
Abstract
Recent evidence for the involvement of zinc in the formation of β-amyloid plaques in the brain in Alzheimer's disease has led to the establishment of new therapeutic strategies for the degenerative disorder based on metal chelation. The present experiment was conducted on a membrane-permeable zinc chelator, clioquinol (CQ), that has shown potential in initial studies on a mouse model of Alzheimer's disease [1]. The degree of chelatable zinc in mice treated with CQ, delivered by two different routes, was measured using complementary protocols for identifying chelatable zinc: 6-methoxy-8-quinolyl-p-toluenesulfonamide (TSQ) histofluorescence, and selenite autometalography. Mice injected intraperitoneally with CQ showed a dramatic reduction in chelatable zinc in brain, testis, and pancreas. In contrast, mice given CQ orally showed no significant change in levels of chelatable zinc in these tissues. This suggests that CQ administered orally to patients with Alzheimer's disease should not significantly perturb chelatable zinc levels in key organs and may be used over long periods without adverse endocrinological and reproductive effects related to zinc deficiency. In contrast, CQ injected intraperitoneally may be used not only as a tool for investigating chelatable zinc pools but also in a clinical context. For example, injected CQ could be employed in situations requiring the rapid buffering of excessive chelatable zinc following ischemic episodes or brain trauma. Thus, our findings indicate that CQ has considerable potential as a versatile scientific and clinical tool used for selective modulation of zinc pools.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Zinc participates in a wide variety of physiological and pathophysiological processes in the body, for instance, as a key component of numerous enzymes and transcription factors (for review see [2]). In addition to this tightly bound zinc, a second, chelatable "pool" of zinc exists which, while smaller, is more physiologically labile. The function of chelatable zinc is largely unknown, although studies of proteins that regulate intracellular levels of chelatable zinc have begun to shed light on its actions in various organs in the body. In the pancreas, for example, where zinc plays a structural role in coordinating insulin incorporation into secretory vesicles in pancreatic β-cells, a number of reports have detailed the role of zinc in the creation of insulin hexamers [3, 4]. Excessive secretion of zinc, however, has been linked to the death of these same cells in a model of type 1 diabetes [5]. Similarly, chelatable zinc in the brain is involved in modulating synaptic activity at excitatory and inhibitory synapses and is also believed to be a key element in events leading to cell death during episodes of excessive release (for review see [6]).
To better understand the functions of zinc and the mechanism(s) by which it acts, a number of approaches, including application of zinc chelators and specific gene knockouts of cellular zinc-regulating elements [7], have been employed. A general limitation of the knockout models is that compensatory mechanisms may be activated which prevent the natural cascade of events that would be expected to occur following the sudden loss of the proteins encoded by the deleted genes. In addition, some of these proteins, are expressed only in one area and not another, for example, metallothionein III [8]. To study the general role of zinc in the body, a zinc-specific chelator is needed that can gain entry to many organs in an in vivo model but does not interfere with the tightly bound cellular zinc pool, such as zinc fingers and numerous catalytic enzymes that are essential for normal cellular function. A zinc chelator such as tetrakis-(2-pyridylmethyl) ethylendediamine (TPEN; K d=2.6×10−16 M), having a strong affinity for zinc, might interfere with protein bound zinc, making it a less likely to meet those criteria.
One possible candidate is the antibiotic, clioquinol (5-chloro-7-iodo-8-hydroxyquinoline; CQ). For many years CQ was banned from clinical practice over questions regarding its contribution to the development of subacute myelo-optico-neuropathy (for review see [9]). Recently however, CQ has gained renewed interest following the demonstration that it can reduce or prevent the formation of Alzheimer's disease-type β-amyloid plaques by chelating zinc [1] and MPTP-induced parkinsonism by chelating iron [10]. In contrast to TPEN, CQ has a relatively weak affinity (K d approx. 1×10−7 M [1]) for zinc and would therefore be expected to interact exclusively with loosely bound, i.e., chelatable zinc. The lipophilic nature of this compound moreover suggests that it will have ready access to organs and tissues throughout the body. Finally, CQ has been shown to possess a low degree of toxicity [1, 11].
Studies of the effects of endogenous zinc in mammals in both physiological and pathophysiological contexts have exploited chelating agents to elucidate the specific role played by this ion (e.g., [12, 13]). CQ has characteristics which have been suggested to make it particularly well suited for both in vivo experimental and clinical applications.
