
Abstract
Nitric oxide (NO) derived from the inducible NO synthase (iNOS) is an important and complex mediator of inflammation in the intestine. Wnt-inducible secreted protein (WISP)-1 (CCN4), a member of the connective tissue growth factor family, is involved in tissue repair. We sought to determine the relationship between iNOS and WISP-1 in colitis. By analyzing human colonic biopsy samples, we showed that the expression of mRNA for both iNOS and WISP-1 was significantly higher in ulcerative colitis samples compared with control tissue. The upregulation of WISP-1 was positively correlated with iNOS expression in two models of colitis, induced by intrarectal trinitrobenzenesulfonic acid (TNBS) or occurring spontaneously in IL-10 deficient mice. Loss of iNOS, studied using iNOS−/− mice in both TNBS-induced and IL-10−/− colitis models, significantly attenuated the colitis-related WISP-1 increase. In human colonic epithelial cell lines, the NO donor, DETA-NONOate, elevated WISP-1 mRNA and protein expression through a β-catenin and CREB-dependent, but Wnt-1-independent, pathway. In addition, NO-induced WISP-1 directly induced secretion of soluble collagen in colonic fibroblast cells. NO increases WISP-1 expression both in vitro and in vivo, suggesting a new role for iNOS and NO in colitis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
NO is a diffusible, free radical gas that mediates cell–cell communication in numerous tissues. NO produced from constitutively expressed isoforms of NO synthase regulates numerous essential functions of the gastrointestinal mucosa. However, NO produced following the upregulation of inducible NOS (iNOS) in epithelial cells has been closely associated with inflammation. Increased iNOS and NO is common both in animals models of intestinal inflammation and in patients with inflammatory bowel disease (IBD) [1–3]. Although there is substantial evidence demonstrating the adverse effects of iNOS-derived NO in inflammation, there are also data to suggest that expression of iNOS plays a beneficial role by participating in the processes of tissue healing and regeneration [4]. Ulcer healing can be accelerated with the administration of NO donors, whereas it is retarded by treatment with NO inhibitors [5, 6]. In iNOS-deficient mice, healing is impaired and it has been suggested that collagen production by fibroblasts is regulated by NO [7–9]. In addition, NO donors were able to promote re-endothelialization through the induction of the angiogenic activity of vascular endothelial growth factor (VEGF) [10, 11].
The factors that determine whether NO is inflammatory or reparative in the intestine are not known. The fate of epithelia that may be exposed to NO could depend upon the underlying state of regulatory pathways. For example, cellular pathways that control intestinal epithelial growth and differentiation can be associated with tumorigenesis given a sufficient burden of genetic mutations. A prime example is the Wnt signaling pathway, which is key in intestinal epithelial differentiation along the crypt–villus axis [12], but which participates in colonic tumorigenesis when one or more members of the pathway are mutated [13]. One member of the Wnt signaling pathway is Wnt-induced secreted protein-1 (WISP-1). WISP-1, also called CCN4, belongs to the CCN (or CTGF/Cyr61/Nov) cysteine-rich protein family [14], members of which can stimulate mitosis, adhesion, extracellular matrix production, migration, and regulate angiogenesis [14]. The function of WISP-1 is complex. On one hand, WISP-1 RNA is commonly overexpressed in colon cancers [15]. On the other hand, there is evidence to suggest that WISP-1 is a tumor suppressor gene [16]. Evidence is more consistent for a role for WISP-1 in the wound healing process [17, 18]. However, it is not known if WISP-1 is upregulated in IBD and whether NO has an effect on WISP-1 expression and function.
In the present studies, we showed that iNOS-derived NO could stimulate WISP-1 expression in colitis. We demonstrated the correlation between iNOS and WISP-1 in IBD and in different murine colitis models. In human colonic epithelial cell lines, NO induced WISP-1 expression through a β-catenin- and CREB-dependent, but Wnt-1-independent, pathway. Furthermore, NO-induced WISP-1 was causally associated with increased collagen synthesis in human intestinal fibroblasts.
Materials and methods
Human samples
Colonic biopsies for RNA extraction were obtained from IBD patients undergoing colonoscopy for diagnostic purposes. The control group was from patients undergoing endoscopy for colon cancer screening. Ethical approval for the use of human samples was obtained from the Calgary Health Region Conjoint Health Research Ethics Board.
Immunohistochemistry
Formalin-fixed and paraffin-embedded colonic biopsy tissue from ulcerative colitis patients was used for immunohistochemistry staining for the expression of WISP-1 protein. Sections (5 µM) of paraffin-embedded tissue were immunostained with primary monoclonal antibodies against WISP-1 (R&D Systems, Minneapolis, MN, USA). Antigen retrieval was done by immersing slides in 0.1 M citrate buffer and microwaving at maximal power for 7 min. A negative control was included by substituting the primary antibody with normal mouse IgG.
