
Abstract
Obesity and type 2 diabetes are the most prevalent metabolic diseases in the western world. Alarmingly, the cluster of pathologies characteristic of obesity-induced disease have started to emerge in children, a phenomenon that up until a decade ago was inconceivable. Hence, the development of new strategies to treat ‘metabolic disease’ is most warranted. Growing evidence suggests that during type 2 diabetes, a state of chronic low-grade inflammation exists in metabolically active tissues such as the liver, adipose tissue and skeletal muscle. This inflammation is often secondary to lipid accumulation in insulin-responsive tissues. Recent studies have focused on the therapeutic potential of ciliary neurotrophic factor (CNTF). CNTF is a pluripotent neurocytokine and, has shown promise as a potential anti-obesogenic therapy. CNTF acts both centrally and peripherally, mimics the biological actions of leptin while overcoming “leptin resistance”, remains effective even after termination of therapy if administered centrally, and appears to reduce inflammatory signaling cascades associated with lipid accumulation in liver and skeletal muscle. The advantages and disadvantages of CNTF as a therapeutic strategy to alleviate obesity-associated diseases will be highlighted in this review.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
It is now estimated that 10% of the world’s population are overweight or obese. Alarmingly, there has been a 75% increase in adult obesity in the last 25 years [1]. In the USA, 20 states have obesity prevalence rates of 15 to 19%, 29 have rates of 20 to 24%, and one has a reported rate of more than 25%. Of major concern, the phenomenon of being overweight or obese is now a significant problem in children, and the incidence is continuing to climb [2]. Rather than using therapeutic intervention, the promotion of lifestyle changes, which include exercise and a healthier diet, should be implemented to treat childhood obesity. An abundant number of disorders directly correlate with obesity. These include glucose intolerance, dyslipidemia, and insulin resistance which may ultimately culminate in pancreatic beta cell failure and type 2 diabetes. There are many current pharmacological drugs to treat the obesity-related disorder type 2 diabetes. These include: (1) thiazolidinediones (TZDs), which function as ligands for the peroxisome proliferator-activated receptor-γ, nuclear receptors controlling adipocyte metabolism, and differentiation; (2) biguanides, which decrease endogenous glucose production via activation of the fuel sensing kinase 5′-AMP activated protein kinase (AMPK), and (3) sulfonylureas, which stimulate insulin secretion. When patients no longer respond to these treatments, insulin is prescribed to control hyperglycemia and arrest the development of diabetic complications [3]. However, the current therapeutic strategies have many disadvantages, not the least being weight gain, particularly when TZDs and insulin are administered. Therefore, the “holy grail” of identifying a drug that is capable of concomitantly decreasing body weight while enhancing insulin action has remained elusive.
Leptin—the obesity breakthrough or not?
When the adipocyte-derived protein leptin and its receptor were first characterized [4–6], it offered an entirely new paradigm in the therapeutic control of obesity because its discovery established a link between a circulating molecule and modification of feeding behavior centrally. More than 10 years on, it is now known that so-called “leptin resistance” occurs in obese subjects [7]. The reasoning for “leptin resistance” is still not fully clear; however, two mechanisms are believed to be at play. Firstly, transport of leptin across the blood–brain barrier may be dysfunctional [8]. Moreover, the second appears to be related to defective signaling through the long isoform of the leptin receptor (LRb). Research from Yoshimura et al. [9] and Hilton and colleagues [10] identified a novel cytokine inducible compound, termed suppressor of cytokine signaling (SOCS-3), that negatively regulated leptin signaling and lead to leptin resistance [11, 12]. In mice that have haploinsufficiency of SOCS3, leptin sensitivity is increased, and these mice are protected from diet-induced obesity [11]. When SOCS3 was selectively ablated in proopiomelanocortin (POMC) expressing neurons of mice, leptin sensitivity was enhanced [13]. This study conclusively demonstrated that POMC-expressing neurons are a major target of leptin and assist in mediating leptins’ beneficial effects. It has been clearly established that phosphorylated Tyr985 of LRb binds SOCS-3 which contributes to the attenuation of LRb signaling. In a recent study by Bjornholm et al. [14], homologous recombination was adopted to replace the WT LRb in mice with a receptor that possesses a mutation at Tyr985 (Tyr → Leu), resulting in a lack of SOCS-3 binding. Interestingly, mice, homologous for the mutation displayed reduced feeding and adiposity and an increased sensitivity to exogenous leptin. Unexpectedly, the phenotype was particularly evident in female mice. Interestingly, in independent studies, heterozygous SOCS-3-deficient and brain SOCS-3-deficient females displayed a more robust leptin-sensitive response compared to males [11, 12]. The increased estrogen levels in females may be the mediator of these effects, as estrogen is known to interact with identical signaling molecules as leptin in the hypothalamus to control energy homeostasis [15]. These results substantiate that the mutation of Tyr985 prevents the activation of an inhibitory Tyr985 dependent LRb signal. It is well documented that leptin mediates a majority of its effects in the central nervous system by reducing the activation of 5′AMP-activated protein kinase (AMPK). In muscle cells that overexpress SOCS3, leptin can no longer activate AMPK and its downstream target acetyl-CoA carboxylase β (ACCβ), which fails to suppress ACC β activity. This ultimately prevents the increase in fatty acid oxidation [16]. It is clear, therefore, that in these systems, SOCS-3 transpires to negate the effects of leptin resulting in leptin resistance (Fig. 1a). As both human and rodent obesity are characterized by increased SOCS-3 [17, 18] and dysfunctional leptin signaling [19, 20], the use of leptin as an antiobesity therapeutic may not be an attractive option [21]. However, gp130 receptor ligands, in particular, ciliary neurotrophic factor (CNTF), may provide an avenue for circumventing leptin resistance, as CNTF is known to have “leptin-like” effects in obesity [7].