The present study assessed the effect of in vivo administration of CQ through two alternative routes on the chelatable zinc pool in three areas rich in chelatable zinc: brain, testis, and pancreas. Our results indicate that CQ administered intraperitoneally dramatically reduces chelatable zinc in these areas. However, CQ administered by mouth did not produce a significant effect on tissue chelatable zinc. The physiological and pathophysiological implications of our findings are discussed.
Materials and methods
CD-1 male mice, 8–10 weeks old, were used throughout this study. The animals were fed ad libitum and maintained under standard laboratory conditions of 24°C and 12–12 h light-dark cycle. Treatment, specimen collection, staining, and quantification were performed in a double-blind manner.
Clioquinol administration and specimen collection
Mice (n=18) were injected intraperitoneally with CQ 30 mg/kg (dissolved in dimethyl sulfoxide) or vehicle. A second set of mice (n=8) were administered CQ 30 mg/kg (emulsified in 2% carboxymethylcellulose) or vehicle per os. The mice were anesthetized 1.5–2 h after injection with Nembutal, and the brain, right testicle, and pancreas were removed and frozen on dry ice. Alternatively, mice from all treatment groups, received an intraperitoneal injection of sodium selenite (20 mg/kg) 30–60 min after the CQ/sham treatment and were killed by cervical dislocation 1 h later. Both orally and intraperitoneally treated animals were killed at the same time, post-administration. All tissues were sectioned frozen (12 µm) on a Minotome cryostat (IEC) and thaw-mounted onto glass slides. A separate group of animals were assessed for effects of CQ after 1 week of daily injections. These animals exhibited no ill effects of the treatment (data not shown).
Selenite autometalography
Brain sections collected from specimens treated with sodium selenite prior to killing were immersed in a developer solution as described previously [14]. After rinsing and fixation the slides were Nissl-counterstained, dehydrated through an ascending series of ethanols to xylene, and mounted with Eukitt (O. Kindler, Germany).
TSQ histofluorescence
Histofluorescence with the zinc-sensitive dye, 6-methoxy-8-quinolyl-p-toluenesulfonamide (TSQ), was carried out as described previously [15]. Tissue images were captured into a PC workstation with a digital camera (SPOT RT, Diagnostic Instruments, Mich., USA) using a UV-2A filter block (330–380 nm; barrier 420), and fluorescent signal density determined using NIH image 1.62 (Wayne Rasband, NINDS, NIH). Data were analyzed by the Mann-Whitney U test (InStat 2.00, GraphPad, Calif., USA). Brain tissue fluorescence was normalized to a control section through the hilus of the hippocampus.
Results
Selenite labeling of synaptic zinc was unambiguous in the vehicle-injected brains (Fig. 1A, C, and E). Intense labeling was observed in the zinc-containing mossy fibers of the hippocampus as well as in layers 2/3 and 5 of the neocortex in dimethyl sulfoxide injected mice. In contrast, following the CQ treatment this labeling was virtually absent (Fig. 1B, D, and F). TSQ histofluorescence confirmed this result (data not shown). This finding indicates that treatment with CQ indeed dramatically reduced the availability of free zinc in major chelatable zinc pools in the brain. In contrast per os CQ treatment, shown previously to reduce the senile plaques in Alzheimer's disease mouse model [1], did not result in significant changes in the brain chelatable zinc stores with respect to vehicle fed controls, as was assessed by both methods used in this study, TSQ histofluorescence or selenite autometalography histochemistry (Figs. 2, 3 respectively).

