
Abstract
Angiotensin (Ang) II is not only generated in the circulation by renin and angiotensin-converting enzyme (ACE) but also is produced locally in numerous organs including kidney, vessels, heart, adrenal gland, eye, testis, and brain. Furthermore, widely distributed mast cells have been shown to be a production site. Local Ang II production process is commonly termed the result of a “tissue” renin–angiotensin system (RAS). Because pharmacological experiments do not easily allow targeting of specific tissues, many novel findings about the functional importance of tissue RAS have been collected from transgenic rodent models. These animals either overexpress or lack RAS components in specific tissues and thereby elucidate their local functions. The data to date show that in most tissues local RAS amplify the actions of circulating Ang II with important implications for physiology and pathophysiology of cardiovascular diseases. This review summarizes the recent findings on the importance of tissue RAS in the most relevant cardiovascular organs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
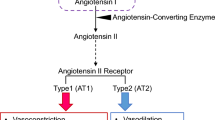
Since its discovery in 1898 [1] and subsequent work thereafter spanning more than half a century, the renin–angiotensin (Ang) system (RAS) was thought to be a hormone system by which the kidney influences systemic cardiovascular regulation. Reacting to changes in renal perfusion pressure, tubular salt content, and the renal sympathetic nerve activity, the juxtaglomerular (JG) cells of the kidney release active renin into the circulation. In the blood, the aspartyl protease proteolytically cleaves the liver-borne angiotensinogen (AOGEN) to form the inactive decapeptide Ang I. The angiotensin-converting enzyme (ACE) further removes two C-terminal amino acids thereby generating Ang II. ACE is a sessile zinc-containing metalloproteinase on endothelial cells. The pulmonary endothelium is a particularly rich source of ACE (Fig. 1). Ang II has two receptors, AT1 and AT2, expressed in many cardiovascular and other tissues. Both receptors belong to the G-protein-coupled receptor class with seven transmembrane domains. The AT1 receptor confers most classical actions of the peptide such as vasoconstriction, aldosterone release from the adrenal zona glomerulosa, salt retention in the renal proximal tubules, and stimulation of the sympathetic nervous system via receptors in the brain. In rodents, which carry two isoforms of the AT1 receptor, AT1A and AT1B, the AT1A receptor mediates most of these actions.

The classical renin–angiotensin system (RAS). The components of the classical RAS, their interactions, the sites of intervention by clinically approved drugs (in red), and the main effects of the RAS in different cardiovascular organs are shown
In addition to the classical RAS components, several new participants have been discovered in recent years. A homolog of ACE, ACE2, was discovered and shown to degrade Ang II yielding Ang-(1-7) (Fig. 2) [2, 3]. Santos et al. discovered that the Mas proto-oncogene is a receptor for this peptide and that the ACE2–Ang-(1–7)–Mas axis is counter-regulating the abovementioned cardiovascular actions of the classical RAS [4, 5]. Furthermore, a protein has recently been discovered, which binds and activates renin and prorenin in tissues, the (pro)renin receptor or (P)RR [6, 7]. The physiological role of these new RAS components is not completely resolved, but, as outlined below, they probably exert considerable impact on local Ang II generation and effect mediation in tissues.

The new renin–angiotensin system (RAS). The newly discovered components of the RAS, such as angiotensin-(1-7), ACE2, Mas, and (P)RR are shown in red
The RAS has been a therapeutic target for cardiovascular diseases since the discovery of the ACE inhibitor captopril about 30 years ago [8]. Later, antagonists for the AT1 receptor were developed [9] and joined the ACE inhibitors as very efficient antihypertensive agents (Fig. 1). Very recently, inhibitors of the rate-limiting enzyme in the RAS, renin, were approved for clinical use [10]. The efficiency of these drugs is partially based on the fact that they not only inhibit the classical RAS in the circulation but also local RAS in tissues [11–14]. In this short update, we will only summarize the data of the last decades and will add some novel aspects, which are mostly based on experiments with transgenic animal models with altered RAS components in single tissues.