Models of murine colitis
Protocols for the use of mice were approved by the University of Calgary Health Sciences Animal Care Committee. All procedures involving mice conformed to the guidelines of the Canadian Council on Animal Care. IL-10 deficient and IL-10/iNOS double-deficient mice were generated as described previously [19]. All mice were genotyped for IL-10 and iNOS by PCR analysis of genomic DNA purified from tail biopsies. TNBS colitis was induced as described previously [20]. Briefly, TNBS (6 mg in 0.1 ml 30% ethanol per mouse) (Sigma, Oakville, ON, USA) or saline (0.1 ml) was administered into the distal colon. Mice were studied on the third or seventh day after TNBS–ethanol or saline administration. Male iNOS knock out (stock # 002609) and corresponding wild-type mice for TNBS-induced colitis were purchased from Jackson Labs (Bar Harbor, ME, USA).
HE staining
The formalin-fixed colon tissue was processed, embedded in paraffin, and sectioned by routine histological techniques. The sections were stained with hematoxylin and eosin prior to evaluation of microscopic signs of inflammation.
Cell culture
The human colon epithelial cell lines RKO, Caco-2, and HT-29 (ATCC, Manassas, VA, USA) were grown in Dulbecco’s modified Eagle’s medium/F12 (Sigma) supplemented with 10% fetal bovine serum (Invitrogen, Burlington, ON, Canada). Stably transfected cells (RKOβ-cat) were generated and maintained as described previously [21]. The human colonic myofibroblast cell line CCD18co (ATCC) was maintained in MEM/EBSS medium with 10% fetal bovine serum as recommended by the manufacturer.
RNA extraction and RT-PCR
Total RNA from human and animal samples were isolated with Trizol® (Invitrogen) according to the manufacturer’s instruction. Total RNA from cell lines was extracted using RNeasy mini-kit (QIAGEN, Valencia, CA, USA). The reverse transcription (RT) reaction and polymerase chain reaction (PCR) were performed as described previously [22].
Real-time PCR
WISP-1 mRNA level in mouse samples were measured by real-time PCR with a QIAGEN Quantitect SYBR green PCR kit as described previously [23]. Specificity of amplification was checked by melt curve analysis. WISP-1 expression was normalized against actin. Change (n-fold) over control levels was determined according to the comparative cycle threshold method as previously described [24].
Protein extraction and Western blot
Whole cell lysate [21] and nuclear fractionation [25] were performed as previously described. The proteins were separated with pre-cast 4–12% SDS/PAGE gel (BioRad Laboratories, Hercules, CA, USA). After transfer, the membranes (PVDF) were blotted for human WISP-1 (Santa Cruz), β-catenin (BD Biosciences Pharmingen), phospho-CREB (S133) (R&D Systems), CREB (Cell Signaling Technology, Beverly, MA, USA), and PARP (Cell Signaling Technology). Bands were visualized with Immobilon™ Western (Millipore, Billerica, MA, USA).
Luciferase assay
β-Catenin-driven luciferase plasmid (TOPFLASH) and WISP-1 promoter-driven luciferase plasmids were used in the present study. The luciferase reporters were transfected into cells with Lipofectamine 2000™ (Invitrogen). The plasmid FOP-FLASH with mutated Tcf-4 sites was used as control in all transfection assays. Wild-type and TBE or CRE site mutated WISP-1 promoter-driven luciferase plasmids were generously provided by Dr. Arnold Levine (Cancer Institute of New Jersey, New Brunswick, NJ, USA). All data were normalized to pTK-RL (Promega, Madison, WI, USA). Relative luciferase activity was measured with Dual-luciferase assay kit (Promega).
Small interfering RNA for β-catenin and CREB
Duplex oligo siRNA was purchased from Dharmacon (Lafayette, CO, USA). The target sequences of siRNA oligonucleotides were: β-catenin (5′-AAG UCC UGU AUG AGU GGG AAC-3′) and CREB (5′-CCU UAG UGC AGC UGC CCA AdTdT-3′). CONTROL™ (Dharmacon) was used as a negative control RNA.
Conditioned medium
Caco-2 cells were treated with DETA-NONOate at 0.05 mM for 24 h. The cell culture medium was collected and used as conditioned medium. The conditioned medium from the cells without NO donor treatment was used as the control. WISP-1 in the conditioned medium was depleted with anti-WISP-1 antibody (2 μg, Santa Cruz) and A/G protein agarose beads (40 μl, Santa Cruz).
Soluble collagen assay
CCD18co myofibroblast cells were starved in serum-free medium for 24 h before being challenged with recombinant WISP-1 (R&D Systems) or conditioned medium. The level of soluble collagen in the medium was measured by Sircol™ assay kit (Biocolor Ltd., County Antrim, UK) according to the manufacturer’s instructions.
