nitrosylation of Cys86 (GTPase domain) and Cys607 (PH domain) by nitric oxide (NO) increases GTPase activity and endocytosis [23]. In a mouse model of kidney disease, cathepsin L induction leads to the cleavage of the cytoplasmic DNM2 at positions 355–360 (Fig. 1b) [24]. Sever et al. identified a cathepsin L cleavage site at positions 355–360 in the middle domain (Fig. 1b). Actin network is then reorganized in renal podocytes leading to filtration impairment and proteinuria [24]. It remains to be determined whether such proteolytic regulation occurs only in pathological context and in other tissues. Finally, it was demonstrated that Ca2+ inhibits DNM2 GTPase activity (IC50 = 150 µM) and receptor-mediated endocytosis in Hela cells [25]. This may have physiological importance in excitable cells like neurons and muscle fibers.
It is still largely unknown how DMN2 expression is regulated. In rat, DNM2 is up-regulated during normal pancreatic development after birth [26] but not in the liver [8] or the brain [27]. In mouse, treatment with opioid agonist results in increased DNM2 protein content in the spinal cord [28] whereas opioid antagonist decreases DNM2 abundance [29]. These changes in the level of DNM2 expression are inversely correlated with opioid receptor density at the plasma membrane, suggestive of feed-back regulation.
DNM2 function
Endocytosis
DNM2 has been implicated in the formation of clathrin-coated pits (Fig. 2) [17]. In the cytosol, DNM2 forms a complex with sorting nexin 9 (SNX9) and fructose-1,6-bisphosphate aldolase [30]. Phosphorylation of SNX9 releases aldolase from the SNX9–DNM2 complex which is now competent for membrane targeting [30, 31]. DNM2 anchorage to the membrane occurs via interaction with PI4,5P2 membrane phosphoinositide [32] and BAR domain proteins, amphiphysin 1, amphiphysin 2, and SNX9 (Table 1) in curved sites of the membranes. DNM2 forms an oligomer helical structure around the neck of the nascent vesicles [17], and GTP hydrolysis is associated with the release of the vesicles. Interestingly, DNM2 co-localizes with clathrin before and during the internalization of the coated vesicle [6] suggesting that DNM2 plays also a role during the maturation of clathrin-coated pits [33].

DNM2 cellular functions. Representation of the multiple cellular localizations reported for DNM2 (in red)
EE early endosome, LE late endosome
DNM2 is also involved in clathrin-independent endocytosis by its participation in the formation of the phagosomes and caveolae [34, 35]. Predescu et al. described a protein complex, including DNM2, intersectin, and SNAP-23 that was important for the internalization of caveolae [36]. In caveolae, DNM2 also interacts with endothelial nitric-oxide synthase (eNOS) in bovine aortic endothelial cells [37] where DNM2 may regulate eNOS activation and the NO signaling cascade [37, 38]. DNM2 also participates in coat-independent endocytosis processes, i.e., micropinocytosis and macropinocytosis, by which fluid droplets and specific membrane components are internalized [39, 40].
Intracellular membrane trafficking
DNM2 is targeted to the Golgi apparatus where it is predominantly localized in the trans-Golgi network (TGN) [41]. Anti-DNM2 antibody injection and over-expression of DNM2 mutants impair vesicle formation from the TGN [42, 43]. Association of DNM2 with cortactin and syndapin 2 is required for trafficking of nascent vesicles from the TGN [44, 45]. DNM2 is also found at the clathrin-coated buds of early endosomes [46] and in late endosomes in Hela cells, located to the tubulo-vesicular appendices [47]. In these two cases, interfering DNM2 mutant impairs the recycling of components from the endosomal system towards the plasma membrane or TGN [46, 47]. These data highlight the role of DNM2 in the secretory pathway and in the sorting of cell components from the Golgi apparatus and endosomal compartment.
Exocytosis
DNM2 may participate in endocytosis–exocytosis coupling as suggested in mouse pancreatic β-cells [48]. However, a role for DNM2 in exocytosis alone has been reported. During cell-mediated killing by natural killer (NK) cells, DNM2 co-localizes with lytic granules after NK cell activation and is required for fusion of the granules with the plasma membrane [49]. Similarly in macrophages, focal exocytosis is blocked after anti-DNM2 antibody microinjection [50], and DNM2 GTPase activity regulates the fusion of secretory vesicles at the plasma membrane [51]. Further studies will be necessary to precisely identify the molecular role played by DNM2 in the exocytosis machinery.
Actin network
Actin-based dynamic processes are crucial for late-stage endocytosis and vesicle formation, and DNM2 interacts with the actin-binding proteins Abp1 (actin-binding protein 1) [52] and cortactin [53, 54]. Abp1 is an Src kinase which provides a physical bridge between the endocytosis machinery and the cortical actin network, and cortactin is a component of the clathrin-mediated endocytosis machinery. However, interaction between DNM2 and the actin cytoskeleton may have another cytoskeletal role such as in the formation of membrane tubules and protrusions. Furthermore, a recent study showed the crucial function played by the DNM2-cortactin complex in the global organization and remodeling of the actomyosin cytoskeleton [55]. In addition, DNM2 is present in cortical ruffles and lamellipodia, both important in cell migration [10, 53]. The supramolecular complex including DNM2, cortactin, and Arp2/3 mediates the reorganization of actin allowing lamellipodia formation at the leading edge of migrating cells [56]. Disruption of DNM2 functions by DNM2-K44A mutant or small interfering RNA (siRNA) inhibits the formation of lamellipodia [57]. Similarly, under PDGF stimulation, DNM2 is concentrated within the leading ruffles of migrating fibroblasts where it co-localizes with cortactin [53]. To allow cell migration, DNM2 participates in disassembly of focal adhesions, as well as β-integrin internalization at the rear of the cell [58, 59]. Additionally, DNM2 is enriched in specialized membrane protrusions such as podosomes and invadipodia. Podosomes represent attachment sites between cells and substratum [60], and invadipodia are focalized matrix degradation sites [61]. Inhibition of DNM2 diminishes the amount of such structures [61]. It has also been shown that DNM2 regulates the formation of actin-stress fibers by interaction with the cell surface heparin sulfate proteoglycan syndecan-4 [62]. Expression of DNM2-mutant, truncated for the PRD domain mediating interaction with cortactin, increases the number of actin-stress fibers, which is associated with abnormal cell shape [53].