Kidney
The first place to surmise a tissue RAS is the kidney because the kidney is the source of the initiating enzyme of the cascade, renin. When the substrate AOGEN and the second enzyme ACE were found to be expressed within the kidney, a local generation of Ang II with physiological importance became a foregone conclusion [15, 16]. Furthermore, early studies detected renin and its messenger RNA (mRNA) [17] outside of the JG cells in the proximal tubules and even in the collecting duct. At these sites, renin is not primarily implicated in the regulation of circulating Ang II levels. Intrarenal Ang II generation is very effective and, under positive feedback control at these renal sites, causes higher local concentrations of the peptide than in the circulation [18–20]. Ang II has numerous functions within the kidney. Besides effects in renal development [21], knockout mice lacking AT1 receptors have shown that Ang II regulates glomerular blood flow, tubular sodium reabsorption, and renin secretion. The local RAS in the kidney may be of high relevance for blood pressure regulation as an amplifier of circulating Ang II actions. In elegant experiments, Crowley et al. [22, 23] showed that AT1 receptors in the kidney are relevant for baseline blood pressure regulation and even more importantly for hypertension induced by Ang II infusion. Bilaterally nephrectomized mice transplanted with one kidney lacking AT1A receptors hardly reacted to chronic Ang II infusion with a blood pressure increase, in contrast to mice lacking AT1A receptors in all tissues except in a transplanted kidney. These mice developed the same increased blood pressure levels as wild-type (transplanted control) mice. Furthermore, the local kidney RAS may be pivotal for renal damage caused by hypertension. We recently showed that mice lacking intrarenal AOGEN synthesis developed less hypertensive damage in the kidney than control mice [24]. Accordingly, mice generating more renal Ang II, either by a transgenic human RAS [25] or by local overexpression of rat AOGEN [26], develop high blood pressure and ample renal injury.
Müller, Luft, and their associates recently shed light on mechanisms involved in Ang II-induced target-organ damage. Using our double-transgenic rat model expressing the human RAS [27], they found that Ang II elicits an inflammatory and immunological response, which leads to interstitial fibrosis, glomerulosclerosis, albuminuria, and finally renal failure [28, 29]. The novel (P)RR protein is implicated in renin- and prorenin-mediated organ damage, both related to and independent of Ang II [6]. The (P)RR is able to activate bound prorenin, thereby facilitating local Ang II generation, but also initiates extracellular-related kinase signaling on its own. The (P)RR has been implicated in the pathogenesis of hypertensive and diabetic kidney damage. Ichihara and coworkers have presented compelling evidence involving a peptide inhibiting the interaction of prorenin with (P)RR. They found that their “decoy” peptide could blunt renal damage induced by diabetes and hypertension [30, 31]. Nevertheless, these data require confirmation in the light of the fact that (P)RR has additional essential functions in cellular physiology [7, 32].
Vascular wall
Almost 40 years ago, Ganten et al. [33] were able to show that renin can be released from splanchnic vessels. Further studies detected AOGEN mRNA and protein in the vessel wall and documented the local generation of Ang II [34]. By direct action on AT1 receptors in vascular smooth muscle cells, Ang II increases vascular tone and blood pressure. However, this classical concept has recently been challenged by the use of T-lymphocyte-deficient mice, which showed a blunted pressor response to low-dose Ang II infusion. These findings by the Harrison laboratory suggest that immune cells may be involved in the local actions of the peptide on vascular tone [35]. Moreover, these mice did not develop the vascular dysfunction and damage normally observed after Ang II infusion. When these data can be confirmed, we will have to accept the fact that the effects of Ang II on the vascular wall are partially mediated by AT1 receptors on T-cells and probably other immune cells. By personal communication, we know that the Müller–Luft laboratory has made similar observations in mice lacking dendritic cells (personal communication).
ACE2, its product Ang-(1-7), and Mas have all been found in the vascular wall [36]. The postulate that (P)RR is responsible for uptake of renin from the circulation into the vessel wall was supported by us in experiments employing a transgenic rat model overexpressing this protein in vascular smooth muscle cells. These (P)RR transgenic animals showed an increased accumulation of prorenin in vessels and elevated blood pressure [37, 38]. Ang-(1-7) is generated in the vascular wall from Ang II by ACE2 and interacts with Mas on endothelial cells [4, 39]. As we could recently show using Mas-deficient mice, this interaction improves endothelial function and reduces blood pressure [40]. Thus, the ACE2–Ang-(1-7)–Mas system is counteracting the classical RAS in the vessel wall. Moreover, using an animal model overexpressing the AT1 receptor only in endothelial cells, Ramchandran et al. [41] demonstrated that Ang II can also act as a vasodilator, when interacting with AT1 on these cells. A similar effect had already been shown for AT2 receptors earlier. Thus, the net cardiovascular effect of angiotensin metabolism in the vascular wall depends on the relative expression of classical and novel components of the RAS in endothelial and smooth muscle cells.