Statistical analysis
Data are presented as the mean ± standard error of the mean. Comparison of more than two groups was made using ANOVA with a post hoc Tukey test. Comparison of two groups was made using Student’s t test for unpaired data. An associated probability (p) value of <0.05 was considered significant.
Results
Overexpression of iNOS and WISP-1 in human colitis
To study the expression of iNOS and WISP-1 in human colitis, colonic biopsies of inflamed tissue were taken from ulcerative colitis (UC) patients. The control group included biopsy samples from patients undergoing colonoscopy for colon cancer screening. First, levels of iNOS and WISP-1 mRNA were measured by semiquantitative RT-PCR. Consistent with previous reports [26], iNOS was significantly upregulated in UC tissue compared with tissue from control patients (Fig. 1a). WISP-1 mRNA was also significantly upregulated in the UC group (Fig. 1a), which was further confirmed by real-time PCR (Fig. 1b). Immunohistochemical staining revealed that WISP-1 was localized in the cytoplasm of epithelial cells (Fig. 1c).

Upregulation of iNOS and WISP-1 in human colitis. a RT-PCR for human iNOS and WISP-1 were performed with the total RNA isolated from biopsy samples. Lanes 1 to 4 are from normal tissue while lanes 5–9 are from ulcerative colitis tissue. Each lane represents biopsy tissue from a different patient. b Real-time PCR for the expression of WISP-1 was performed with the same samples and compared to the expression of actin. Data are shown as the mean ± SEM of four to five samples. *p < 0.05 compared to the normal tissue group. c Immunohistochemistry was performed to determine the expression of WISP-1 in human colon tissue. Scale bar = 50 µm
TNBS-induced colitis
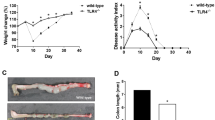
We next assessed whether overexpression of iNOS could cause upregulation of WISP-1 using two different animal models of colitis. First, colitis was induced with intracolonic administration of trinitrobenzenesulfonic acid (TNBS) in ethanol to wild-type C57Bl/6 mice as we previously described [20]. Colonic tissue from each animal was processed for hematoxylin and eosin (HE) staining to confirm the induction of inflammation. Tissues from TNBS-treated groups exhibited histological signs of inflammation (Fig. 2a), including disruption of mucosal architecture, mucosal thickening, and dilation of blood vessels. Both iNOS (Fig. 2b) and WISP-1 (Fig. 2c) mRNA were significantly elevated 7 days after administration of TNBS. Although there was no significant histological difference between wild-type and iNOS−/− mice (Fig. 2a), iNOS deficiency in iNOS knockout mice significantly blocked colitis-related WISP-1 expression (Fig. 2c).

Inducible NOS-dependent WISP-1 expression in the TNBS mouse model of colitis. a HE staining of colon tissue from wild-type and iNOS knockout mice after saline or TNBS administration. Each micrograph was taken at the same magnification (×100, scale bar = 100 µm), so the level of mucosal thickening in the TNBS groups is evident. The levels of b iNOS and c WISP-1 mRNA expression were measured by real-time PCR. *p < 0.05, **p < 0.01 compared with wild-type control group. #p < 0.05 compared with wild-type TNBS group
IL-10 deficiency-induced colitis
In the IL10 deficiency-induced colitis model, mice were assessed at age of 3 months, a time point when spontaneous, chronic intestinal inflammation has developed [19]. HE staining revealed the presence of inflammation in IL10−/− mice (Fig. 3a), as indicated by a large inflammatory cell infiltrate, mucosal thickening, and loss of mucosal architecture. Real-time PCR analysis revealed that both iNOS (Fig. 3b) and WISP-1 (Fig. 3c) mRNA were significantly increased in IL10−/− mice compared to the wild-type group. As described previously [19], the absence of iNOS in IL10−/−/iNOS−/− mice did not change the degree of inflammation compared to IL10−/− littermates (Fig. 3a). However, as with TNBS-induced colitis, the loss of iNOS significantly reduced colitis-related WISP-1 expression (Fig. 3c) in this model.