Microtubule network and MTOC
DNM2 interacts with microtubules [17, 63], and the binding region was located to the PRD [5, 64]. It was shown that down-regulation of DNM2 by siRNA increases the amount of acetylated tubulin, a more stable form of tubulin in microtubules and reduces their growing capacity [63], suggesting that DNM2 may regulate the polymerization–depolymerization equilibrium of microtubules. Through its interaction with microtubules, DNM2 appears involved in Golgi apparatus cohesion [63]. Moreover, DNM2 has been identified as a component of the centrosome, the main microtubule organizing center (MTOC), where it binds to γ-tubulin [65]. The centrosome consists of a pair of centrioles embedded in a filamentous pericentriolar matrix, where γ-tubulin is essential for microtubule nucleation. The function played by DNM2 at the centrosome is still unknown, but DNM2 silencing by siRNA suggests a role in centrosome splitting [65]. Likewise, participation of DNM2 in all the phases of mitosis has also been reported. DNM2 is detected in the two MTOC during early prophase, along the mitotic spindle during metaphase and in the spindle midzone region during anaphase and early telophase [66]. Thereafter, DNM2 is accumulated at the intracellular bridge where the final separation occurs. The time required for separation of the two daughter cells is longer in DNM2 knock-out cells [40]. Taken together, these data suggest that DNM2 may regulate microtubule-dependent processes by acting on microtubule dynamics and organization.
Apoptosis
In order to establish a stable Hela cell line over-expressing DNM2 isoform 2, Fish et al. have reported a significant cell toxicity in dividing cells [67]. The cytotoxicity occurred via induction of apoptosis by a p53-dependent mechanism. Similar results were gained in vascular smooth muscle cells [68]. The capacity to trigger apoptosis appears DNM2-specific as DNM1 over-expression does not induce apoptosis [67]. The GTPase domain of DNM2 is crucial to induce apoptosis [69]. In addition, a point mutation (p.I684K) in the DNM2 GED enhances the apoptosis induction by the wild-type DNM2 suggesting that GED negatively regulates this DNM2 function [69]. Mitochondria are key players in apoptosis and, interestingly, DNM2 has been detected in isolated mitochondria from bovine lymphoblastoid BL-3 cells [70]. However, to our knowledge, such localization has not been reported in other cell lines or tissues. DNM2 also regulates the apoptosis-inducing Fas–Fas ligand pathway by facilitating the transport of Fas from the trans-Golgi network to the plasma membrane [71].
Specific functions of DNM2 isoforms
In a cultured rat epithelial cell line (clone 9), both DNM2 isoforms 1 and 3 show punctuate labeling of clathrin heavy chain-positive or heavy chain-negative structures, but only isoform 1, with the GEIL sequence in the MD, appears located to the Golgi apparatus [10]. These data suggest a role for the GEIL sequence in targeting to the Golgi apparatus. However, cell-type specificity probably exists, as isoforms without the GEIL sequence were also shown to be targeted to the Golgi apparatus in MDCK cells [43], 3T3L1 adipocytes [72], and fibroblastoid-like cells derived from mouse embryonic stem cells [40]. Nevertheless, this possible differential localization argues for distinct functions. Indeed, in clone 9 cells, the K44A mutants of isoforms 2 and 4 are able to inhibit fluid-phase endocytosis, whereas the mutant forms of isoforms 1 and 3 do not [39] and are also more potent inhibitors of clathrin-mediated endocytosis. Similarly, in a hepatocyte cell line, the K44A-isoform 1 inhibits caveolae-dependent internalization, but not the other K44A mutant isoforms [73]. In fibroblastoid-like cells derived from mouse embryonic stem cells, isoforms 2 and 4 are the most efficient at rescuing export from the Golgi in DNM2 knock-out cells [40]. Altogether, these data suggest a preferential involvement of isoforms 1 and 3 in clathrin- and caveolae-dependent endocytosis, whereas isoforms 2 and 4 participate in uncoated endocytosis and trafficking from the Golgi apparatus. However, cell-type specificity also occurs as the four isoforms exhibit a similar subcellular distribution in 3T3L1 adipocytes, and dominant negative mutants of each isoform similarly affect basal and insulin-stimulated GLUT4 trafficking [72].
DNM2 and human diseases
Mutations in the DNM2 gene cause rare forms of the Charcot–Marie-Tooth peripheral neuropathy (CMT) [2, 74–77] and autosomal dominant centronuclear myopathy (CNM) [3, 11, 78–80]. The 19 reported heterozygous mutations affect only the MD, the PH domain, and the GED (Fig. 1b). DNM2-related CNM is a slowly progressive congenital myopathy characterized by frequent centrally located nuclei in muscle fibers. The most common clinical features are delayed motor milestones, facial and generalized muscle weakness, ptosis, and ophthalmoplegia [81]. Nevertheless, the severity of DNM2-CNM is variable, ranging from severe neonatal to mild late-onset forms. DNM2-CMT is a peripheral neuropathy characterized by progressive muscle weakness and atrophy. DNM2 mutations can cause axonal CMT (CMT2) and dominant intermediate CMT (DI-CMT-B). In some CMT patients, neuropathy is associated with neutropenia [2, 75, 77] but this association has not been described in DNM2-CNM patients. Clinical overlap could exist in some patients [81], but the majority of patients are affected by a tissue-specific disorder. No clear genotype–phenotype relationship can be generated, except for the de novo mutations located in the C-terminal part of the PH domain, which are all associated with a severe neonatal CNM phenotype [98]. In these patients, the phenotype progressively improves, suggesting compensatory mechanisms.
More recently, the DNM2 gene has been described as a susceptibility gene for late-onset Alzheimer disease [82], and DNM2 expression was found to be decreased in the brains of these patients [83]. Cognitive impairments have been reported in some CNM patients harboring the p.E368Q [78], p.R465W [84; Family 1], and p.R369Q [84; Families 2 and 3] DNM2 mutations. Future studies will be necessary to determine the prevalence of central nervous system involvement in DNM2-related diseases.