Heart
Local Ang II production in the heart has been observed about 20 years ago [42, 43]. While cardiac AOGEN and ACE expression was unequivocally shown, the expression of renin is disputed. In bilaterally nephrectomized pigs, cardiac renin activity was reduced to minute amounts, which argues against local renin expression [44]. Probably, (P)RR or other renin binding proteins are responsible for the uptake of the enzyme from the circulation into the heart where it initiates Ang II generation [45]. Another source of renin may be mast cells which carry and release renin from their granules and which invade the heart in particular after myocardial infarction. Mast-cell-derived renin was found to be pivotal for activating a cardiac RAS leading via AT1A receptors to increased local norepinephrine release via cardiac neurons. The result was malignant rhythm disturbances [46].
Cardiac fibroblasts and myocytes express AT1 and AT2 receptors. Ang II was found to exhibit growth-promoting effects in the heart more than 30 years ago [47]. Furthermore, in the heart [48], these effects were thought to be most relevant by inducing hypertrophy and fibrosis. An interplay between AT1 and AT2 receptors in the heart has been described [49]. However, recent evidence suggests that this paradigm must be revised [50]. In the experiments with transplanted AT1A-deficient kidneys already mentioned above, the extent of cardiac hypertrophy correlated solely with the blood pressure of the transplanted mice and not with the presence or absence of AT1A receptors in the heart [23]. Moreover, in most transgenic animal models with increased generation of Ang II locally in the heart, either by overexpression of AOGEN, ACE, or a protein releasing the peptide, no hypertrophy was detected, as long as the animals remained normotensive [51–53]. However, in some cases, increased fibrosis and an augmented hypertrophic response to increased afterload was reported [51, 52]. The same was true for some, but not all, transgenic rat and mouse models overexpressing the AT1 receptor in cardiomyocytes [54, 55]. Some cardiac AT1 overexpression models developed cardiac hypertrophy, if an interaction with the epidermal growth factor receptor (EGFR) was possible [56–58]. Furthermore, in hypertensive mice lacking local AOGEN generation in the heart, cardiac hypertrophy and fibrosis was attenuated [24]. How can these data be reconciled into a “unifying theory” about Ang II and cardiac hypertrophy? Probably, locally produced Ang II alone is not sufficient for hypertrophy but it maybe for fibrosis induction. Pressure-induced cardiac hypertrophy appears to require an interaction between Ang II and the EGFR. In this pathway, AT1B or AT2 receptors may compensate for the absence of AT1A. This role of the cardiac RAS may explain the therapeutic effectiveness of RAS inhibitors in the amelioration of hypertensive end-organ damage often exceeding their efficacy in blood pressure control in patients.
Another component of the classical RA(A)S, aldosterone, has gained therapeutic interest in particular in cardiac diseases. Mineralocorticoid receptor antagonists turned out to decrease the risk after myocardial infarction [59, 60]. The underlying pathophysiological mechanisms, however, are not yet completely understood.
Brain
The concept of tissue RAS in general was coined after the discovery of local Ang II generation in the brain [61, 62]. However, the identity of the synthesizing enzyme as being true renin is still under discussion and other enzymes have been postulated to be responsible for Ang II generation in the brain [63].
Due to the blood–brain barrier, most Ang II receptors, which are expressed at multiple sites in the brain, cannot be reached by circulating Ang II. To activate these sites, Ang II needs to be synthesized from locally expressed AOGEN by brain-derived ACE and renin. Exceptions are the circumventricular organs (CVO), where a fenestrated endothelium allows the sensing of the hormonal status in the circulation including the systemic Ang II levels by AT1 receptors expressed there in high amounts. Activation of these receptors leads to increases in blood pressure, thirst, and salt appetite.