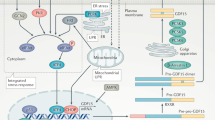
CNTF and leptin signaling pathways involved in regulating Jak/Stat signaling and the negative effects of SOCS-3 expression. (a) When leptin binds the leptin receptor b (LRb), Jak2 also binds LRb at its intracellular binding domain and is phosphorylated. This, in turn, phosphorylates STAT3 which is bound to Tyr1138 of the LRb. STAT3 acts as a critical transcription factor for SOCS-3 and other STAT3-dependent genes in the nucleus. When SOCS-3 protein expression increases, it inhibits leptin signaling by binding to the LRb at its Src homology phosphatase-2 (SHP2) domain (Tyr985) to inhibit JAK tyrosine kinase activity. CNTF can signal by firstly binding the CNTFRα (b) or IL-6Rα (c). After recruitment of the LIFRβ and gp130β receptors, JAK/STAT signaling occurs on the intracellular domains of the LIFRβ and gp130β receptors. As with the LRb, SOCS-3 can inhibit JAK/STAT signaling on the LIFRβ and gp130β receptor, by binding the SHP2/Tyr974 and SHP2/Tyr759 binding sites of each receptor, respectively. It is currently hypothesized that CNTF may overcome SOCS-3 inhibition because the gp130β receptor has three additional STAT-3 binding sites compared to the LRb. In addition, it is known that the C-terminal SOCS box recruits ubiquitin transferases to mediate the degradation of receptor Jak complexes. Adapted from [67] and [68]
Ciliary neurotrophic factor/glycoprotein 130 (gp130) receptor signaling
Approximately, a quarter of a century ago, CNTF was identified as a factor which promoted survival of chick ciliary ganglion neurons [22]. Ten years later, CNTF was purified and cloned from sciatic nerves [23, 24]. In addition to its pro-survival functions [25], CNTF encourages the differentiation of sympathetic neurons and glial progenitor cells into astrocytes. To mediate its effects, CNTF binds the CNTF receptor (α receptor; Fig. 1b). This event then leads to heterodimerization of glycoprotein 130 (gp130) and the leukemia inhibitory factor receptor (LIFR; β receptors). Both gp130 and the LIFR enable downstream signaling through the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway [26]. Of critical biological significance is the fact that CNTF may also utilize the interleukin-6 receptor (IL-6R) as an α-receptor ([27]; Fig. 1c). Another gp130 ligand, interleukin-6, has also been the topic of much research relating to obesity; however, this review will focus on CNTF-mediated effects on obesity-related disorders. It is important to note differences between CNTF and IL-6 binding epitopes. Both IL-6 and CNTF each possess three binding epitopes. Interleukin-6 may bind the IL-6R at the site I epitope and gp130 at the site II and III epitopes. In contrast, CNTF may bind either the IL-6R or CNTFR at the site I epitope, gp130 at site II, and the LIFR at the site III epitope [28].
Expression of the CNTF receptor-α (CNTFR) is most abundant in the tissue of the nervous system; however, it is expressed in numerous peripheral tissues including skeletal muscle [25, 29]. The CNTFR-α expression levels in both cultured preadipocytes/mature adipocytes and adipose tissue in vivo will be discussed in detail later in this review. Interestingly, in skeletal muscle, the CNTFR is considerably lower in expression when compared with the IL-6Rα [16]. Direct effects of CNTF in skeletal muscle include dedifferentiation of human myoblasts into multipotent progenitor cells [30]. In addition, CNTF promotes muscle strength [31]. It was first discovered that CNTF possessed antiobesogenic characteristics when amytrophic lateral sclerosis patients were treated with CNTF in an effort to attenuate disease progression [32]. Remarkably, CNTF-treated patients underwent marked weight loss. Since this study, a vast number of rodent studies have further substantiated the antiobesogenic properties of CNTF and the human recombinant variant of CNTF, Axokine® (CNTFAx15) [25].