Pathophysiological hypotheses
The DNM2 mutations identified so far in CNM and CMT are heterozygous missense mutations or small deletions (Fig. 1). We have shown that DNM2 transcript, protein expression, and localization are normal in fibroblasts from CNM patients [3, 11]. These data are in agreement with DNM2 mutants having a dominant negative effect, resulting in a loss of function of DNM2 in endocytosis [11] or in microtubule-related functions [63] (see below).
Membrane trafficking and signaling pathway hypothesis
In addition to the DNM2 mutations in autosomal dominant CNM, mutations in the BIN1 gene encoding amphiphysin 2, a partner of DNM2 in the endocytic process, cause the autosomal recessive form of the disease [85]. This suggests that endocytic impairment is a potential pathomechanism of autosomal CNM. Indeed, impairment of clathrin-mediated endocytosis was reported in cultured cells expressing CNM- or CMT-DNM2 mutants [2, 11, 63]. Among these studies, one CMT-mutant was unable to block the uptake of transferrin, a marker of clathrin-mediated receptor endocytosis [63]. Nevertheless, the transferrin-containing compartment was not located to the perinuclear region after 30 min of incubation showing that its intracellular trafficking was impaired by the CMT-mutant. The crucial question which remains to be explored is how a defect in endocytosis can alter the cell function, especially in a tissue-specific manner. On one hand, inhibition of DNM2-dependent trafficking may lead to a decrease in receptor stimulated signaling as shown for the MAPK ERK1/2 pathway [11]. On the other hand, DNM2 mutations may lead to a prolonged half-life of various proteins at the cell surface due to a defect in protein removal, as shown for the GLUT4 glucose transporter [72]. A deregulation of glucose transport in patients with DNM2 mutations could have a strong impact on muscle fibers given their high glucose consumption.
To date, the impact of disease-associated DNM2 mutants on other DNM2-dependent membrane trafficking processes, especially in endosomal and Golgi pathways, has not been studied. We cannot exclude a participation of these pathways in the pathomechanisms of DNM2-related disorders.
Cytoskeleton impairment and its putative role on nuclear positioning
In DNM2-CNM, the majority of patients harbor a mutation in the MD, which is essential for the centrosomal localization of DNM2 and for its interaction with γ-tubulin [65]. Previous results in skin fibroblasts indicate that transfected GFP-DNM2-mutants fail to correctly target to the centrosome, suggesting that DNM2 mutations might cause CNM by interfering with centrosomal functions [3]. In addition, CMT-related DNM2 mutants can disorganize the microtubule cytoskeleton [2], and one particular CMT-mutant was shown to impair microtubule-dependent membrane transport [63]. In addition to their roles in intracellular trafficking, the microtubule and actin networks regulate cellular architecture including nuclear positioning [86, 87]. Thus, cytoskeletal impairment may play a role in the abnormal central location of the nuclei in the muscle fibers in CNM. In CMT, DNM2 mutations could also induce a destabilization of the microtubule network leading to abnormal axonal transport and protein trafficking, a pathophysiological mechanism described previously in various forms of CMT [88].
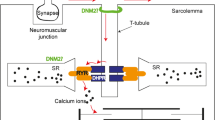
T-tubule hypothesis in CNM
The X-linked recessive form of CNM (also called XLMTM for X-linked myotubular myopathy) is due to mutations in the MTM1 gene encoding the myotubularin, and the autosomal recessive and dominant CNMs result from mutations of amphiphysin 2 and DNM2, respectively [89]. The muscle specific isoform of amphiphysin 2 is concentrated at T-tubules in mouse and drosophila and is involved in the organization of this plasma membrane invagination acting in excitation–contraction coupling [90, 91]. Myotubularin is also located to the T-tubules in mouse [92] and zebrafish [93], and knock-down of myotubularin in these species leads to disorganization of the T-tubule system, reduction in Ca2+ release from the sarcoplasmic reticulum, and defect in excitation–contraction coupling [93, 94]. In addition, abnormal localization of T-tubule markers was shown in muscle biopsies from BIN1-CNM and MTM1-CNM patients [85, 93], suggesting that an excitation–contraction coupling impairment due to T-tubule dysfunction could be a common pathomechanism leading to muscle weakness in CNMs. Future studies are necessary to explore this hypothesis in the DNM2-CNM in which no morphological abnormalities of the T-tubule system have been reported to date.
Concluding remarks and open questions
Given the numerous distinct functions in which the ubiquitously expressed DNM2 is involved, the identification of pathophysiological mechanisms will be a challenge. The phenotypes encountered in CNM and CMT patients could be due to impairment of the various functions of the protein. To date, there is no explanation for the tissue-specific impact of the DNM2-mutations in human diseases. DNM2 is engaged in numerous protein–protein interactions (Table 1), but these interactions in muscles and nerves are largely unexplored. Another unresolved question is whether each particular mutation can similarly affect the functions of the four DNM2 isoforms. Finally, whereas some data emerge on the impact of disease-related DNM2 mutations on the microtubule network, their impact on the actin cytoskeleton is totally unknown. Future developments of animal models will certainly be useful to better determine the main functions of DNM2 in vivo, especially in skeletal muscle and nerves where membrane trafficking displays unique cell length dependent characteristics.