However, there is now increasing evidence that the transduction of the signals from the CVO to physiological outputs such as release of vasopressin or activation of the sympathetic nervous system requires a local RAS in areas of the brain inside the blood–brain barrier. Concordantly, transgenic mice with increased Ang II generation only in the brain became hypertensive and exhibited increased salt appetite [64–66]. Even more convincing were studies in which Ang II generation was specifically decreased in the brain. Transgenic rats expressing an antisense RNA against AOGEN only in astrocytes, TGR(ASrAOGEN), were suitable tools for studying this issue [67]. The animals showed reduced blood pressure, sympathetic nervous system activity, and vasopressin release, as well as a blunted response to increased circulating Ang II [67–69]. In a more sophisticated approach, the groups of Sigmund and Davisson generated mice expressing human AOGEN in the whole brain except the subfornical organ (SFO). The investigators locally injected an adenovirus, which deleted the AOGEN transgene [70]. These animals showed a blunted pressor response to intracerebroventricular human renin infusion, indicating that the SFO is of pivotal importance for the central pressor effect of Ang II. When in transgenic animals carrying human renin and human AOGEN, the local expression of AOGEN in the SFO was ablated in the same way; water intake decreased. This observation provides evidence that this brain region is also essential for the drinking control exerted by Ang II [71].
Adrenal gland
Forty years ago, renin and later its mRNA was discovered in the adrenal gland [72, 73]. The gland already begins to express renin in high amounts during embryogenesis in parallel to the kidney [74]. In contrast to the heart (see above), adrenal renin concentration is even upregulated after bilateral nephrectomy, indicating independence of the adrenal RAS from the systemic one [75]. The functions of the adrenal RAS may include modulation of aldosterone secretion in conjunction with the circulating Ang II. This conclusion is supported by the drastically altered steroidogenesis in TGR(mREN2)27 rats with a stimulated adrenal RAS in the presence of normal plasma Ang II levels [76, 77]. Interestingly, the adrenal gland expresses also a cytoplasmic form of renin called renin A [78, 79]. When the renin A isoform is overexpressed in transgenic rats, aldosterone synthesis is stimulated [80]. The adrenal RAS may serve as an amplification system for the effects of the circulating RAS on steroidogenesis because Ang II can induce renin release from adrenocortical cells [81]. Furthermore, a role of Ang II in adrenal development is implicated by the early embryonic expression of renin in this organ [82], but as yet no conclusive evidence was provided. However, growth-promoting, but probably not proliferative, effects of the locally generated Ang II are of major importance for the adjustment of the size of adrenal glomerulosa to physiological needs [83].
Conclusions
The local generation of Ang II has been demonstrated for all tissues relevant for cardiovascular control. These tissue RAS play important roles in the functional regulation of the respective organs mostly conveying and amplifying the effects of circulating Ang II. Thereby, they modulate cardiovascular parameters and influence—mostly accelerate—the pathogenesis of cardiovascular diseases. Thus, tissue RAS form the basis for the understanding of the extraordinary therapeutic efficiency of drugs inhibiting the RAS, such as ACE inhibitors, AT1 antagonists, and the newly developed renin inhibitors.