Direct effects of CNTF action on the brain
Gloaguen et al. [33] reported that the CNTFRα and the LRb were co-localized in the hypothalamic region of the brain involved in the regulation of energy balance. In addition, systemic administration of both CNTF and leptin activated genes in the arcuate nuclei, suggesting that both cytokines were capable of anorexogenic neuronal signaling. Moreover, they found that administration of CNTF in leptin resistance models of obesity, namely, ob/ob, db/db, and high-fat-fed mice, resulted in reduced feeding, body weight, and insulin levels. Lambert et al. [34] also demonstrated that CNTFAx15 reversed the obese phenotype in leptin-resistant rodent models and importantly failed to induce fever which quite often occurs with cytokine treatment [35]. Interestingly, subsequent studies have reported the presence [36, 37] or absence [16] of fever or increases in pro-inflammatory gene expression after treatment with CNTF or CNTFAx15.
In the study by Lambert et al. [34], mice maintained a decreased body weight after the CNTF treatment was discontinued. It has only become evident in the last 2 years, exactly how CNTF or CNTFAx15 prevented weight gain after the cessation of treatment. It has been shown that centrally administered CNTF leads to proliferation of cells in the hypothalamus of mice [38]. In proof-of-principle experiments, administration of the mitotic blocker cytosine-β-d-aribinofuranoside with CNTF prevented hypothalamic neurogenesis, and this eventuated in an increase in weight gain. Thus, a benefit of central administration of CNTF or CNTF analogs is the ability to remain effective after therapy has been terminated. This prolonged effect may provide scope to trial drugs that could be used in a cyclic manner in the treatment of obesity. Further studies are warranted to fully elucidate the pathophysiological significance of CNTF-mediated hypothalamic neurogenesis.
While it has been known that CNTF exerts anorexic effects via activation of neurons in the arcuate nuclei of the hypothalamus for some years, the precise subset of cells that CNTF acts on in this region of the brain has remained elusive until recently. A transgenic mouse with a selective ablation of the gp130 receptor in anorexigenic proopiomelanocortin (POMC) expressing neurons \({\left( {{\text{gp130}}^{{\Delta {\text{POMC}}}} {\text{mice}}} \right)}\) was engineered [39]. The \({\text{gp130}}^{{\Delta {\text{POMC}}}} \) mice and littermate control mice displayed similar phenotypes when fed a normal chow or high-fat diet. When CNTF was administered centrally, the effect of centrally administered CNTF was abolished in \({\text{gp130}}^{{\Delta {\text{POMC}}}} \) mice compared with littermate control mice. This conclusively identified the precise neuronal pathway that CNTF uses in the hypothalamus. CNTF has also been shown to mediate effects on orexigenic NPY hypothalamic neurons. Interestingly, hypothalamic NPY mRNA expression was markedly decreased in CNTF-treated rats compared to their control counterparts [40]. In addition, NPY-induced feeding was considerably reduced in CNTF-treated animals. The authors concluded that CNTF-induced anorexia is partly due to reduced NPY supply. Hence, CNTF has effects on both POMC and NPY hypothalamic neurons.
It has recently been demonstrated that part of the central action of CNTF is due to reduced AMPK activation. When CNTFAx15 was administered intracerebroventricularly (ICV) and intraperitoneally (IP), AMPK α2 activity in the hypothalamus was reduced [41]. In addition, ICV treatment with CNTFAx15, promoted phosphorylation of STAT3, reduced phosphorylation of AMPK and ACC in the arcuate nucleus, induced hypophagia, and decreased body weight of mice fed a standard and/or high fat chow [41]. In conclusion, CNTF or CNTF analogs mediate hypothalamic control of energy balance by specific activation of POMC neurons in the arcuate nuclei via reduced AMPK activation. In addition, CNTF appears unique as its neurotrophic properties result in hypothalamic neurogenesis, allowing for the possibility of cyclic treatment regimes in the treatment of obesity (see Fig. 2). However, the neurotrophic effects of CNTF, which could possibly result in unwanted side effects, coupled with the observations that CNTF can activate inflammatory gene expression in the brain [36, 37], may limit the efficacy of CNTF as a centrally acting therapeutic agent.