Micrographs illustrating selenite autometalography distribution from three distinct brain regions. Cerebral cortex (A, B), hippocampus (C, D) and olfactory bulb (E, F) of sham vs. CQ (intraperitoneally) treated animals, respectively. Note the virtual elimination of synaptic zinc in all regions following CQ treatment. II/III, IV, V cortical layers 2/3, 4, and 5, respectively; DG dentate gyrus; p pyramidal layer; arrows CA3 mossy fibers; Cx cortex; Gr granule cell layer; g glomerular cell layer. Bar 250 µm

TSQ histofluorescence in the CA3 mossy fibers (A, B; asterisks) and in the hilus (C, D; h) of the dentate gyrus following sham (A, C) vs. CQ (oral) treatment (B, D). No difference in fluorescence was detected in oral CQ vs. sham-treated brains. Bar 40 µm

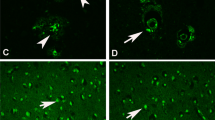
Selenite autometalography histochemical labeling of hippocampus in orally treated sham control (A) and CQ subjects (B). No difference in histochemically detectable zinc is evident in the two sections. Bar 100 µm
Secretion of zinc by pancreatic β-cells has been suggested to be involved in cell damage, similar to the toxic effects of zinc on neurons [5]. As shown in Fig. 4, intraperitoneal CQ had a dramatic effect on detectable tissue zinc also in the pancreas. The intense fluorescence observed in pancreatic islets from control subjects (Fig. 4A) was reduced to undetectable levels in the treated mice (Fig. 4B). Thus the effect of CQ on pancreatic free zinc may indicate its utility as a tool for preventing or reducing such damage. Orally administered CQ on the other hand had no significant effect on islet zinc in comparison with control mice (Fig. 5).

TSQ fluorescence in pancreas (A, B) and testis (C, D) of sham vs. CQ (intraperitoneally) treated mice, respectively. Free zinc is not discernible following CQ treatment. TSQ histofluorescence is shown in a single pancreatic islet (A) and following CQ (B). Bar 100 µm

TSQ fluorescence in pancreas (A, B) and testis (C, D) of (oral) sham vs. CQ treated mice, respectively. Oral CQ treatment had no measurable effect on chelatable zinc in these tissues. Two pancreatic islets and a single pancreatic islet are shown in Fig. 4A and B, respectively. Bar 100 µm
The role of zinc in the maturation and function of testis is well documented (for review see [2]). Indeed, in control mice, strong zinc fluorescence was frequently present throughout the basal-apical extent of the seminiferous tubules (Fig. 4C). Chelatable zinc in the seminiferous tubules of the testis was virtually eliminated by intraperitoneal CQ but, again, not by oral CQ (Fig. 4D, 5D). Chelatable zinc was also reduced significantly in the testosterone-producing Leydig cells in the interstitial tissue located between the tubules by intraperitoneal treatment with CQ, but not by CQ per os treatment.
Quantitative analysis of fluorescence intensity of TSQ in tissue sections obtained from animals treated with CQ was compared to sham control animals in the above tested organs where intense zinc homeostasis occurs. While vehicle-treated animals exhibited a normal distribution of zinc in all the areas analyzed, a virtual absence of free zinc was observed in mice injected with CQ, although not in animals treated orally. These data are summarized in Fig. 6.

Densitometric values for TSQ fluorescence in hippocampus, pancreas, and testis of sham-treated, and CQ-treated animals injected intraperitoneally (A) vs. oral gavage (B). Note the lack of detectable fluorescence in all tissues sampled for animals treated intraperitoneally with CQ. Columns, error bars represent averages (n=6) and standard deviations, respectively. Brain regions represent data normalized to a control hippocampal hilus. *P<0.01, Mann-Whitney U test
Discussion
Methodology
The present study assessed the effects of CQ on tissue chelatable zinc stores 1.5–2 h after administration. This time was chosen based on studies indicating rapid absorption and clearance rates of this agent [16, 17]. Subtle differences in histochemical zinc detection in the brain have been described in the literature with regard to one or another method. For this reason free zinc was assayed in the brain using two different methods: TSQ histofluorescence and selenite autometalography. Both techniques are widely employed to demonstrate this transition metal in its loosely bound and chelatable form [14, 15]. In the present case the results of the two methods were nearly identical, further supporting our conclusions regarding the effects of CQ.
The principle finding of the present work is that CQ, injected intraperitoneally but not ingested orally, rapidly targets chelatable zinc stores in brain and in peripheral tissues in mice. How long this effect lasts is not currently known. To our knowledge, this is the first report on the efficacy of CQ with respect to chelatable zinc in vivo. The fact that CQ is currently being investigated as a potential therapeutic agent because of its ability to inhibit the formation of Alzheimer's disease type β-amyloid plaques in a transgenic mouse model underscores the importance of our results linking CQ to chelatable zinc. Indeed, there is strong evidence that the mechanism by which CQ, administrated per os, represses plaque formation is by its chelating effects on zinc in the plaques [1]. Nevertheless, questions about the nature of CQ's chelating activity per se have not been addressed in vivo. CQ administered per os is well suited for its proposed use in preventing accumulation of amyloid plaques in Alzheimer's disease. The findings presented in this study suggest that the reduction in the β-amyloid plaque load following exposure to this agent results from minute changes in the (protein-metal complex protein + metal) equilibrium rather than from a gross effect on chelatable zinc stores. Thus the effect shown in the present study is probably a local zinc probe blocking effect (via chelation) rather than actual removal of this ion from tissue. Indeed, it has been shown previously that CQ treatment does not decrease tissue zinc levels [1, 18]. Our finding further suggests that CQ may well find wider application in situations requiring more robust chelating actions when administered through a non-oral route.