Reference
Tigerstedt R, Bergman PG (1898) Niere und Kreislauf. Skand Arch Physiol 8:223–271
Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ (2000) A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275:33238–33243
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S (2000) A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 87:E1–E9
Santos RA, Simoes e Silva AC, Maric C, Silva DMR, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SVB, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss HP, Speth R, Walther T (2003) Angiotensin-(1-7) is an endogenous ligand for the G-protein coupled receptor Mas. Proc Natl Acad Sci U S A 100:8258–8263
Santos RA, Ferreira AJ (2007) Angiotensin-(1-7) and the renin–angiotensin system. Curr Opin Nephrol Hypertens 16:122–128
Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD (2002) Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109:1417–1427
Burckle C, Bader M (2006) Prorenin and its ancient receptor. Hypertension 48:549–551
Ferguson RK, Turini GA, Brunner HR, Gavras H, McKinstry DN (1977) A specific orally active inhibitor of angiotensin-converting enzyme in man. Lancet 1:775–778
Wong PC, Barnes TB, Chiu AT, Christ DD, Duncia JV, Herblin WF, Timmermans PBMWM (1991) Losartan (DuP 753), an orally active nonpeptide angiotensin II receptor antagonist. Cardiovasc Drug Rev 9:317–339
Staessen JA, Li Y, Richart T (2006) Oral renin inhibitors. Lancet 368:1449–1456
Bader M, Peters J, Baltatu O, Müller DN, Luft FC, Ganten D (2001) Tissue renin–angiotensin systems: new insights from experimental animal models in hypertension research. J Mol Med 79:76–102
Sakai K, Sigmund CD (2005) Molecular evidence of tissue renin–angiotensin systems: a focus on the brain. Curr Hypertens Rep 7:135–140
Fleming I, Kohlstedt K, Busse R (2006) The tissue renin–angiotensin system and intracellular signalling. Curr Opin Nephrol Hypertens 15:8–13
Paul M, Poyan MA, Kreutz R (2006) Physiology of local renin–angiotensin systems. Physiol Rev 86:747–803
Gomez RA, Lynch KR, Chevalier RL, Everett AD, Johns DW, Wilfong N, Peach MJ, Carey RM (1988) Renin and angiotensinogen gene expression and intrarenal renin distribution during ACE inhibition. Am J Physiol 254:900–906
Schulz WW, Hagler HK, Buja LM, Erdos EG (1988) Ultrastructural localization of angiotensin I-converting enzyme (EC 3.4.15.1) and neutral metalloendopeptidase (EC 3.4.24.11) in the proximal tubule of the human kidney. Lab Invest 59:789–797
Moe OW, Ujiie K, Star RA, Miller RT, Widell J, Alpern RJ, Henrich WL (1993) Renin expression in renal proximal tubule. J Clin Invest 91:774–779
Campbell DJ, Lawrence AC, Towrie A, Kladis A, Valentijn AJ (1991) Differential regulation of angiotensin peptide levels in plasma and kidney of the rat. Hypertension 18:763–773
Navar LG, Harrison Bernard LM, Imig JD, Wang CT, Cervenka L, Mitchell KD (1999) Intrarenal angiotensin II generation and renal effects of AT(1) receptor blockade. J Am Soc Nephrol 10:S266–S272
Kobori H, Ozawa Y, Satou R, Katsurada A, Miyata K, Ohashi N, Hase N, Suzaki Y, Sigmund CD, Navar LG (2007) Kidney-specific enhancement of ANG II stimulates endogenous intrarenal angiotensinogen in gene-targeted mice. Am J Physiol Renal Physiol 293:F938–F945
Matsusaka T, Miyazaki Y, Ichikawa I (2002) The renin angiotensin system and kidney development. Annu Rev Physiol 64:551–561
Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, Spurney RF, Kim HS, Smithies O, Le TH, Coffman TM (2005) Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin–angiotensin system. J Clin Invest 115:1092–1099
Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM (2006) Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A 103:17985–17990
Kang N, Walther T, Tian XL, Bohlender J, Fukamizu A, Ganten D, Bader M (2002) Reduced hypertension-induced end-organ damage in mice lacking cardiac and renal angiotensinogen synthesis. J Mol Med 80:359–366