Pathways by which CNTF mediates weight loss and insulin sensitivity. CNTF functions centrally via gp130 receptor signaling in proopiomelanocortin (POMC) expressing neurons in the hypothalamus to reduce AMPK activation. In addition, CNTF increases neurogenesis in the arcuate nuclei. Ultimately, hypophagia prevails. In peripheral organs/tissues such as skeletal muscle, CNTF upregulates AMPK activation, which eventuates in increased fatty acid oxidation. CNTF acts to decrease steatosis of the liver and lipid build up in skeletal muscle. The promotion of fatty acid oxidation and lowered lipid accumulation (diacyglyceride and ceramide) in liver and skeletal muscle decreases the activation of serine threonine kinases (JNK and IKK) and the transcription of SCD-1 in liver to improve lipid induced insulin resistance
CNTF: activity outside of the central nervous system
Two in vitro studies have eluded to the fact that CNTF may directly act on cells originating from peripheral tissues in a centrally independent manner. In the first study, CNTF was shown to stimulate STAT3, MAPK, Akt, and p70S6K in brown adipocytes [42]. The second study also demonstrated that CNTF may directly activate cultured 3T3-L1 preadipocytes and mature adipocytes as evidenced by STAT3 phosphorylation [43]. The authors of the latter study noted that the activation of cultured mature adipocytes by CNTF occurred despite the fact these adipocytes do not express the CNTFRα. This further highlighted that the IL-6Rα may serve as an α receptor for CNTF in adipocytes in vitro [27]. Interestingly, in contrast to the aforementioned in vitro results, the CNTFRα is highly expressed in the adipose tissue of numerous rodent models of obesity [43]. As CNTF is known to promote insulin sensitivity, the authors of this study hypothesized that the upregulation of the CNTFRα in adipose tissue of obese rodents may be a compensatory mechanism to increase insulin sensitivity. In addition, when wild-type mice fed a normal chow were treated with CNTF, phosphorylation of STAT3 occurred in skeletal muscle and adipose tissue. This observation also suggested that CNTF could exert peripheral, centrally independent actions, but it still remained to be eliminated that intraperitoneal administration of CNTF was not ultimately acting centrally. To test whether central administration of CNTF could result in activation of AMPK in murine skeletal muscle, CNTF was administered either IP or ICV, and skeletal muscle was dissected. Intracerebroventricular delivery of CNTF failed to have any effect on skeletal muscle. However, intraperitoneal administration of CNTF promoted activation of STAT3 and AMPK in red gastrocnemius muscle [16]. This was the first report that proved that CNTF could act in a centrally independent manner. In addition, numerous markers of fatty acid oxidation were elevated at the mRNA level in skeletal muscle after intraperitoneal administration of CNTF only. Although it may be inferred that increased insulin sensitivity and weight loss would result after the increased fatty acid oxidation, it still needs to be formally demonstrated. Chronic administration of CNTF to mice both intracerebroventricularly and intraperitoneally, followed by body weight measurements and glucose/insulin tolerance testing, will fulfill this aim. In the aforementioned study, the authors were able to also determine precise pathways for CNTF action in peripheral tissues such as skeletal muscle. CNTF promoted fatty acid oxidation in skeletal muscle in an AMPK-dependent manner. This was concluded when the CNTF-mediated increase in fatty acid oxidation was abrogated when skeletal muscle cells were infected with an AMPK-dominant negative adenovirus. Of great importance, insulin signal transduction and insulin action was restored in the skeletal muscle of mice treated with CNTF for 7 days compared with sham-treated pair-fed animals on a high fat diet. Unlike leptin, CNTF promoted phosphorylation of STAT3, AMPK, and ACC and increased fatty acid oxidation in skeletal muscle from mice fed a high-fat diet [16]. These results pointed to CNTF or CNTF analogs acting as anti-obesity targets [44] and eluded to a possible mechanism whereby gp130 ligands may overcome leptin resistance. The gp130β receptors and LRb are strikingly similar with regard to numerous aspects of their carboxyl domains. Of note, however, there are some critical differences. Similar to the LRb, where SOCS-3 can bind the SHP-2 domain, SOCS-3 can inhibit Jak/Stat signaling on the human and mouse gp130 receptor, by binding the SHP2/Tyr759 or Tyr757 binding site, respectively. It should be noted that the LRb receptor has 1 STAT-3 binding site (human: Tyr 1138), whereas, gp130 has 4 STAT-3 binding sites (human: Tyr 767, 814, 905, and 915) in their cytoplasmic domains. Therefore, it appears that CNTF can overcome SOCS-3 inhibition of receptor signaling because the gp130 receptor has an additional 3 STAT binding sites. A recent murine study has indicated that the four STAT3 binding domains on the gp130 receptor appear critical for promoting the beneficial metabolic effects in skeletal muscle after CNTF binding (Fig. 1b and c). This was clearly shown when \({\text{gp130}}^{{\Delta {\text{STAT}}}} \) and wild-type mice were treated with CNTF. The \({\text{gp130}}^{{\Delta {\text{STAT}}}} \)mice lack the STAT-3 binding sites in the cytoplasmic domain of gp130. Interestingly, activation of STAT3, AMPK, and ACC failed to occur in \({\text{gp130}}^{{\Delta {\text{STAT}}}} \) mice and culminated in an absence of fatty acid oxidation, which was in direct contrast to wild-type mice [16].