The principle of using chelators to explore the significance of metal ions or metal-specific detection methods in biology is not novel. Dithizone, for example, was previously employed in the determination of the Timm's and selenite methods [19, 20, 21, 22, 23]. Diethylthiocarbamate, an active metabolite of disulfiram (Antabuse), has been used in a similar manner [24] and has also been used to demonstrate zinc-dependent functions in various paradigms (e.g., [25, 26, 27, 28]). EDTA, a membrane impermeable chelator still used in clinical practice to treat heavy metal intoxication (for review see [12]), has also been employed to assess various zinc-mediated physiological processes (e.g., [29, 30, 31]). Recently, EDTA was used to assess the role of synaptically released zinc in induction of long-term potentiation [32]. The present study suggests that CQ, by virtue of its differential effects on various zinc pools, can be exploited for both research and therapeutic purposes. This is of particular relevance in light of the current interest in the role of metal chelation as a therapeutic strategy for neurodegenerative diseases [1, 10].
Linking cerebral effects exerted by CQ to synaptic zinc
Under pathological conditions, for example, brain ischemia, zinc is massively released from forebrain glutamatergic terminals (for review see [6]) and possibly from intracellular stores [30] and is believed to mediate cellular processes leading to neuronal death (for review see [33]). In our hands, intraperitoneal CQ treatment rapidly and thoroughly reduced the levels of chelatable zinc, suggesting the utility of such a strategy in treating this type of acute event. Indeed, increased cell survival in vitro and in vivo after treatment with zinc chelators has been reported previously (e.g., [34, 35, 36]), although these findings remain inconclusive and require further investigation [25, 28]. Moreover, not all chelators are equal; a clear if paradoxical advantage of CQ over other membrane-permeable zinc chelators is its relatively low affinity for zinc (with respect, for example, to TPEN). This would diminish the chances that it would affect zinc bound to functionally important proteins, for example, zinc fingers.
Implications of zinc chelation in peripheral tissues
In the pancreas zinc may play a dual role. On the one hand, free zinc is known to be involved in synthesis and storage of insulin, is co-secreted with this hormone [37, 38], and exerts an insulomimetic effect through a mechanism that is not well understood (for review see [39]). On the other hand, chelatable zinc is highly toxic to β-cells in streptozotocin-induced diabetes [5]. Our finding that CQ reduces free zinc in the islets of Langerhans may suggest a clinical application for this antibiotic in preventing zinc-related degeneration of β-cells [39]. The fact that oral CQ did not cause a measurable reduction in chelatable zinc in the pancreas is reassuring, for example, in the context of its potential use as a therapeutic agent in the long-term treatment of Alzheimer's disease.
Zinc is essential for proper development of the male reproductive system. Zinc ions in the testis are found in distinct cell types in the seminiferous tubule [40, 41, 42] and are associated with specific stages of spermatogenesis [43]. However, a key question that remains unsolved is whether it is the strongly bound or the free-chelatable zinc that exerts these physiological functions. We have shown that intraperitoneal CQ sharply reduces chelatable zinc in testis, both in the seminiferous tubules and in the endocrine Leydig cells. Thus CQ appears to be an excellent tool for analyzing the specific role of the chelatable zinc pool in spermatogenesis and in other functions of the testis. On the other hand, orally administered CQ does not appear to significantly reduce testicular chelatable zinc, thus reducing the risk of compromised testicular function (e.g., [42]) in the context of long-term treatment in this manner.
Our overall findings suggest that with respect to the effects on tissue zinc, the agent, administered orally, can be safely employed. Intraperitoneally injected CQ, in contrast, could have a dramatic effect on the bioavailability of chelatable zinc in vivo. This effect illustrates the potential utility of CQ as a versatile and selective tool, either for removal of zinc from plaques or for fast elimination of chelatable zinc pools. The latter may have implications, as mentioned above, for short-term treatment in acute conditions such as stroke, acute brain trauma, and epilepsy, in which massive zinc release is associated with neurodegeneration [6, 33]. An advantage of such an approach is that such acute and short treatment would avoid the side effects of the CQ that are generally manifested after long-term treatment [9].