Lavoie JL, Lake-Bruse KD, Sigmund CD (2004) Increased blood pressure in transgenic mice expressing both human renin and angiotensinogen in the renal proximal tubule. Am J Physiol Renal Physiol 286:F965–F971
Sachetelli S, Liu Q, Zhang SL, Liu F, Hsieh TJ, Brezniceanu ML, Guo DF, Filep JG, Ingelfinger JR, Sigmund CD, Hamet P, Chan JS (2006) RAS blockade decreases blood pressure and proteinuria in transgenic mice overexpressing rat angiotensinogen gene in the kidney. Kidney Int 69:1016–1023
Ganten D, Wagner J, Zeh K, Bader M, Michel JB, Paul M, Zimmermann F, Ruf P, Hilgenfeldt U, Ganten U, Kaling M, Bachmann S, Fukamizu A, Mullins JJ, Murakami K (1992) Species specificity of renin kinetics in transgenic rats harboring the human renin and angiotensinogen genes. Proc Natl Acad Sci U S A 89:7806–7810
Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J, Heidecke H, Haller H, Zenke M, Luft FC (2002) Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol 161:1679–1693
Park JK, Fischer R, Dechend R, Shagdarsuren E, Gapeljuk A, Wellner M, Meiners S, Gratze P, Al Saadi N, Feldt S, Fiebeler A, Madwed JB, Schirdewan A, Haller H, Luft FC, Muller DN (2007) p38 mitogen-activated protein kinase inhibition ameliorates angiotensin II-induced target organ damage. Hypertension 49:481–489
Ichihara A, Kaneshiro Y, Takemitsu T, Sakoda M, Nakagawa T, Nishiyama A, Kawachi H, Shimizu F, Inagami T (2006) Contribution of nonproteolytically activated prorenin in glomeruli to hypertensive renal damage. J Am Soc Nephrol 17:2495–2503
Ichihara A, Hayashi M, Kaneshiro Y, Suzuki F, Nakagawa T, Tada Y, Koura Y, Nishiyama A, Okada H, Uddin MN, Nabi AH, Ishida Y, Inagami T, Saruta T (2004) Inhibition of diabetic nephropathy by a decoy peptide corresponding to the “handle” region for nonproteolytic activation of prorenin. J Clin Invest 114:1128–1135
Bader M (2007) The second life of the (Pro)renin receptor. J Renin Ang Ald System 8:205–208
Ganten D, Hayduk K, Brecht HM, Boucher R, Genest J (1970) Evidence of renin release or production in splanchnic territory. Nature 226:551–552
Müller DN, Hilgers KF, Bohlender J, Lippoldt A, Wagner J, Fischli W, Ganten D, Mann JFE, Luft FC (1995) Effects of human renin in the vasculature of rats transgenic for human angiotensinogen. Hypertension 26:272–278
Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG (2007) Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204:2449–2460
Alenina N, Xu P, Rentzsch B, Bader M (2008) Genetically altered animal models for Mas and angiotensin-(1-7). Exp Physiol (in press)
Burckle C, Danser AHJ, Müller DN, Garrelds IM, Gasc J-M, Plehm R, Peters J, Bader M, Nguyen G (2006) Elevated blood pressure and heart rate in human renin receptor transgenic rats. Hypertension 47:552–556
Batenburg WW, Krop M, Garrelds IM, de Vries R, de Bruin RJA, Burckle C, Mueller DN, Bader M, Nguyen G, Danser AHJ (2007) Prorenin is the endogenous agonist of the (pro)renin receptor. Binding kinetics of renin and prorenin in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor. J Hypertens 25:2441–2453
Sampaio WO, Henrique dC, Santos RA, Schiffrin EL, Touyz RM (2007) Angiotensin-(1–7) counterregulates angiotensin II signaling in human endothelial cells. Hypertension 50:1093–1098
Xu P, Goncalves ACC, Todiras M, Rabelo LA, Sampaio WO, Moura MM, Santos SS, Luft FC, Bader M, Gross V, Alenina N, Santos RA (2008) Endothelial dysfunction and elevated blood pressure in Mas gene-deleted mice. Hypertension 51:574–580
Ramchandran R, Takezako T, Saad Y, Stull L, Fink B, Yamada H, Dikalov S, Harrison DG, Moravec C, Karnik SS (2006) Angiotensinergic stimulation of vascular endothelium in mice causes hypotension, bradycardia, and attenuated angiotensin response. Proc Natl Acad Sci U S A 103:19087–19092
Lindpaintner K, Jin M, Wilhelm MJ, Suzuki F, Linz W, Schoelkens BA, Ganten D (1988) Intracardiac generation of angiotensin and its physiologic role. Circulation 77(suppl 1):I–18–I-23
Dzau VJ (1987) Implications of local angiotensin production in cardiovascular physiology and pharmacology. Am J Cardiol 59:59A–65A