A hallmark of insulin resistance is the accumulation of lipid intermediates in peripheral organs such as skeletal muscle and liver [45, 46]. Stress kinases such as c-jun terminal amino kinase (JNK), which attenuates insulin signaling, may be activated by the production of fatty acid metabolites within insulin-responsive tissues [47–49]. Therefore, it is of prime importance that CNTF treatment of mice fed a high-fat diet greatly decreased the build up of lipid in skeletal muscle and the activation of serine kinase cascades [16]. In this same study, CNTF treatment promoted insulin sensitivity of mice fed a high-fat diet as evidenced by increased glucose uptake and insulin signaling in skeletal muscle. In support of the aforementioned murine studies, it was also shown that rats infused with lipid and treated with CNTFAx15 also displayed increased insulin responsiveness and decreased activation of JNK in skeletal muscle and diminished JNK and NFκB in the liver [50]. This was linked with lowered fat accumulation in skeletal muscle and liver. CNTF has also been shown to lower the degree of hepatic steatosis in conjunction with increases in liver function, liver insulin signaling, and metabolic rate, in db/db mice, which were administered CNTFAx15 for 10 days [51]. This same group also documented increases in uncoupling protein 1 (UCP1) mRNA levels in brown adipose tissue of mice treated with CNTF [52]. In an independent study, Liu et al. [53, 54] also demonstrated that 30 days of recombinant human ciliary neurotrophic factor (rhCNTF) administration to obese diabetic KK-Ay mice resulted in marked reductions in body weight, blood glucose, perirenal fat mass, serum-free fatty acids, and pancreatic islet triglycerides. Enhanced expression of UCP-1, NRF-1 (nuclear respiratory factor-1) and TFam (mitochondrial transcription factor A) was observed in brown adipose tissue after 3 days of rhCNTF administration in KK-Ay mice. In addition, rhCNTF treatment increased the activity of mitochondrial complex IV, which suggests that mitochondrial respiration was increased. This study has highlighted that upregulation of NRF-1 and TFam may contribute to increased UCP-1 expression after rhCNTF treatment. These latter observations are consistent with the fact that CNTF may upregulate peroxisome proliferator-activated receptor γ coactivator 1α (Ppargc1a) mRNA and protein expression in skeletal muscle [16] and brown adipose tissue [54]. Importantly, increased AMPK activity can directly phosphorylate PGC-1α to increase its activity [55], and a number of studies have recently implicated defective mitochondria in the etiology of insulin resistance and type 2 diabetes [56–59]. In summary, CNTF clearly acts centrally, and recent studies demonstrate that this gp130 ligand promotes insulin sensitivity and fatty acid oxidation in peripheral tissues in a centrally independent manner as depicted in Fig. 2.
Are mutations in human CNTF/CNTFRα functional?
A limited number of mutations occur in either the CNTF [60] or the CNTFRα gene [61]. As CNTF is clearly implicated in energy balance, several researchers have aimed to assess whether the CNTF or CNTFRα gene mutations are associated with body mass in humans. Whether the null mutation of the CNTF gene is associated with body weight in humans is equivocal, as the mutation in the CNTF gene appears to have little association with early onset obesity [62], but is associated with a 10-kg increase in body weight in older male Caucasians [63]. Most importantly, the C174T polymorphism in exon 9 of the CNTFRα gene correlated with fat-free mass in both sexes [61].
Efficacy of human clinical trials using CNTFAx15
As previously discussed, CNTFAx15, the human recombinant variant of CNTF, has been developed under the name Axokine®. The results from a phase II clinical trial were reported 3 years ago [63]. All subjects in this clinical trial had an average BMI of ~41. Interestingly, the weight of the control patients remained steady state, while the patients administered Axokine® lost 3–4 kg after 84 days. Unfortunately, patients administered high doses of Axokine® experienced nausea, and numerous subjects developed neutralizing anti-CNTFAx15 antibodies. Added to this, a follow-up study eluded that patients treated with Axokine® had gained weight [63]. It appears, therefore, that the follow up study using Axokine® has revealed somewhat disappointing results.