What is the pharmacological basis for the striking differences between the per os and intraperitoneal effects of CQ on chelatable zinc pools? Dramatic differences in tissue/blood CQ accumulation following oral vs. intraperitoneal administration in vivo have been described previously [17]. This phenomenon has been attributed to several factors, including lower absorption rates and first pass liver metabolism reported for per os treatment [44]. CQ injected intraperitoneally appears to pass directly into the blood stream over the large surface area represented by the peritoneum. Thus the marked difference in the CQ accumulation may explain the dramatic difference in its chelation potential.
Conclusions
In the present study we show that CQ dramatically reduces chelatable zinc following short-term intraperitoneal treatment in brain, pancreas, and testis. CQ administered orally, in contrast, had little or no effect. These data suggest that CQ may find wide application both as a research tool to elucidate the physiological role of chelatable zinc in various systems and as a therapeutic agent in the context of syndromes in which zinc plays an operative role, for example, epilepsy, stroke, and possibly diabetes. Moreover, our findings provide further support for the safety of using CQ as an oral agent to treat Alzheimer's disease patients without apparent chelatable zinc physiology related consequences.
Abbreviations
- CQ :
-
Clioquinol
- TPEN :
-
Tetrakis-(2-pyridylmethyl) ethylendediamine
- TSQ :
-
6-Methoxy-8-quinolyl-p-toluenesulfonamide
References
Cherny RA, et al (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30:665–676
Vallee BL, Falchuk KH (1993) The biochemical basis of zinc physiology. Physiol Rev 73:79–118
Bentley G, et al (1976) Structure of insulin in 4-zinc insulin. Nature 261 166–168
Dodson G, Steiner D (1998) The role of assembly in insulin's biosynthesis. Curr Opin Struct Biol 8:189–194
Kim BJ, et al (2000) Zinc as a paracrine effector in pancreatic islet cell death. Diabetes 49:367–372
Choi DW, Koh JY (1998) Zinc and brain injury. Annu Rev Neurosci 21:347–375
Cole TB, et al (1999) Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci U S A 96:1716–1721
Palmiter RD, et al (1992) MT-III, a brain-specific member of the metallothionein gene family. Proc Natl Acad Sci U S A 89:6333–6337
Tateishi J (2000) Subacute myelo-optico-neuropathy: clioquinol intoxication in humans and animals. Neuropathology 20 [Suppl]:S20–S24
Kaur D, et al (2003) Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron 37:899–909
Regland B, et al (2001) Treatment of Alzheimer's disease with clioquinol. Dement Geriatr Cogn Disord 12:408–414
Aposhian HV, et al (1995) Mobilization of heavy metals by newer, therapeutically useful chelating agents. Toxicology 97:23–38
Aballay A, et al (1995) Zn2+ depletion blocks endosome fusion. Biochem J 312:919–923
Danscher G (1982) Exogenous selenium in the brain. A histochemical technique for light and electron microscopical localization of catalytic selenium bonds. Histochemistry 76:281–293
Frederickson CJ, et al (1987) A quinoline fluorescence method for visualizing and assaying the histochemically reactive zinc (bouton zinc) in the brain. J Neurosci Methods 20:91–103
Toyokura Y, Takasu T, Matsuoka O (1975) Experimental studies utilizing radionuclide-labelled clioquinol as tracer in vivo. Jpn J Med Sci Biol 28 [Suppl]:79–86
Ogata M, et al (1973) Accumulation of clioquinol in mice. Lancet I:1325
Yassin MS, et al (2000) Changes in uptake of vitamin B (12) and trace metals in brains of mice treated with clioquinol. J Neurol Sci 173:40–44
Danscher G (1984) Similarities and differences in the localization of metals in the rat brain after treatment with sodium sulphide and sodium selenite. In Frederickson CJ, Howell GA, Kasarskis EJ (eds) Neurobiology of zinc. Liss, New York, pp 229–242
Danscher G (1984) Do the Timm sulphide silver method and the selenium method demonstrate zinc in the brain? In: Frederickson CJ, Howell GA, Kasarskis EJ (eds) Neurobiology of zinc. Liss, New York, pp 273–287