Danser AH, van Kats JP, Admiraal PJ, Derkx FH, Lamers JM, Verdouw PD, Saxena PR, Schalekamp MA (1994) Cardiac renin and angiotensins. Uptake from plasma versus in situ synthesis. Hypertension 24:37–48
Peters J, Farrenkopf R, Clausmeyer S, Zimmer J, Kantachuvesiri S, Sharp MG, Mullins JJ (2002) Functional significance of prorenin internalization in the rat heart. Circ Res 90:1135–1141
Mackins CJ, Kano S, Seyedi N, Schafer U, Reid AC, Machida T, Silver RB, Levi R (2006) Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J Clin Invest 116:1063–1070
Gill GN, Ill CR, Simonian MH (1977) Angiotensin stimulation of bovine adrenocortical cell growth. Proc Natl Acad Sci U S A 74:5569–5573
Schelling P, Ganten D, Speck G, Fischer H (1979) Effects of angiotensin II and angiotensin II antagonist saralasin on cell growth and renin in 3T3 and SV3T3 cells. J Cell Physiol 98:503–514
Senbonmatsu T, Ichihara S, Price E Jr., Gaffney FA, Inagami T (2000) Evidence for angiotensin II type 2 receptor-mediated cardiac myocyte enlargement during in vivo pressure overload. J Clin Invest 106:R25–R29
Reudelhuber TL, Bernstein KE, Delafontaine P (2007) Is angiotensin II a direct mediator of left ventricular hypertrophy? Time for another look. Hypertension 49:1196–1201
van Kats JP, Methot D, Paradis P, Silversides DW, Reudelhuber TL (2001) Use of a biological peptide pump to study chronic peptide hormone action in transgenic mice. Direct and indirect effects of angiotensin II on the heart. J Biol Chem 276:44012–44017
Tian XL, Pinto YM, Costerousse O, Franz WM, Lippoldt A, Hoffmann S, Unger T, Paul M (2004) Over-expression of angiotensin converting enzyme-1 augments cardiac hypertrophy in transgenic rats. Hum Mol Genet 13:1441–1450
Xiao HD, Fuchs S, Campbell DJ, Lewis W, Dudley SC Jr., Kasi VS, Hoit BD, Keshelava G, Zhao H, Capecchi MR, Bernstein KE (2004) Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am J Pathol 165:1019–1032
Hoffmann S, Krause T, van Geel PP, Willenbrock R, Pagel I, Pinto YM, Buikema H, Van Gilst WH, Lindschau C, Paul M, Inagami T, Ganten D, Urata H (2001) Overexpression of the human angiotensin II type 1 receptor in the rat heart augments load induced cardiac hypertrophy. J Mol Med 79:601–608
Hein L, Stevens ME, Barsh GS, Pratt RE, Kobilka BK, Dzau VJ (1997) Overexpression of angiotensin AT1 receptor transgene in the mouse myocardium produces a lethal phenotype associated with myocyte hyperplasia and heart block. Proc Natl Acad Sci U S A 94:6391–6396
Paradis P, Dali-Youcef N, Paradis FW, Thibault G, Nemer M (2000) Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc Natl Acad Sci U S A 97:931–936
Zhai P, Yamamoto M, Galeotti J, Liu J, Masurekar M, Thaisz J, Irie K, Holle E, Yu X, Kupershmidt S, Roden DM, Wagner T, Yatani A, Vatner DE, Vatner SF, Sadoshima J (2005) Cardiac-specific overexpression of AT1 receptor mutant lacking G alpha q/G alpha i coupling causes hypertrophy and bradycardia in transgenic mice. J Clin Invest 115:3045–3056
Zhai P, Galeotti J, Liu J, Holle E, Yu X, Wagner T, Sadoshima J (2006) An angiotensin II type 1 receptor mutant lacking epidermal growth factor receptor transactivation does not induce angiotensin II-mediated cardiac hypertrophy. Circ Res 99:528–536
Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J (1999) The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 341:709–717
Connell JM, Mackenzie SM, Freel EM, Fraser R, Davies E (2008) A lifetime of aldosterone excess: long-term consequences of altered regulation of aldosterone production for cardiovascular function. Endocr Rev (in press)
Ganten D, Minnich JL, Granger P, Hayduk K, Brecht HM, Barbeau A, Boucher R, Genest J (1971) Angiotensin-forming enzyme in brain tissue. Science 173:64–65
Phillips MI, Mann JFE, Haebara H, Hoffman WE, Dietz R, Schelling P, Ganten D (1977) Lowering of hypertension by central saralasin in the absence of plasma renin. Nature 270:445–447
Bader M, Ganten D (2002) Editorial: it’s renin in the brain. Circ Res 90:8–10