Abbreviations
- PI4,5P2:
-
phophatidylinositol 4,5-bisphosphate
- PI3,4,5P3:
-
phophatidylinositol 3,4,5-triphosphate
- PI3,4P2:
-
phophatidylinositol 3,4-bisphosphate
- PI4P:
-
phophatidylinositol 4-monophosphate
- PI3P:
-
phophatidylinositol 3-monophosphate
- LPA:
-
lysophosphatidic acid
- GLUT4:
-
glucose transporter 4
- TGN:
-
trans-Golgi network
- BAR:
-
Bin1/Amphiphysin/RVS167
References
Heymann JA, Hinshaw JE (2009) Dynamins at a glance. J Cell Sci 122:3427–3431
Zuchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, De Jonghe P, Merory J, Oliveira SA, Speer MC, Stenger JE, Walizada G, Zhu D, Pericak-Vance MA, Nicholson G, Timmerman V, Vance JM (2005) Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot–Marie-Tooth disease. Nat Genet 37:289–294
Bitoun M, Maugenre S, Jeannet PY, Lacène E, Ferrer X, Laforêt P, Martin JJ, Laporte J, Lochmuller H, Beggs AH, Fardeau M, Eymard B, Romero NB, Guicheney P (2005) Mutations in dynamin 2 cause dominant centronuclear myopathy. Nature Genet 37:1207–1209
Sontag JM, Fykse EM, Ushkaryov Y, Liu JP, Robinson PJ, Sudhof TC (1994) Differential expression and regulation of multiple dynamins. J Biol Chem 269:4547–4554
Lin HC, Barylko B, Achiriloaie M, Albanesi JP (1997) Phosphatidylinositol (4, 5)-bisphosphate-dependent activation of dynamins I and II lacking the proline/arginine-rich domains. J Biol Chem 272:25999–26004
Rappoport JZ, Simon SM (2003) Real-time analysis of clathrin-mediated endocytosis during cell migration. J Cell Sci 116:847–855
Artalejo CR, Elhamdani A, Palfrey HC (2002) Sustained stimulation shifts the mechanism of endocytosis from dynamin-1-dependent rapid endocytosis to clathrin- and dynamin-2-mediated slow endocytosis in chromaffin cells. Proc Natl Acad Sci USA 99:6358–6363
Cook TA, Urrutia R, McNiven MA (1994) Identification of dynamin 2, an isoform ubiquitously expressed in rat tissues. Proc Natl Acad Sci USA 91:644–648
Diatloff-Zito C, Gordon AJ, Duchaud E, Merlin G (1995) Isolation of an ubiquitously expressed cDNA encoding human dynamin II, a member of the large GTP-binding protein family. Gene 163:301–306
Cao H, Garcia F, McNiven MA (1998) Differential distribution of dynamin isoforms in mammalian cells. Mol Biol Cell 9:2595–2609
Bitoun M, Durieux AC, Prudhon B, Bevilacqua JA, Herledan A, Sakanyan V, Urtizberea A, Cartier L, Romero NB, Guicheney P (2009) Dynamin 2 mutations associated with human diseases impair clathrin-mediated receptor endocytosis. Hum Mutat 30:1419–1427
Smirnova E, Shurland DL, Newman-Smith ED, Pishvaee B, van der Bliek AM (1999) A model for dynamin self-assembly based on binding between three different protein domains. J Biol Chem 274:14942–14947
Chen YJ, Zhang P, Egelman EH, Hinshaw JE (2004) The stalk region of dynamin drives the constriction of dynamin tubes. Nat Struct Mol Biol 11:574–575
Dong J, Misselwitz R, Welfle H, Westermann P (2000) Expression and purification of dynamin II domains and initial studies on structure and function. Protein Expr Purif 20:314–323
Klein DE, Lee A, Frank DW, Marks MS, Lemmon MA (1998) The pleckstrin homology domains of dynamin isoforms require oligomerization for high affinity phosphoinositide binding. J Biol Chem 273:27725–27733
Sever S, Muhlberg AB, Schmid SL (1999) Impairment of dynamin’s GAP domain stimulates receptor-mediated endocytosis. Nature 398:481–486
Warnock DE, Baba T, Schmid SL (1997) Ubiquitously expressed dynamin-II has a higher intrinsic GTPase activity and a greater propensity for self-assembly than neuronal dynamin-I. Mol Biol Cell 8:2553–2562
Eccleston JF, Binns DD, Davis CT, Albanesi JP, Jameson DM (2002) Oligomerization and kinetic mechanism of the dynamin GTPase. Eur Biophys J 31:275–282
Soulet F, Yarar D, Leonard M, Schmid SL (2005) SNX9 regulates dynamin assembly and is required for efficient clathrin-mediated endocytosis. Mol Biol Cell 16:2058–2067
Solomaha E, Palfrey HC (2005) Conformational changes in dynamin on GTP binding and oligomerization reported by intrinsic and extrinsic fluorescence. Biochem J 391:601–611
Shajahan AN, Timblin BK, Sandoval R, Tiruppathi C, Malik AB, Minshall RD (2004) Role of Src-induced dynamin-2 phosphorylation in caveolae-mediated endocytosis in endothelial cells. J Biol Chem 279:20392–20400
Efendiev R, Yudowski GA, Zwiller J, Leibiger B, Katz AI, Berggren PO, Pedemonte CH, Leibiger IB, Bertorello AM (2002) Relevance of dopamine signals anchoring dynamin-2 to the plasma membrane during Na+, K+-ATPase endocytosis. J Biol Chem 277:44108–44114
Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V (2007) Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. J Cell Sci 120:492–501
Sever S, Altintas MM, Nankoe SR, Moller CC, Ko D, Wei C, Henderson J, del Re EC, Hsing L, Erickson A, Cohen CD, Kretzler M, Kerjaschki D, Rudensky A, Nikolic B, Reiser J (2007) Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest 117:2095–2104
Cousin MA, Robinson PJ (2000) Ca(2+) influx inhibits dynamin and arrests synaptic vesicle endocytosis at the active zone. J Neurosci 20:949–957