Inducible NOS-dependent WISP-1 expression in the IL10−/− mouse model of colitis. a HE staining of colon tissue from wild-type, IL10−/−, and IL10−/−/iNOS−/− double knockout mice. Scale bar = 100 µm. The levels of b iNOS and c WISP-1 mRNA in colonic tissue from wild-type, IL10−/−, and IL10−/−/iNOS−/− mice were measured by real-time PCR. *p < 0.05 compared to wild-type mice, #p < 0.05 compared to IL10−/− mice. Data are shown as the mean ± SEM of four to eight samples
NO donors induce WISP-1 expression
To further determine if NO can upregulate WISP-1 and, if so, by what mechanism, the human colonic epithelial cell lines, RKO, CaCo-2, and HT29, were used. In RKO cell lines, NO donor, DETA-NONOate, increased WISP-1 mRNA expression in a dose-dependent manner as determined by real-time PCR (Fig. 4a). The expression of WISP-1 mRNA was induced as early as 2 h and lasted to 24 h (Fig. 4b). Western blot showed that exposure of cells to the NO donor also upregulated WISP-1 at the protein level (Fig. 4c). The same effect was observed in Caco-2 and HT-29 cells and with a structurally dissimilar NO donor, sodium nitroprusside (data not shown). Pretreatment with either the DNA synthesis inhibitor (actinomycin D) or the protein synthesis inhibitor (cycloheximide) completely blocked NO-induced WISP-1 upregulation (Fig. 4d). These results suggest that NO upregulated WISP-1 expression at the transcriptional level in a dose- and time-dependent manner in human colon epithelial cell lines.

NO induces WISP-1 expression in human colonic cell lines. a In RKO cells, the level of human WISP-1 mRNA was measured by real-time PCR after treatment with the NO donor, DETA-NONOate, at 0.05 and 0.25 mM for 24 h. Data are shown as the mean ± SEM of at least three different samples. *p < 0.05 compared to 0 mM control group. b Time course of NO-induced WISP-1 expression following exposure of RKO cells to DETA-NONOate at 0.25 mM. Data are shown as the mean ± SEM of at least three different samples. *p < 0.05 compared to 0 h group. c HT-29 cells were collected for lysis after treatment with DETA-NONOate for 24 h. The protein expression of WISP-1 was measured by Western blot with an anti-WISP-1 antibody. The blot represents three independent experiments. d RKO cells were pretreated with actinomycin D (2 μg/ml) or cycloheximide (2 μg/ml) for 1 h and then stimulated with 0.25 mM DETA-NONOate for 24 h. The mRNA levels of WISP-1 and actin were measured by real-time PCR. Data are shown as the mean ± SEM of three to four samples. *p < 0.05 compared with vehicle group (Veh)
β-catenin mediates NO-induced WISP-1 expression
To elucidate the mechanism by which NO induced upregulation of WISP-1, we studied the role of β-catenin, which has been shown to regulate the transcription of WISP-1 in other systems [27]. Constitutively overexpressed β-catenin in the RKO-based, stably transfected cell line, RKOβ-cat, resulted in significantly higher levels of WISP-1 mRNA expression (Fig. 5a), indicating that WISP-1 is a β-catenin-responsive gene in RKO cells.

β-Catenin mediates NO-induced WISP-1 expression. a The protein level of β-catenin (Western blot) was measured in RKO and RKO β-catenin stable transfectant (RKOβ-cat) cells (left). The level of WISP-1 mRNA expression in RKO and RKOβ-cat cells was measured by real-time PCR (right). Data are shown as mean ± SEM of three to five samples. *p < 0.05 compared with RKO group. b The level of β-catenin in nuclear fractions was measured by Western blot after DETA-NONOate (0.25 mM) treatment. Poly(ADP-ribosyl) transferase (PARP) was used as a loading control. The experiment has been repeated three times independently. c The transcriptional activity of β-catenin was measured by luciferase assay with a β-catenin-driven luciferase reporter (TOPFLASH). Plasmid TK-RL was transfected with TOPFLASH and used as an internal control. *p < 0.05 compared with control group. d The protein level of β-catenin was knocked down by the transfection of siRNA targeted against β-catenin (siRNAβ-cat). A non-specific siRNA was used as a control (c-RNA). e After transfection with siRNAβ-cat or c-RNA, RKO cells were challenged with DETA-NONOate (0.25 mM) and then the level of WISP-1 mRNA was assessed by real-time PCR. *p < 0.05 compared with c-RNA only group, #p < 0.05 compared with c-RNA with DETA-NONOate treatment group
Treatment with DETA-NONOate not only significantly elevated the level of nuclear β-catenin compared to untreated cells (Fig. 5b) but also increased β-catenin transcriptional activity as determined using a β-catenin-driven luciferase reporter (TOPFLASH) assay (Fig. 5c). Most importantly, β-catenin siRNA (Fig. 5d) significantly reduced NO-induced WISP-1 expression compared to control RNA group (Fig. 5e). Furthermore, inactivating mutations of all three Tcf-binding elements on the WISP-1 promoter (TBE mut) completely abolished NO-induced WISP-1 transcriptional activation (Fig. 6d). Taken together, these data strongly support the conclusion that the NO donor activated β-catenin, which then mediated WISP-1 expression in these colonic epithelial cells.