Morimoto S, Cassell MD, Beltz TG, Johnson AK, Davisson RL, Sigmund CD (2001) Elevated blood pressure in transgenic mice with brain-specific expression of human angiotensinogen driven by the glial fibrillary acidic protein promoter. Circ Res 89:365–372
Morimoto S, Cassell MD, Sigmund CD (2002) Glia- and neuron-specific expression of the renin–angiotensin system in brain alters blood pressure, water intake, and salt preference. J Biol Chem 277:33235–33241
Lochard N, Silversides DW, van Kats JP, Mercure C, Reudelhuber TL (2003) Brain-specific restoration of angiotensin II corrects renal defects seen in angiotensinogen-deficient mice. J Biol Chem 278:2184–2189
Schinke M, Baltatu O, Böhm M, Peters J, Rascher W, Bricca G, Lippoldt A, Ganten D, Bader M (1999) Blood pressure reduction and diabetes insipidus in transgenic rats deficient in brain angiotensinogen. Proc Natl Acad Sci U S A 96:3975–3980
Baltatu O, Janssen BJ, Bricca G, Plehm R, Monti J, Ganten D, Bader M (2001) Alterations in blood pressure and heart rate variability in transgenic rats with low brain angiotensinogen. Hypertension 37:408–413
Baltatu O, Silva JA Jr., Ganten D, Bader M (2000) The brain renin–angiotensin system modulates angiotensin II-induced hypertension and cardiac hypertrophy. Hypertension 35:409–412
Sinnayah P, Lazartigues E, Sakai K, Sharma RV, Sigmund CD, Davisson RL (2006) Genetic ablation of angiotensinogen in the subfornical organ of the brain prevents the central angiotensinergic pressor response. Circ Res 99:1125–1131
Sakai K, Agassandian K, Morimoto S, Sinnayah P, Cassell MD, Davisson RL, Sigmund CD (2007) Local production of angiotensin II in the subfornical organ causes elevated drinking. J Clin Invest 117:1088–1095
Ryan JW (1967) Renin-like enzyme in the adrenal gland. Science 158:1589–1590
Ganten D, Ganten U, Kubo S, Granger P, Nowaczynski W, Boucher R, Genest J (1974) Influence of sodium, potassium and pituitary hormones on iso-renin in rat adrenal gland. Am J Physiol 227:224–229
Jones CA, Sigmund CD, McGowan RA, Kane-Haas CM, Gross KW (1990) Expression of murine renin genes during fetal development. Mol Endocrinol 4:375–383
Naruse M, Inagami T (1982) Markedly elevated specific renin levels in the adrenal gland in genetically hypertensive rats. Proc Natl Acad Sci U S A 79:3295–3299
Mullins JJ, Peters J, Ganten D (1990) Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature 344:541–544
Sander M, Bader M, Djavidani B, Maser Gluth C, Vecsei P, Mullins J, Ganten D, Peters J (1992) The role of the adrenal gland in hypertensive transgenic rat TGR(mREN2)27. Endocrinology 131:807–814
Bader M, Ganten D (2000) Regulation of renin: new evidence from cultured cells and genetically modified mice. J Mol Med 78:130–139
Clausmeyer S, Stürzebecher R, Peters J (1999) An alternative transcript of the rat renin gene can result in a truncated prorenin that is transported into adrenal mitochondria. Circ Res 84:337–344
Peters J, Wanka H, Peters B, Hoffmann S (2008) A renin transcript lacking exon 1 encodes for a non secretory intracellular renin that increases aldosterone production in transgenic rats. J Cell Mol Med (in press)
Peters J, Münter K, Bader M, Hackenthal E, Mullins JJ, Ganten D (1993) Increased adrenal renin in transgenic hypertensive rats, TGR(mREN2)27, and its regulation by cAMP, angiotensin II, and calcium. J Clin Invest 91:742–747
Sequeira Lopez ML, Pentz ES, Nomasa T, Smithies O, Gomez RA (2004) Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell 6:719–728
Otis M, Campbell S, Payet MD, Gallo-Payet N (2007) The growth-promoting effects of angiotensin II in adrenal glomerulosa cells: an interactive tale. Mol Cell Endocrinol 273:1–5
Acknowledgment
The Deutsche Forschungsgemeinschaft supported the work of the authors. We thank Friedrich Luft for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bader, M., Ganten, D. Update on tissue renin–angiotensin systems. J Mol Med 86, 615–621 (2008). https://doi.org/10.1007/s00109-008-0336-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-008-0336-0