Danscher G, et al (1985) The dithizone, Timm's sulphide silver and the selenium methods demonstrate a chelatable pool of zinc in CNS. A proton activation (PIXE) analysis of carbon tetrachloride extracts from rat brains and spinal cords intravitally treated with dithizone. Histochemistry 83:419–422
Haug FM, Danscher G (1971) Effect of intravital dithizone treatment on the Timm sulfide silver pattern of rat brain. Histochemie 27:290–299
Otsuka N, Ibata Y (1966) Quantitative changes of zinc content in the hippocampal region after dithizone administration. Arch Histol Jpn 27:419–424
Danscher G, Haug FM, Fredens K (1973) Effect of diethyldithiocarbamate (DEDTC) on sulphide silver stained boutons. Reversible blocking of Timm's sulphide silver stain for "heavy" metals in DEDTC treated rats (light microscopy). Exp Brain Res 16:521–532
Lees GJ, Cuajungco MP, Leong W (1998) Effect of metal chelating agents on the direct and seizure-related neuronal death induced by zinc and kainic acid. Brain Res 799:108–117
Sorensen, et al MB (1999) Chelation of intracellular zinc ions affects human sperm cell motility. Mol Hum Reprod 5:338–341
Varea E, et al (2001) Imaging synaptic zinc release in living nervous tissue. J Neurosci Methods 110:57–63
Dominguez MI, et al (2003) Zinc chelation during non-lesioning overexcitation results in neuronal death in the mouse hippocampus. Neuroscience 116:791–806
Frederickson RE, Frederickson CJ, Danscher G (1990) In situ binding of bouton zinc reversibly disrupts performance on a spatial memory task. Behav Brain Res 38:25–33
Lee JY, et al (2000) Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J Neurosci 20:RC79
Frederickson CJ, et al (2002) Depletion of intracellular zinc from neurons by use of an extracellular chelator in vivo and in vitro. J Histochem Cytochem 50:1659–1662
Li Y, et al (2001) Induction of mossy fiber ->Ca3 long-term potentiation requires translocation of synaptically released Zn2+. J Neurosci 21:8015–8025
Weiss JH, Sensi SL, Koh JY (2000) Zn (2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci 21:395–401
Lee JM, et al (2002) Zinc translocation accelerates infarction after mild transient focal ischemia. Neuroscience 115:871–878
Park JA, et al (2000) Co-induction of p75NTR and p75NTR-associated death executor in neurons after zinc exposure in cortical culture or transient ischemia in the rat. J Neurosci 20:9096–9103
Suh SW, et al (2000) Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res 852:268–273
Zalewski PD, et al (1994) Video image analysis of labile zinc in viable pancreatic islet cells using a specific fluorescent probe for zinc. J Histochem Cytochem 42:877–884
Gee KR, et al (2002) Detection and imaging of zinc secretion from pancreatic beta-cells using a new fluorescent zinc indicator. J Am Chem Soc 124:776–778
Chausmer AB (1998) Zinc, insulin and diabetes. J Am Coll Nutr 17:109–115
Stoltenberg M, et al (1997) Autometallographic demonstration of zinc ions in rat sperm cells. Mol Hum Reprod 3:763–767
Stoltenberg M, et al (1996) Histochemical localization of zinc ions in the epididymis of the rat. Histochem J 28:173–185
Sorensen MB, et al (1998) Histochemical tracing of zinc ions in the rat testis. Mol Hum Reprod 4:423–428
Wong WY, et al (2000) Male factor subfertility: possible causes and the impact of nutritional factors. Fertil Steril 73:435–442
Watanabe S, et al (1973) Distribution of clioquinol in rats with hepatic dysfunction. Lancet II:681–682
Acknowledgements
We are grateful to Prof. Z. Ben-Zvi for expertise in applying the oral gavage technique. This work was supported by the German-Israel Binational Foundation (Grant #10588099) and the Israel Science Foundation (Grant #456) to I.S; The National Institutes of Health (Grants # NS042882, NS041682) to C.F.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nitzan, Y.B., Sekler, I., Frederickson, C.J. et al. Clioquinol effects on tissue chelatable zinc in mice. J Mol Med 81, 637–644 (2003). https://doi.org/10.1007/s00109-003-0462-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-003-0462-7