CREB mediates NO-induced WISP-1 expression. a The activated form of CREB in the nuclear fraction was measured by Western blot with anti-phospho-CREB (S133) antibody in RKO and HT-29 cells. Total CREB level was measured as the control. The same experiment was repeated at least three times. b To block the activation of CREB, siRNA targeted against CREB (siRNA CREB ) was transfected into RKO cells. The protein level of CREB was assessed by Western blot. A non-specific siRNA was used a control RNA (c-RNA). c After transfection with siRNACREB or c-RNA, RKO cells were challenged with DETA-NONOate (0.25 mM), and the level of WISP-1 mRNA was measured by real-time PCR. Data are shown as the mean ± SEM of three samples. *p < 0.05 compared with c-RNA in the group not exposed to DETA-NONOate. #p < 0.05 compared with c-RNA in the DETA-NONOate treated group. d HT-29 cells were transfected with wild-type (WT), Tcf-binding element mutated (TBE mut), or cAMP response element mutated (CRE mut) WISP-1-driven luciferase reporter. On the third day after transfection, the cells were treated with DETA-NONOate (0.5 mM) for 24 h and the cells were harvested for luciferase assay. Ptk-RL was used as internal control. Data are shown as the mean ± SEM of three samples. *p < 0.05 compared with wild-type group without DETA-NONOate. #p < 0.05 compared with wild-type with DETA-NONOate-treated group
CREB mediates NO-induced WISP-1 expression
Since previous studies had shown that the cyclic AMP response element (CRE) on the WISP-1 promoter is critical for WISP-1 expression [27], we examined the involvement of CRE binding protein (CREB) in the response to DETA-NONOate. We showed that the level of activated phospho-CREB (ser133) was significantly elevated after stimulation with the NO donor (Fig. 6a). CREB siRNA not only knocked down the level of CREB (Fig. 6b) but also completely inhibited the DETA-NONOate-induced WISP-1 expression compared to cells transfected with control RNA (Fig. 6c). Furthermore, as with β-catenin, an inactivating mutation of CRE on the WISP-1 promoter completely inhibited NO-induced WISP-1 activation (Fig. 6d). Thus, these studies suggest that, along with β-catenin, activation of CREB is necessary for NO-induced WISP-1 expression in colonic epithelial cells.
Wnt-1 does not mediate NO-induced WISP-1 expression
Since WISP-1 is known as a Wnt-1-regulated protein, we measured the level of Wnt-1 mRNA in the IL-10 deficiency colitis model, based on the hypothesis that if Wnt-1 was playing a role in inflammation-induced WISP-1 expression then Wnt-1 mRNA expression would be increased in colitic vs. control tissue. Real-time PCR showed that the level of Wnt-1 was not increased, but rather was decreased in the IL-10−/− colitis group (Wnt-1:actin ratio, 1.89E−5 ± 0.08, n = 5) compared to the control group (Wnt-1:actin ratio, 1.12 ± 0.49, n = 5). However, this apparent difference was not statistically significant due to the variation among samples. In the TNBS-induced colitis model, there was also no significant difference between control and colitis groups (data not shown). Taken together, these data suggested that NO induced WISP-1 expression through a Wnt-1-independent pathway.
WISP-1 increases the secretion of soluble collagen in colon myofibroblast cells
To test whether NO-induced WISP-1 expression had a direct functional effect on colonic myofibroblast cells, human colonic myofibroblast cells, CCD18Co, were incubated with conditioned medium collected from DETA-NONOate-treated Caco-2 cells. Conditioned medium increased the soluble collagen secreted by myofibroblast cells compared with the control group (Fig. 7a). Direct exposure of the myofibroblast cells to DETA-NONOate had no effect on collagen secretion (data not shown). Depletion of WISP-1 from the conditioned medium by WISP-1 antibody significantly blocked the conditioned medium-induced collagen expression (Fig. 7a). Furthermore, recombinant WISP-1 mimicked the effect of conditioned medium and induced collagen secretion at nanomolar concentration (5 and 25 nM) (Fig. 7b). These data strongly suggest that WISP-1 expression induced by exposure to NO in epithelial cells increased collagen secretion in myofibroblast cells.

The effect of WISP-1 on colonic fibroblast cells. a Human CCD18co colonic fibroblast cells were treated with medium only (Control), medium with DETA-NONOate (NO-Con), conditioned medium (CM), or conditioned medium pre-cleared with anti-WISP1 antibody (CM + WISP1-Ab) for 24 h. Then the supernatant was collected for collagen assay. b The levels of soluble collagen were measured after the treatment with recombinant WISP-1 at the concentrations of 5 nM (WISP1–5) or 25 nM (WISP1–25). Data are shown as the mean ± SEM of four samples. *p < 0.05, **p < 0.01 compared with the control group. #p < 0.05 compared with the CM group
Discussion
CCN family proteins stimulate a variety of biological activities, including cell proliferation, angiogenesis, and the production of collagen and fibronectin [14, 17, 28, 29]. Connective tissue growth factor (CTGF), one member of the CCN family, plays an important role in the regulation of mucosal repair and fibrosis in IBD [30]. A recent study showed that colonic mRNA of CTGF was 5-fold higher in 89% of Crohn’s disease patients compared to normal controls [31]. Our studies now demonstrate that another CCN family member, WISP-1/CCN4, is upregulated in IBD and, more importantly, we show that WISP-1 expression is induced by iNOS-derived NO during chronic inflammation and that NO-induced WISP-1 causes the secretion of collagen from colonic fibroblast cells. The present study provides a novel mechanism to explain how NO is involved in the resolution of colitis.