Cook TA, Mesa KJ, Gebelein BA, Urrutia RA (1996) Upregulation of dynamin II expression during the acquisition of a mature pancreatic acinar cell phenotype. J Histochem Cytochem 44:1373–1378
Cook T, Mesa K, Urrutia R (1996) Three dynamin-encoding genes are differentially expressed in developing rat brain. J Neurochem 67:927–931
Zhang Q, Purohit V, Yoburn BC (2005) Continuous opioid agonist treatment dose-dependently regulates mu-opioid receptors and dynamin-2 in mouse spinal cord. Synapse 56:123–128
Rajashekara V, Patel CN, Patel K, Purohit V, Yoburn BC (2003) Chronic opioid antagonist treatment dose-dependently regulates mu-opioid receptors and trafficking proteins in vivo. Pharmacol Biochem Behav 75:909–913
Lundmark R, Carlsson SR (2004) Regulated membrane recruitment of dynamin-2 mediated by sorting nexin 9. J Biol Chem 279:42694–42702
Lundmark R, Carlsson SR (2005) Expression and properties of sorting nexin 9 in dynamin-mediated endocytosis. Methods Enzymol 404:545–556
Zoncu R, Perera RM, Sebastian R, Nakatsu F, Chen H, Balla T, Ayala G, Toomre D, De Camilli PV (2007) Loss of endocytic clathrin-coated pits upon acute depletion of phosphatidylinositol 4, 5-bisphosphate. Proc Natl Acad Sci USA 104:3793–3798
Loerke D, Mettlen M, Yarar D, Jaqaman K, Jaqaman H, Danuser G, Schmid SL (2009) Cargo and dynamin regulate clathrin-coated pit maturation. PLoS Biol 7:e57
Gold ES, Underhill DM, Morrissette NS, Guo J, McNiven MA, Aderem A (1999) Dynamin 2 is required for phagocytosis in macrophages. J Exp Med 190:1849–1856
Henley JR, Krueger EW, Oswald BJ, McNiven MA (1998) Dynamin-mediated internalization of caveolae. J Cell Biol 141:85–99
Predescu SA, Predescu DN, Timblin BK, Stan RV, Malik AB (2003) Intersectin regulates fission and internalization of caveolae in endothelial cells. Mol Biol Cell 14:4997–5010
Cao S, Yao J, McCabe TJ, Yao Q, Katusic ZS, Sessa WC, Shah V (2001) Direct interaction between endothelial nitric-oxide synthase and dynamin-2. Implications for nitric-oxide synthase function. J Biol Chem 276:14249–14256
Sanchez FA, Rana R, Kim DD, Iwahashi T, Zheng R, Lal BK, Gordon DM, Meininger CJ, Duran WN (2009) Internalization of eNOS and NO delivery to subcellular targets determine agonist-induced hyperpermeability. Proc Natl Acad Sci USA 106:6849–6853
Cao H, Chen J, Awoniyi M, Henley JR, McNiven MA (2007) Dynamin 2 mediates fluid-phase micropinocytosis in epithelial cells. J Cell Sci 120:4167–4177
Liu YW, Surka MC, Schroeter T, Lukiyanchuk V, Schmid SL (2008) Isoform and splice-variant specific functions of dynamin-2 revealed by analysis of conditional knock-out cells. Mol Biol Cell 19:5347–5359
Maier O, Knoblich M, Westermann P (1996) Dynamin II binds to the trans-Golgi network. Biochem Biophys Res Commun 223:229–233
Jones SM, Howell KE, Henley JR, Cao H, McNiven MA (1998) Role of dynamin in the formation of transport vesicles from the trans-Golgi network. Science 279:573–577
Kreitzer G, Marmorstein A, Okamoto P, Vallee R, Rodriguez-Boulan E (2000) Kinesin and dynamin are required for post-Golgi transport of a plasma-membrane protein. Nat Cell Biol 2:125–127
Cao H, Weller S, Orth JD, Chen J, Huang B, Chen JL, Stamnes M, McNiven MA (2005) Actin and Arf1-dependent recruitment of a cortactin–dynamin complex to the Golgi regulates post-Golgi transport. Nat Cell Biol 7:483–492
Kessels MM, Dong J, Leibig W, Westermann P, Qualmann B (2006) Complexes of syndapin II with dynamin II promote vesicle formation at the trans-Golgi network. J Cell Sci 119:1504–1516
van Dam EM, Stoorvogel W (2002) Dynamin-dependent transferrin receptor recycling by endosome-derived clathrin-coated vesicles. Mol Biol Cell 13:169–182
Nicoziani P, Vilhardt F, Llorente A, Hilout L, Courtoy PJ, Sandvig K, van Deurs B (2000) Role for dynamin in late endosome dynamics and trafficking of the cation-independent mannose 6-phosphate receptor. Mol Biol Cell 11:481–495
Min L, Leung YM, Tomas A, Watson RT, Gaisano HY, Halban PA, Pessin JE, Hou JC (2007) Dynamin is functionally coupled to insulin granule exocytosis. J Biol Chem 282:33530–33536
Arneson LN, Segovis CM, Gomez TS, Schoon RA, Dick CJ, Lou Z, Billadeau DD, Leibson PJ (2008) Dynamin 2 regulates granule exocytosis during NK cell-mediated cytotoxicity. J Immunol 181:6995–7001
Di A, Nelson DJ, Bindokas V, Brown ME, Libunao F, Palfrey HC (2003) Dynamin regulates focal exocytosis in phagocytosing macrophages. Mol Biol Cell 14:2016–2028
Jaiswal JK, Rivera VM, Simon SM (2009) Exocytosis of post-Golgi vesicles is regulated by components of the endocytic machinery. Cell 137:1308–1319
Kessels MM, Engqvist-Goldstein AE, Drubin DG, Qualmann B (2001) Mammalian Abp1, a signal-responsive F-actin-binding protein, links the actin cytoskeleton to endocytosis via the GTPase dynamin. J Cell Biol 153:351–366
McNiven MA, Kim L, Krueger EW, Orth JD, Cao H, Wong TW (2000) Regulated interactions between dynamin and the actin-binding protein cortactin modulate cell shape. J Cell Biol 151:187–198
Schafer DA, Weed SA, Binns D, Karginov AV, Parsons JT, Cooper JA (2002) Dynamin2 and cortactin regulate actin assembly and filament organization. Curr Biol 12:1852–1857