Fine tuning CNTF as an antiobesity therapy
As discussed, the IL6Rα is a promiscuous receptor for both IL-6 and CNTF ([16, 27] and Fig. 1c). However, IL-6 and CNTF still possess a greater degree of binding affinity for their specific α receptor. A vast number of studies have been conducted in an effort to ascertain the role that IL-6 plays in type 2 diabetes or insulin resistance. Currently, interleukin-6 has been implicated in both the promotion of insulin sensitivity [64] and resistance [65]. In addition, a major disadvantage of IL-6 therapy lies in the fact that sustained immunostimulation may occur. It is of considerable interest that the IL-6Rα is much more highly expressed in peripheral tissues such as skeletal muscle [16] compared with the CNTFRα. In addition, while CNTF delivered ICV seems to result in upregulation of inflammatory gene expression in the brain [36, 37], this does not appear to be the case in peripheral tissue [16]. Together, one potential therapeutic strategy may be to design a gp130 chimera that is “CNTF-like” in action, with a greater binding affinity for the IL-6Rα and which specifically targets peripheral tissue such as skeletal muscle and adipose. In fact, receptor recognition sites of gp130 cytokines are organized as exchangeable modules and various chimeras, where the site III loop of IL-6 has been substituted for the site III loop of CNTF have previously been reported [66]. The site III loop is situated on the C-terminal end of the protein and is the region which binds the receptor [64]. Whether “designer gp130 receptor ligands” may indeed prove to be the “holy grail” as an antiobesity drug remains to be tested.
References
Flegal KM, Carroll MD, Ogden CL, Johnson CL (2002) Prevalence and trends in obesity among US adults, 1999–2000. JAMA 288:1723–1727
Mascie-Taylor CG, Karim E (2003) The burden of chronic disease. Science 302:1921–1922
Giorgino F, Laviola L, Leonardini A (2005) Pathophysiology of type 2 diabetes: rationale for different oral antidiabetic treatment strategies. Diabetes Res Clin Pract 68(Suppl1):S22–S29
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432
Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM (1995) Weight-reducing effects of the plasma protein encoded by the obese gene. Science 269:543–546
Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Wool EA, Monroe CA, Tepper RI (1995) Identification and expression cloning of a leptin receptor, OB-R. Cell 83:1263–1271
Flier JS (2004) Obesity wars: molecular progress confronts an expanding epidemic. Cell 116:337–350
Oh-I S, Shimizu H, Sato T, Uehara Y, Okada S, Mori M (2005) Molecular mechanisms associated with leptin resistance: n−3 polyunsaturated fatty acids induce alterations in the tight junction of the brain. Cell Metab 1:331–341
Yoshimura A, Ohkubo T, Kiguchi T, Jenkins NA, Gilbert DJ, Copeland NG, Hara T, Miyajima A (1995) A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J 14:2816–2826
Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ (1997) A family of cytokine-inducible inhibitors of signaling. Nature 387:917–921
Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS (2004) Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med 10:734–738
Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A (2004) Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med 10:739–743
Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS (2006) Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab 4:123–132
Bjornholm M, Munzberg H, Leshan RL, Villanueva EC, Bates SH, Louis GW, Jones JC, Ishida-Takahashi R, Bjorbaek C, Myers Jr MG (2007) Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J Clin Invest 117:1354–1360
Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, Leranth C, Toran-Allerand D, Priest CA, Roberts JL, Gao XB, Mobbs C, Shulman GI, Diano S, Horvath TL (2007) Anorectic estrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med 13:89–94
Watt MJ, Dzamko N, Thomas WG, Rose-John S, Ernst M, Carling D, Kemp BE, Febbraio MA, Steinberg GR (2006) CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat Med 12:541–548
Ueki K, Kondo T, Tseng YH, Kahn CR (2004) Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc Natl Acad Sci USA 101:10422–10427
Steinberg GR, Parolin ML, Heigenhauser GJ, Dyck DJ (2002) Leptin increases FA oxidation in lean but not obese human skeletal muscle: evidence of peripheral leptin resistance. Am J Physiol Endocrinol Metab 283:E187–E192
Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, Caro JF (1996) Serum immunoreactive-leptin concentrations in normal-weight and obese humans. New Engl J Med 334:292–295
Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS (1995) Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med 1:1311–1314
Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, DePaoli AM, O’Rahilly S (2002) Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest 110:1093–1103