We have shown that expression of both iNOS and WISP-1 is upregulated in colonic biopsies from patients with ulcerative colitis, compared to biopsies from control patients receiving endoscopy for colon cancer screening. These data are consistent with the well-accepted concept that IBD is associated with an increase in the expression of iNOS in the epithelium [32]. Our data indicated that epithelial cells are the source of WISP-1 in colitis. These are consistent with previous studies that WISP-1 is expressed in epithelial cells in colon cancer [15].
To demonstrate a causal association between iNOS and WISP-1 expression, two different mouse models of colitis were used. In studies using iNOS−/− mice, we demonstrated that loss of function of iNOS was able to block the WISP-1 expression associated with inflammation in TNBS and IL10−/− models of colitis. We and others previously showed that loss of iNOS did not affect the development of colitis in either the TNBS [33] or IL10−/− [19] models of colitis. Thus, the abolition of WISP-1 upregulation in colitic iNOS−/− mice was not due to decreased inflammation.
Because we showed that iNOS and WISP-1 expression was primarily epithelial, we used colonic epithelial cell lines to determine the mechanism underlying NO-induced WISP-1 expression. Using the NO donor, DETA-NONOate, we showed that NO transcriptionally increased WISP-1 expression through a β-catenin and CREB-dependent pathway in colonic epithelial cells. The fact that knockdown of either β-catenin or CREB or inactivating mutations of β-catenin or CREB binding sites on the WISP-1 promoter completely blocked NO-induced WISP-1 expression suggests that both must be present for expression to occur. Indeed, we have previously shown that there is a physical interaction between β-catenin and CREB that is required for cyclooxygenase-2 expression induced by activation of protease-activated receptor-2 [34]. A similar association may be triggered by NO. However, at present, it is not clear how NO acts to induce either β-catenin transcriptional activity or CREB phosphorylation. Our findings do not imply that all β-catenin-mediated transcription is dependent upon activation of CREB. Indeed, there are examples of β-catenin-mediated effects that are CREB independent [35].
Although Wnt-1 is known as an important gene that regulates WISP-1 expression [27], the current study suggested that NO increased the expression of WISP-1 through a Wnt-1-independent pathway in TNBS and IL-10−/− colitis models. Most recently, You et al. showed that while there is significantly increased expression of mRNA for several Wnt genes, including the canonical Wnt-3a, in ulcerative colitis, there is no change in the expression of several canonical Wnt target genes in IBD patients [36]. Considering our in vitro studies, it is likely that NO directly induced signaling pathways that regulate WISP-1 expression in colonic epithelial cells, although we cannot rule out the possibility that NO is inducing the expression of a canonical Wnt protein, which is then mediating the transcription of WISP-1. More work will be required to conclusively determine the role of Wnt-associated genes in the response to NO.
While understanding the mechanism underlying NO-induced WISP-1 expression is important, we further sought to demonstrate a functional link between NO and WISP-1. WISP-1 has been shown to stimulate cell proliferation [27] as well as the healing process [17]. However, little is known about the role of WISP-1 in the resolution of inflammatory conditions such as colitis. Matrix remodeling, including the deposition of collagen, is key to post-inflammatory tissue repair, and WISP-1 has been shown to induce collagen release from cardiac fibroblasts [18]. We hypothesized that one of the roles of NO-induced WISP-1 could be to stimulate collagen synthesis in intestinal fibroblasts. In the present study, we showed that NO-induced WISP-1 in epithelial cells increased the level of soluble collagen released from CCD18Co colonic myofibroblasts in culture. Conditioned medium from epithelial cells exposed to DETA-NONOate caused an increase in collagen release from fibroblast cells, and this was blocked by treating the conditioned medium with a neutralizing antibody against WISP-1. To ensure this was not due to DETA-NONOate, or one of its metabolites, in the conditioned medium, we showed that non-conditioned medium (i.e., not incubated with epithelial cells) containing DETA-NONOate did not induce collagen synthesis by fibroblasts.