Mooren OL, Kotova TI, Moore AJ, Schafer DA (2009) Dynamin2 GTPase and cortactin remodel actin filaments. J Biol Chem 284:23995–24005
Krueger EW, Orth JD, Cao H, McNiven MA (2003) A dynamin-cortactin-Arp2/3 complex mediates actin reorganization in growth factor-stimulated cells. Mol Biol Cell 14:1085–1096
Schlunck G, Damke H, Kiosses WB, Rusk N, Symons MH, Waterman-Storer CM, Schmid SL, Schwartz MA (2004) Modulation of Rac localization and function by dynamin. Mol Biol Cell 15:256–267
Ezratty EJ, Partridge MA, Gundersen GG (2005) Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat Cell Biol 7:581–590
Vassilieva EV, Gerner-Smidt K, Ivanov AI, Nusrat A (2008) Lipid rafts mediate internalization of beta1-integrin in migrating intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 295:G965–G976
Ochoa GC, Slepnev VI, Neff L, Ringstad N, Takei K, Daniell L, Kim W, Cao H, McNiven M, Baron et a (2000) A functional link between dynamin and the actin cytoskeleton at podosomes. J Cell Biol 150:377–389
Baldassarre M, Pompeo A, Beznoussenko G, Castaldi C, Cortellino S, McNiven MA, Luini A, Buccione R (2003) Dynamin participates in focal extracellular matrix degradation by invasive cells. Mol Biol Cell 14:1074–1084
Yoo J, Jeong MJ, Cho HJ, Oh ES, Han MY (2005) Dynamin II interacts with syndecan-4, a regulator of focal adhesion and stress-fiber formation. Biochem Biophys Res Commun 328:424–431
Tanabe K, Takei K (2009) Dynamic instability of microtubules requires dynamin 2 and is impaired in a Charcot–Marie-Tooth mutant. J Cell Biol 185:939–948
Hamao K, Morita M, Hosoya H (2009) New function of the proline rich domain in dynamin-2 to negatively regulate its interaction with microtubules in mammalian cells. Exp Cell Res 315:1336–1345
Thompson HM, Cao H, Chen J, Euteneuer U, McNiven MA (2004) Dynamin 2 binds gamma-tubulin and participates in centrosome cohesion. Nat Cell Biol 6:335–342
Thompson HM, Skop AR, Euteneuer U, Meyer BJ, McNiven MA (2002) The large GTPase dynamin associates with the spindle midzone and is required for cytokinesis. Curr Biol 12:2111–2117
Fish KN, Schmid SL, Damke H (2000) Evidence that dynamin-2 functions as a signal-transducing GTPase. J Cell Biol 150:145–154
Kashiwakura Y, Watanabe M, Kusumi N, Sumiyoshi K, Nasu Y, Yamada H, Sawamura T, Kumon H, Takei K, Daida H (2004) Dynamin-2 regulates oxidized low-density lipoprotein-induced apoptosis of vascular smooth muscle cell. Circulation 110:3329–3334
Soulet F, Schmid SL, Damke H (2006) Domain requirements for an endocytosis-independent, isoform-specific function of dynamin-2. Exp Cell Res 312:3539–3545
Atapattu DN, Albrecht RM, McClenahan DJ, Czuprynski CJ (2008) Dynamin-2-dependent targeting of Mannheimia haemolytica leukotoxin to mitochondrial cyclophilin D in bovine lymphoblastoid cells. Infect Immun 76:5357–5365
Ivanov VN, Ronai Z, Hei TK (2006) Opposite roles of FAP-1 and dynamin in the regulation of Fas (CD95) translocation to the cell surface and susceptibility to Fas ligand-mediated apoptosis. J Biol Chem 281:1840–1852
Kao AW, Yang C, Pessin JE (2000) Functional comparison of the role of dynamin 2 splice variants on GLUT-4 endocytosis in 3T3L1 adipocytes. Am J Physiol Endoc M 278:E825–E831
Yao Q, Chen J, Cao H, Orth JD, McCaffery JM, Stan RV, McNiven MA (2005) Caveolin-1 interacts directly with dynamin-2. J Mol Biol 348:491–501
Fabrizi GM, Ferrarini M, Cavallaro T, Cabrini I, Cerini R, Bertolasi L, Rizzuto N (2007) Two novel mutations in dynamin-2 cause axonal Charcot–Marie-Tooth disease. Neurology 69:291–295
Bitoun M, Stojkovic T, Prudhon B, Maurage CA, Latour P, Vermersch P, Guicheney P (2008) A novel mutation in the dynamin 2 gene in a Charcot–Marie-Tooth type 2 patient: clinical and pathological findings. Neuromuscul Disord 18:334–338
Gallardo E, Claeys KG, Nelis E, Garcia A, Canga A, Combarros O, Timmerman V, De Jonghe P, Berciano J (2008) Magnetic resonance imaging findings of leg musculature in Charcot–Marie-Tooth disease type 2 due to dynamin 2 mutation. J Neurol 255:986–992
Claeys KG, Zuchner S, Kennerson M, Berciano J, Garcia A, Verhoeven K, Storey E, Merory JR, Bienfait HM, Lammens M, Nelis E, Baets J, De Vriendt E, Berneman ZN, De Veuster I, Vance JM, Nicholson G, Timmerman V, De Jonghe P (2009) Phenotypic spectrum of dynamin 2 mutations in Charcot–Marie-Tooth neuropathy. Brain 132:1741–1752
Echaniz-Laguna A, Nicot AS, Carre S, Franques J, Tranchant C, Dondaine N, Biancalana V, Mandel JL, Laporte J (2007) Subtle central and peripheral nervous system abnormalities in a family with centronuclear myopathy and a novel dynamin 2 gene mutation. Neuromuscul Disord 17:955–959
Bitoun M, Bevilacqua JA, Prudhon B, Maugenre S, Taratuto AL, Monges S, Lubieniecki F, Cances C, Uro-Coste E, Mayer M, Fardeau M, Romero NB, Guicheney P (2007) Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann Neurol 62:666–670
Bitoun M, Bevilacqua JA, Eymard B, Prudhon B, Fardeau M, Guicheney P, Romero NB (2009) A new centronuclear myopathy phenotype due to a novel dynamin 2 mutation. Neurology 72:93–95