Adler R, Landa KB, Manthorpe M, Varon S (1979) Cholinergic neuronotrophic factors: intraocular distribution of trophic activity for ciliary neurons. Science 204:1434–1436
Lin LF, Mismer D, Lile JD, Armes LG, Butler ET 3rd, Vannice JL, Collins F (1989) Purification, cloning and expression of ciliary neurotrophic factor (CNTF). Science 246:1023–1025
Stöckli KA, Lottspeich F, Sendtner M, Masiakowski P, Carroll P, Gotz R, Lindholm D, Thoenen H (1989) Molecular cloning, expression and regional distribution of rat ciliary neurotrophic factor. Nature 342:920–923
Sleeman MW, Anderson KD, Lambert PD, Yancopoulos GD, Wiegand SJ (2000) The ciliary neurotrophic factor and its receptor, CNTFRα. Pharm Acta Helv 74:265–272
Davis S, Aldrich TH, Valenzuela DM, Wong VV, Furth ME, Squinto SP, Yancopoulos GD (1991) The receptor for ciliary neurotrophic factor. Science 253:59–63
Schuster B, Kovaleva M, Sun Y, Regenhard P, Matthews V, Grotzinger J, Rose-John S, Kallen KJ (2003) Signalling of human ciliary neurotrophic factor (CNTF) revisited: the interleukin-6 (IL-6) receptor can serve as an α-receptor for CNTF. J Biol Chem 278:9528–9535
Kallen K-J, Grotzinger J, Rose-John S (2000) New perspectives on the design of cytokines and growth factors. Trends Biotechnol 18:455–461
Davis S, Aldrich TH, Ip NY, Stahl N, Scherer S, Farruggelia T, DiStefano PS, Curtis R, Panayotatos N, Gascan H (1993) Released form of CNTF receptor alpha component as a soluble mediator of CNTF responses. Science 259:1736–1739
Chen X, Mao Z, Liu S, Liu H, Wang X, Wu H, Wu Y, Zhao T, Fan W, Li Y, Yew DT, Kindler PM, Li L, He Q, Qian L, Wang X, Fan M (2005) Dedifferentiation of adult human myoblasts induced by ciliary neurotrophic factor in vitro. Mol Biol Cell 16:3140–3151
Guillet C, Auguste P, Mayo W, Kreher P, Gascan H (1999) Ciliary neurotrophic factor is a regulator of muscular strength in aging. J Neurosci 19:1257–1262
ALS CNTF Treatment Study Group (1996) A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rhCNTF) in amyotrophic lateral sclerosis. Neurology 46:1244–1249
Gloaguen I, Costa P, Demartis A, Lazzaro D, Di Marco A, Graziani R, Paonessa G, Chen F, Rosenblum CI, Van der Ploeg LH, Cortese R, Ciliberto G, Laufer R (1997) Ciliary neurotrophic factor corrects obesity and diabetes associated with leptin deficiency and resistance. Proc Natl Acad Sci USA 94:6456–6461
Lambert PD, Anderson KD, Sleeman MW, Wong V, Tan J, Hijarunguru A, Corcoran TL, Murray JD, Thabet KE, Yancopoulos GD, Wiegand SJ (2001) Ciliary neurotrophic factor activates leptin-like pathways and reduces body fat, without cachexia or rebound weight gain, even in leptin-resistant obesity. Proc Natl Acad Sci USA 98:4652–4657
Kalra SP (2001) Circumventing leptin resistance for weight control. Proc. Natl Acad Sci USA 98:4279–4281
Kelly JF, Elias CF, Lee CE, Ahima RS, Seeley RJ, Bjorbaek C, Oka T, Saper CB, Flier JS, Elmquist JK (2004) Ciliary neurotrophic factor and leptin induce distinct patterns of immediate early gene expression in the brain. Diabetes 53:911–920
Prima V, Tennant M, Gorbatyuk OS, Muzyczka N, Scarpace PJ, Zolotukhin S (2004) Differential modulation of energy balance by leptin, ciliary neurotrophic factor, and leukemia inhibitory factor gene delivery: microarray deoxyribonucleic acid-chip analysis of gene expression. Endocrinology 145:2035–2045
Kokoeva MV, Yin H, Flier JS (2005) Neurogenesis in the hypothalamus of adult mice: potential role in energy balance. Science 310:679–683
Janoschek R, Plum L, Koch L, Munzberg H, Diano S, Shanabrough M, Muller W, Horvath TL, Bruning JC (2006) gp130 signaling in proopiomelanocortin neurons mediates the acute anorectic response to centrally applied ciliary neurotrophic factor. Proc Natl Acad Sci USA 103:10707–10712
Xu B, Dube MG, Kalra PS, Farmerie WG, Kaibara A, Moldawer LL, Martin D, Kalra SP (1998) Anorectic effects of the cytokine, ciliary neurotrophic factor, are mediated by hypothalamic neuropeptide Y: Comparison with leptin. Endocrinology 139:466–473
Steinberg GR, Watt MJ, Fam BC, Proietto J, Andrikopoulos S, Allen AM, Febbraio MA, Kemp BE (2006) Ciliary neurotrophic factor suppresses hypothalamic AMP-kinase signaling in leptin-resistant obese mice. Endocrinology 147:3906–3914
Ott V, Fasshauer M, Dalski A, Klein HH, Klein J (2002) Direct effects of ciliary neurotrophic factor on brown adipocytes: evidence for a role in peripheral regulation of energy homeostasis. J Endocrinol 173:R1–R8
Zvonic S, Cornelius P, Stewart WC, Mynatt RL, Stephens JM (2003) The regulation and activation of ciliary neurotrophic factor signaling proteins in adipocytes. J Biol Chem 278:2228–2235
Ahima RS (2006) Overcoming insulin resistance with CNTF. Nat Med 12:511–512
Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI (1999) Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 42:113–116
Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA, Bogardus C, Jenkins AB, Storlien LH (1997) Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 46:983–988
Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim DW, Liu ZX, Soos TJ, Cline GW, O’Brien WR, Littman DR, Shulman GI (2004) PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest 114:823–827
Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE (2005) Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 11:183–190
Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS (2003) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Watt MJ, Hevener A, Lancaster GI, Febbraio MA (2006) Ciliary neurotrophic factor prevents acute lipid-induced insulin resistance by attenuating ceramide accumulation and phosphorylation of c-Jun N-terminal kinase in peripheral tissues. Endocrinology 147:2077–2085
Sleeman MW, Garcia K, Liu R, Murray JD, Malinova L, Moncrieffe M, Yancopoulos GD, Wiegand SJ (2003) Ciliary neurotrophic factor improves diabetic parameters and hepatic steatosis and increases basal metabolic rate in db/db mice. Proc Natl Acad Sci USA 100:14297–14302
Bluher S, Moschos S, Bullen J Jr, Kokkotou E, Maratos-Flier E, Wiegand SJ, Sleeman MW, Mantzoros CS (2004) Ciliary neurotrophic factorAx15 alters energy homeostasis, decreases body weight, and improves metabolic control in diet-induced obese and UCP1-DTA mice. Diabetes 53:2787–2796
Liu Q-S, Wang Q-J, Du G-H, Zhu S-Y, Gao M, Zhang L, Zhu J-M, Cao J-F (2007) Recombinant human ciliary neurotrophic factor reduces weight partly by regulating nuclear respiratory factor 1 and mitochondrial transcription factor A. Eur J Pharmacol 563:77–82
Liu Q-S, Gao M, Zhu S-Y, Li S-J, Zhang L, Wang Q-J, Du G-H (2007) The novel mechanism of recombinant human ciliary neurotrophic factor on the anti-diabetes activity. Basic Clin Pharmacol Toxicol 101:78–84
Jäger S, Handschin C, St-Pierre J, Spiegelman BM (2007) AMP AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA 104:12017–12022
Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI (2003) Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300:1140–1142
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. New Engl J Med 350:664–671
Mootha V, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273
Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 100:8466–8471
Takahashi R, Yokoji H, Misawa H, Hayashi M, Hu J, Deguchi T (1994) A null mutation in the human CNTF gene is not causally related to neurological diseases. Nat Genet 7:79–84
Roth SM, Schrager MA, Ferrell RE, Riechman SE, Metter EJ, Lynch NA, Lindle RS, Hurley BF (2001) CNTF genotype is associated with muscular strength and quality in humans across the adult age span. J Appl Physiol 90:1205–1210
Munzberg H, Tafel J, Busing B, Hinney A, Ziegler A, Mayer H, Siegfried W, Matthaei S, Greten H, Hebebrand J, Hamann A (1998) Screening for variability in the ciliary neurotrophic factor (CNTF) gene: no evidence for association with human obesity. Exp Clin Endocrinol Diabetes 106:108–112
Ettinger MP, Littlejohn TW, Schwartz SL, Weiss SR, McIlwain HH, Heymsfield SB, Bray GA, Roberts WG, Heyman ER, Stambler N, Heshka S, Vicary C, Guler HP (2003) Recombinant variant of ciliary neurotrophic factor for weight loss in obese adults: a randomized, dose-ranging study. JAMA 289:1826–1832
Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, Prelovsek O, Hohnen-Behrens C, Watt MJ, James DE, Kemp BE, Pedersen BK, Febbraio MA (2006) Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 55:2688–2697
Klover PJ, Zimmers TA, Koniaris LG, Mooney RA (2003) Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes 52:2784–2789
Kallen KJ, Grotzinger J, Lelievre E, Vollmer P, Aasland D, Renne C, Mullberg J, Myer zum Buschenfelde KH, Gascan H, Rose-John S (1999) Receptor recognition sites of cytokines are organized as exchangeable modules. Transfer of the leukemia inhibitory factor receptor-binding site from ciliary neurotrophic factor to interleukin-6. J Biol Chem 274:11859–11867
Peelman F, Couturier C, Dam J, Zabeau L, Tavernier J, Jockers R (2006) Techniques: New pharmacological perspectives for the leptin receptor. Trends Pharmacol Sci 27:218–225
Ernst M, Jenkins BJ (2004) Acquiring signaling specificity from the receptor gp130. Trends Genet 20:23–32
Acknowledgements
The support from the National Health and Medical Research Council of Australia (NHMRC), The Australian Research Council, and The Diabetes Australia Research Trust is gratefully acknowledged. VBM is supported, in part, by a Baker Heart Research Institute Early Career Scientist (ECS) Grant; MAF is supported by a Principal Research Fellowship from the NHMRC.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Matthews, V.B., Febbraio, M.A. CNTF: a target therapeutic for obesity-related metabolic disease?. J Mol Med 86, 353–361 (2008). https://doi.org/10.1007/s00109-007-0286-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-007-0286-y