In summary, we have demonstrated that WISP-1/CCN4 is upregulated in human colitis and have shown, using a variety of models, that the overexpression of WISP-1 is mediated by iNOS and NO through β-catenin- and CREB-dependent, and Wnt-1-independent, transcription pathways in colonic epithelial cells (Fig. 8). Our findings reveal a novel mechanism by which NO may be involved in the resolution of inflammation and the tissue repair in colitis.

Diagram of signaling pathways responsible for nitric oxide (NO)-induced WISP-1 expression and function. Inflammatory stimuli induce the expression of inducible NO synthase (NO) by colonic epithelial cells. NO stimulates WISP-1 expression that is dependent upon β-catenin (β-cat) and cAMP response element (CRE) binding protein (CREB) transcriptional activity. WISP-1 stimulates myofibroblast production of collagen, which participates in repair processes associated with the resolution of inflammation. Tcf, T cell factor, binding site for β-catenin
References
Middleton SJ, Shorthouse M, Hunter JO (1993) Increased nitric oxide synthesis in ulcerative colitis. Lancet 341:465–466
Boughton-Smith NK, Evans SM, Hawkey CJ, Cole AT, Balsitis M, Whittle BJ, Moncada S (1993) Nitric oxide synthase activity in ulcerative colitis and Crohn’s disease. Lancet 342:338–340
Lundberg JO, Hellstrom PM, Lundberg JM, Alving K (1994) Greatly increased luminal nitric oxide in ulcerative colitis. Lancet 344:1673–1674
McCafferty DM, Mudgett JS, Swain MG, Kubes P (1997) Inducible nitric oxide synthase plays a critical role in resolving intestinal inflammation. Gastroenterology 112:1022–1027
Konturek SJ, Brzozowski T, Majka J, Pytko-Polonczyk J, Stachura J (1993) Inhibition of nitric oxide synthase delays healing of chronic gastric ulcers. Eur J Pharmacol 239:215–217
Elliott SN, McKnight W, Cirino G, Wallace JL (1995) A nitric oxide-releasing nonsteroidal anti-inflammatory drug accelerates gastric ulcer healing in rats. Gastroenterology 109:524–530
Shi HP, Efron DT, Most D, Tantry US, Barbul A (2000) Supplemental dietary arginine enhances wound healing in normal but not inducible nitric oxide synthase knockout mice. Surgery 128:374–378
Witte MB, Thornton FJ, Efron DT, Barbul A (2000) Enhancement of fibroblast collagen synthesis by nitric oxide. Nitric Oxide 4:572–582
Yamasaki K, Edington HD, McClosky C, Tzeng E, Lizonova A, Kovesdi I, Steed DL, Billiar TR (1998) Reversal of impaired wound repair in iNOS-deficient mice by topical adenoviral-mediated iNOS gene transfer. J Clin Invest 101:967–971
Ziche M, Morbidelli L, Masini E, Granger H, Geppetti P, Ledda F (1993) Nitric oxide promotes DNA synthesis and cyclic GMP formation in endothelial cells from postcapillary venules. Biochem Biophys Res Commun 192:1198–1203
Xiong M, Elson G, Legarda D, Leibovich SJ (1998) Production of vascular endothelial growth factor by murine macrophages: regulation by hypoxia, lactate, and the inducible nitric oxide synthase pathway. Am J Pathol 153:587–598
Pinto D, Gregorieff A, Begthel H, Clevers H (2003) Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev 17:1709–1713
van Es JH, Barker N, Clevers H (2003) You Wnt some, you lose some: oncogenes in the Wnt signaling pathway. Curr Opin Genet Dev 13:28–33
Brigstock DR (2003) The CCN family: a new stimulus package. J Endocrinol 178:169–175
Pennica D, Swanson TA, Welsh JW, Roy MA, Lawrence DA, Lee J, Brush J, Taneyhill LA, Deuel B, Lew M, Watanabe C, Cohen RL, Melhem MF, Finley GG, Quirke P, Goddard AD, Hillan KJ, Gurney AL, Botstein D, Levine AJ (1998) WISP genes are members of the connective tissue growth factor family that are up-regulated in Wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc Natl Acad Sci USA 95:14717–14722
Hashimoto Y, Shindo-Okada N, Tani M, Nagamachi Y, Takeuchi K, Shiroishi T, Toma H, Yokota J (1998) Expression of the Elm1 gene, a novel gene of the CCN (connective tissue growth factor, Cyr61/Cef10, and neuroblastoma overexpressed gene) family, suppresses in vivo tumor growth and metastasis of K-1735 murine melanoma cells. J Exp Med 187:289–296
French DM, Kaul RJ, D’Souza AL, Crowley CW, Bao M, Frantz GD, Filvaroff EH, Desnoyers L (2004) WISP-1 is an osteoblastic regulator expressed during skeletal development and fracture repair. Am J Pathol 165:855–867