Fischer D, Herasse M, Bitoun M, Barragán-Campos HM, Chiras J, Laforêt P, Fardeau M, Eymard B, Guicheney P, Romero NB (2006) Characterization of the muscle involvement in dynamin 2- related centronuclear myopathy. Brain 129:1463–1469
Aidaralieva NJ, Kamino K, Kimura R, Yamamoto M, Morihara T, Kazui H, Hashimoto R, Tanaka T, Kudo T, Kida T, Okuda J, Uema T, Yamagata H, Miki T, Akatsu H, Kosaka K, Takeda M (2008) Dynamin 2 gene is a novel susceptibility gene for late-onset Alzheimer disease in non-APOE-epsilon4 carriers. J Hum Genet 53:296–302
Kamagata E, Kudo T, Kimura R, Tanimukai H, Morihara T, Sadik MG, Kamino K, Takeda M (2009) Decrease of dynamin 2 levels in late-onset Alzheimer’s disease alters Abeta metabolism. Biochem Biophys Res Commun 379:691–695
Jeannet PY, Bassez G, Eymard B, Laforet P, Urtizberea JA, Rouche A, Guicheney P, Fardeau M, Romero NB (2004) Clinical and histologic findings in autosomal centronuclear myopathy. Neurology 62:1484–1490
Nicot AS, Toussaint A, Tosch V, Kretz C, Wallgren-Pettersson C, Iwarsson E, Kingston H, Garnier JM, Biancalana V, Oldfors A, Mandel JL, Laporte J (2007) Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet 39:1134–1139
Morris NR (2003) Nuclear positioning: the means is at the ends. Curr Opin Cell Biol 15:54–59
Starr DA, Han M (2003) ANChors away: an actin based mechanism of nuclear positioning. J Cell Sci 116:211–216
Shy ME (2004) Charcot–Marie-Tooth disease: an update. Curr Opin Neurol 17:579–585
Jungbluth H, Wallgren-Pettersson C, Laporte J (2008) Centronuclear (myotubular) myopathy. Orphanet J Rare Dis 3:26–38
Lee E, Marcucci M, Daniell L, Pypaert M, Weisz OA, Ochoa GC, Farsad K, Wenk MR, De Camilli P (2002) Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297:1193–1196
Razzaq A, Robinson IM, McMahon HT, Skepper JN, Su Y, Zelhof AC, Jackson AP, Gay NJ, O’Kane CJ (2001) Amphiphysin is necessary for organization of the excitation-contraction coupling machinery of muscles, but not for synaptic vesicle endocytosis in drosophila. Genes Dev 15:2967–2979
Buj-Bello A, Fougerousse F, Schwab Y, Messaddeq N, Spehner D, Pierson CR, Durand M, Kretz C, Danos O, Douar AM, Beggs AH, Schultz P, Montus M, Denefle P, Mandel JL (2008) AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum Mol Genet 17:2132–2143
Dowling JJ, Vreede AP, Low SE, Gibbs EM, Kuwada JY, Bonnemann CG, Feldman EL (2009) Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet 5:e1000372
Al-Qusairi L, Weiss N, Toussaint A, Berbey C, Messaddeq N, Kretz C, Sanoudou D, Beggs AH, Allard B, Mandel JL, Laporte J, Jacquemond V, Buj-Bello A (2009) T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc Natl Acad Sci USA 106:18763–18768
Onabajo OO, Seeley MK, Kale A, Qualmann B, Kessels M, Han J, Tan TH, Song W (2008) Actin-binding protein 1 regulates B cell receptor-mediated antigen processing and presentation in response to B cell receptor activation. J Immunol 180:6685–6695
Nakanishi A, Abe T, Watanabe M, Takei K, Yamada H (2008) Dynamin 2 cooperates with amphiphysin 1 in phagocytosis in sertoli cells. Acta Med Okayama 62:385–391
Kojima C, Hashimoto A, Yabuta I, Hirose M, Hashimoto S, Kanaho Y, Sumimoto H, Ikegami T, Sabe H (2004) Regulation of Bin1 SH3 domain binding by phosphoinositides. EMBO J 23:4413–4422
Turpin E, Russo-Marie F, Dubois T, de Paillerets C, Alfsen A, Bomsel M (1998) In adrenocortical tissue, annexins II and VI are attached to clathrin coated vesicles in a calcium-independent manner. Biochim Biophys Acta 1402:115–130
Lu HA, Sun TX, Matsuzaki T, Yi XH, Eswara J, Bouley R, McKee M, Brown D (2007) Heat shock protein 70 interacts with aquaporin-2 and regulates its trafficking. J Biol Chem 282:28721–28732
Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF (2006) Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 52:445–459
Tosoni D, Cestra G (2009) CAP (Cbl associated protein) regulates receptor-mediated endocytosis. FEBS Lett 583:293–300
Lie PP, Xia W, Wang CQ, Mruk DD, Yan HH, Wong CH, Lee WM, Cheng CY (2006) Dynamin II interacts with the cadherin- and occludin-based protein complexes at the blood-testis barrier in adult rat testes. J Endocrinol 191:571–586
Kim YN, Bertics PJ (2002) The endocytosis-linked protein dynamin associates with caveolin-1 and is tyrosine phosphorylated in response to the activation of a noninternalizing epidermal growth factor receptor mutant. Endocrinology 143:1726–1731
Singleton PA, Salgia R, Moreno-Vinasco L, Moitra J, Sammani S, Mirzapoiazova T, Garcia JG (2007) CD44 regulates hepatocyte growth factor-mediated vascular integrity. Role of c-Met, Tiam1/Rac1, dynamin 2, and cortactin. J Biol Chem 282:30643–30657
Bruzzaniti A, Neff L, Sanjay A, Horne WC, De Camilli P, Baron R (2005) Dynamin forms a Src kinase-sensitive complex with Cbl and regulates podosomes and osteoclast activity. Mol Biol Cell 16:3301–3313
Hartig SM, Ishikura S, Hicklen RS, Feng Y, Blanchard EG, Voelker KA, Pichot CS, Grange RW, Raphael RM, Klip A, Corey SJ (2009) The F-BAR protein CIP4 promotes GLUT4 endocytosis through bidirectional interactions with N-WASp and dynamin-2. J Cell Sci 122:2283–2291