Colston JT, de la Rosa SD, Koehler M, Gonzales K, Mestril R, Freeman GL, Bailey SR, Chandrasekar B (2007) Wnt-induced secreted protein-1 is a prohypertrophic and profibrotic growth factor. Am J Physiol Heart Circ Physiol 293:H1839–H1846
McCafferty DM, Sihota E, Muscara M, Wallace JL, Sharkey KA, Kubes P (2000) Spontaneously developing chronic colitis in IL-10/iNOS double-deficient mice. Am J Physiol Gastrointest Liver Physiol 279:G90–G99
MacNaughton WK, Lowe SS, Cushing K (1998) Role of nitric oxide in inflammation-induced suppression of secretion in a mouse model of acute colitis. Am J Physiol 275:G1353–G1360
Wang H, MacNaughton WK (2005) Overexpressed beta-catenin blocks nitric oxide-induced apoptosis in colonic cancer cells. Cancer Res 65:8604–8607
Buresi MC, Schleihauf E, Vergnolle N, Buret A, Wallace JL, Hollenberg MD, MacNaughton WK (2001) Protease-activated receptor-1 stimulates Ca2+-dependent Cl− secretion in human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 281:G323–G332
Ivory CP, Keller K, Chadee K (2006) CpG-oligodeoxynucleotide is a potent adjuvant with an Entamoeba histolytica Gal-inhibitable lectin vaccine against amoebic liver abscess in gerbils. Infect Immun 74:528–536
Bas A, Forsberg G, Hammarstrom S, Hammarstrom ML (2004) Utility of the housekeeping genes 18S rRNA, beta-actin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase-polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand J Immunol 59:566–573
Nath N, Kashfi K, Chen J, Rigas B (2003) Nitric oxide-donating aspirin inhibits {beta}-catenin/T cell factor (TCF) signaling in SW480 colon cancer cells by disrupting the nuclear {beta}-catenin-TCF association. Proc Natl Acad Sci USA 100:12584–12589
Middleton SJ, Shorthouse M, Hunter JO (1993) Increased nitric oxide synthesis in ulcerative colitis. Lancet 341:465–466
Xu L, Corcoran RB, Welsh JW, Pennica D, Levine AJ (2000) WISP-1 is a Wnt-1- and beta-catenin-responsive oncogene. Genes Dev 14:585–595
Brigstock DR (2002) Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61). Angiogenesis 5:153–165
Chen CC, Mo FE, Lau LF (2001) The angiogenic factor Cyr61 activates a genetic program for wound healing in human skin fibroblasts. J Biol Chem 276:47329–47337
Dammeier J, Brauchle M, Falk W, Grotendorst GR, Werner S (1998) Connective tissue growth factor: a novel regulator of mucosal repair and fibrosis in inflammatory bowel disease. Int J Biochem Cell Biol 30:909–922
di Mola FF, Di Sebastiano P, Gardini A, Innocenti P, Zimmermann A, Buchler MW, Friess H (2004) Differential expression of connective tissue growth factor in inflammatory bowel disease. Digestion 69:245–253
Singer II, Kawka DW, Scott S, Weidner JR, Mumford RA, Riehl TE, Stenson WF (1996) Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology 111:871–885
McCafferty DM, Miampamba M, Sihota E, Sharkey KA, Kubes P (1999) Role of inducible nitric oxide synthase in trinitrobenzene sulphonic acid induced colitis in mice. Gut 45:864–873
Wang H, Wen S, Bunnett NW, Leduc R, Hollenberg MD, MacNaughton WK (2008) Proteinase-activated receptor-2 induces cyclooxygenase-2 expression through β-catenin and cyclic AMP-response element-binding protein. J Biol Chem 283:809–815
Mostany R, Valdizan EM, Pazos A (2008) A role for nuclear beta-catenin in SNRI antidepressant-induced hippocampal cell proliferation. Neuropharmacology 55:18–26
You J, Nguyen AV, Albers CG, Lin F, Holcombe RF (2008) Wnt pathway-related gene expression in inflammatory bowel disease. Dig Dis Sci 53:1013–1019
Acknowledgements
This research was supported by the Canadian Institutes of Health Research (CIHR). Hongying Wang was the recipient of a post-doctoral award from the Canadian Association of Gastroenterology, the CIHR and Crohn’s and Colitis Foundation of Canada. Donna-Marie McCafferty was a CIHR Scholar at the time this work was done. Rui Zhang was supported by a CIHR group grant. The authors thank Dr. Arnold Levine for his generous donation of the wild-type and mutated WISP-1 luciferase reporters.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Wang, H., Zhang, R., Wen, S. et al. Nitric oxide increases Wnt-induced secreted protein-1 (WISP-1/CCN4) expression and function in colitis. J Mol Med 87, 435–445 (2009). https://doi.org/10.1007/s00109-009-0445-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-009-0445-4