Zhao L, Shi X, Li L, Miller DJ (2007) Dynamin 2 associates with complexins and is found in the acrosomal region of mammalian sperm. Mol Reprod Dev 74:750–757
Okamoto PM, Herskovits JS, Vallee RB (1997) Role of the basic, proline-rich region of dynamin in Src homology 3 domain binding and endocytosis. J Biol Chem 272:11629–11635
Cao S, Yao J, Shah V (2003) The proline-rich domain of dynamin-2 is responsible for dynamin-dependent in vitro potentiation of endothelial nitric-oxide synthase activity via selective effects on reductase domain function. J Biol Chem 278:5894–5901
Sengar AS, Wang W, Bishay J, Cohen S, Egan SE (1999) The EH and SH3 domain Ese proteins regulate endocytosis by linking to dynamin and Eps15. EMBO J 18:1159–1171
Kamioka Y, Fukuhara S, Sawa H, Nagashima K, Masuda M, Matsuda M, Mochizuki N (2004) A novel dynamin-associating molecule, formin-binding protein 17, induces tubular membrane invaginations and participates in endocytosis. J Biol Chem 279:40091–40099
Tsujita K, Suetsugu S, Sasaki N, Furutani M, Oikawa T, Takenawa T (2006) Coordination between the actin cytoskeleton and membrane deformation by a novel membrane tubulation domain of PCH proteins is involved in endocytosis. J Cell Biol 172:269–279
Kharbanda S, Saleem A, Yuan Z, Emoto Y, Prasad KV, Kufe D (1995) Stimulation of human monocytes with macrophage colony-stimulating factor induces a Grb2-mediated association of the focal adhesion kinase pp 125FAK and dynamin. Proc Natl Acad Sci USA 92:6132–6136
Yoon SY, Koh WS, Lee MK, Park YM, Han MY (1997) Dynamin II associates with Grb2 SH3 domain in Ras transformed NIH3T3 cells. Biochem Biophys Res Commun 234:539–543
Gorska MM, Cen O, Liang Q, Stafford SJ, Alam R (2006) Differential regulation of interleukin 5-stimulated signaling pathways by dynamin. J Biol Chem 281:14429–14439
Xin X, Rabiner CA, Mains RE, Eipper BA (2009) Kalirin12 interacts with dynamin. BMC Neurosci 10:61
Bhattacharya R, Kang-Decker N, Hughes DA, Mukherjee P, Shah V, McNiven MA, Mukhopadhyay D (2005) Regulatory role of dynamin-2 in VEGFR-2/KDR-mediated endothelial signaling. FASEB J 19:1692–1694
Rasmussen RK, Rusak J, Price G, Robinson PJ, Simpson RJ, Dorow DS (1998) Mixed-lineage kinase 2-SH3 domain binds dynamin and greatly enhances activation of GTPase by phospholipid. Biochem J 335:119–124
Krendel M, Osterweil EK, Mooseker MS (2007) Myosin 1E interacts with synaptojanin-1 and dynamin and is involved in endocytosis. FEBS Lett 581:644–650
Pizzato M, Helander A, Popova E, Calistri A, Zamborlini A, Palu G, Gottlinger HG (2007) Dynamin 2 is required for the enhancement of HIV-1 infectivity by Nef. Proc Natl Acad Sci USA 104:6812–6817
Icking A, Matt S, Opitz N, Wiesenthal A, Muller-Esterl W, Schilling K (2005) NOSTRIN functions as a homotrimeric adaptor protein facilitating internalization of eNOS. J Cell Sci 118:5059–5069
Wan KF, Sambi BS, Frame M, Tate R, Pyne NJ (2001) The inhibitory gamma subunit of the type 6 retinal cyclic guanosine monophosphate phosphodiesterase is a novel intermediate regulating p42/p44 mitogen-activated protein kinase signaling in human embryonic kidney 293 cells. J Biol Chem 276:37802–37808
Park JB, Lee CS, Lee HY, Kim IS, Lee BD, Jang IH, Jung YW, Oh YS, Han MY, Jensen ON, Roepstorff P, Suh PG, Ryu SH (2004) Regulation of phospholipase D2 by GTP-dependent interaction with dynamin. Adv Enzyme Regul 44:249–264
Okamoto PM, Gamby C, Wells D, Fallon J, Vallee RB (2001) Dynamin isoform-specific interaction with the shank/ProSAP scaffolding proteins of the postsynaptic density and actin cytoskeleton. J Biol Chem 276:48458–48465
Bruzzaniti A, Neff L, Sandoval A, Du L, Horne WC, Baron R (2009) Dynamin reduces Pyk2 Y402 phosphorylation and SRC binding in osteoclasts. Mol Cell Biol 29:3644–3656
Lundmark R, Carlsson SR (2003) Sorting nexin 9 participates in clathrin-mediated endocytosis through interactions with the core components. J Biol Chem 278:46772–46781
Haberg K, Lundmark R, Carlsson SR (2008) SNX18 is an SNX9 paralog that acts as a membrane tubulator in AP-1-positive endosomal trafficking. J Cell Sci 121:1495–1505
Rufer AC, Rumpf J, von Holleben M, Beer S, Rittinger K, Groemping Y (2009) Isoform-selective interaction of the adaptor protein Tks5/FISH with Sos1 and dynamins. J Mol Biol 390:939–950
Bertelsen V, Breen K, Sandvig K, Stang E, Madshus IH (2007) The Cbl-interacting protein TULA inhibits dynamin-dependent endocytosis. Exp Cell Res 313:1696–1709
Gomez TS, Hamann MJ, McCarney S, Savoy DN, Lubking CM, Heldebrant MP, Labno CM, McKean DJ, McNiven MA, Burkhardt JK, Billadeau DD (2005) Dynamin 2 regulates T cell activation by controlling actin polymerization at the immunological synapse. Nat Immunol 6:261–270
Acknowledgements
We thank Dr. Rachel Peat and Dr. Edgar Gomes for helpful advice. Anne-Cécile Durieux was the recipient of a fellowship from the Association Française contre les Myopathies (AFM).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Durieux, AC., Prudhon, B., Guicheney, P. et al. Dynamin 2 and human diseases. J Mol Med 88, 339–350 (2010). https://doi.org/10.1007/s00109-009-0587-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-009-0587-4