
Abstract
B7-H1 is a recently identified member of the B7 family molecules. Upon ligation to its receptors on T cells it regulates activation and differentiation of T cells. B7-H1 preferentially costimulates IL-10 production in resting T cells and further induces the apoptosis of activated T cells. PD-1 is a receptor of B7-H1 and is shown to mediate the inhibition of activated T cell response, presumably by inhibiting cell cycle progression. The expression of B7-H1 protein is limited to macrophage lineage of cells in normal tissues, although its mRNA transcription is found in a broad range of tissues. In contrast, B7-H1 is abundant in various human cancers. The tumor-associated B7-H1 increases apoptosis of antigen specific T cells, leading to growth of immunogenic tumor growth in vivo. Current data suggest that B7-H1 regulates the organ-specific tolerance in normal tissue and may contribute to immune evasion by cancers. Selective manipulation of B7-H1 pathway thus aids in the design of new regimens in the treatment of human autoimmune disease and the control of malignant cancers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Modulation of immune responses in tumor sites is a critical mechanism attributing to tumor evasion. There is accumulating evidence for the presence of tumor-specific T cells with phenotypes indicating that they have been previously activated or underwent programmed cell death [1, 2, 3], yet tumor growth is not controlled. In addition, adoptive immunotherapy using in vitro activated T cells, although some T cells were capable of localizing tumors, has not been satisfactory [4, 5]. It appears there is a microenvironment with immune privilege around cancers that protects cancer cells from immune destruction [3, 6, 7, 8]. Both soluble factors and membrane-bound molecules, including transforming growth factor β, interleukin (IL) 10, prostaglandin E2, Fas, tumor necrosis factor related apoptosis inducing ligand (TRAIL), and RACS1, have been found to be upregulated in tumor sites, which potentially inhibit immune responses. We refer to several excellent reviews for these findings [9, 10, 11]. In this contribution we focus our analysis in a recently discovered B7-H1 pathway and its role in the evasion of tumor immunity.
Structure and expression of B7-H1
By searching molecules sharing homology with the immunoglobulin V and C domains of B7-1 (CD80) and B7-2 (CD86) among the human cDNA expressed sequence tags in NCBI database, we identified the third molecule of the B7 family designated as B7-homolog 1 (B7-H1) [12]. Human B7-H1 gene is localized on human chromosome 9p24. The extracellular domain of human B7-H1 amino acid sequence shares higher homology to B7-1 (20%) and B7-2 (15%). The open reading frame of the B7-H1 gene encodes a putative type I transmembrane protein of 290 amino acids consisting of a IgV-like domain, a IgC-like domain, a hydrophobic transmembrane domain, and a cytoplasmic tail of 30 amino acids [12]. Four structural cysteines that are apparently involved in forming the disulfide bonds of the IgV and IgC domains are well conserved in the B7-H1 molecule. Interestingly, the intracellular domain of B7-H1 also consists of one potential site for protein kinase C phosphorylation. In addition, the intracellular domain of B7-H1 shares 16% identity with that of B7-2 (only 6% with B7-1), which has been shown to deliver a "reverse" signal to B cells after triggering with specific monoclonal antibodies (mAb) [13]. It is thus possible that B7-H1 could also transmit signals. In fact we have recently demonstrated that ligation of B7-H1 by immobilized programmed death (PD) 1, a receptor for B7-H1, or mAb against B7-H1 in the presence of anti-CD3, resulted in the activation of naive T cells and apoptosis of activated T cells [14].
Northern blot analysis revealed that the expression of human B7-H1 mRNA is abundant in heart, skeletal muscle, placenta and lung, and also detectable in thymus, spleen, kidney and liver, whereas B7-H1 mRNA is not detectable in brain, colon, small intestine, and peripheral blood mononuclear cells [12]. Freshly isolated T and B cells express negligible levels of B7-H1 while approx. 15% of CD14+ monocytes constitutively express B7-H1. Upon activation, the majority of CD3+ T cells and nearly 100% of CD14+ monocytes express B7-H1, but only approx. 5% of CD19+ B cells express B7-H1, although B cells are activated with cell surface Ig cross-linking. Interestingly, some 50% dendritic cells originated from monocytes in the presence of granulocyte-macrophage colony-stimulating factor and IL-4 in vitro also express B7-H1, and the level of expression could be upregulated to virtually 100% upon the maturation process (Dong et al. unpublished data). Immunohistochemistry studies demonstrate that expression of human B7-H1 protein is limited to the macrophage-derived cells, such as Kupffer cells in liver, macrophages in lung, and histocytes in the paracortical region of tonsil [15]. Consistent with the pattern of mRNA expression, B7-H1 protein is also highly expressed on both the syncytiotrophoblasts and extravillous cytotrophoblast [16].
In sharp contrast to constitutive expression of mRNA, there is no detectable B7-H1 protein in the normal parenchyma of liver, lung, pancreas, uterus, kidney, colon, breast, muscles, and lymphocytes in tonsil [15], suggesting that the expression of B7-H1 is posttranscriptionally controlled. This is consistent with the upregulation of cell surface B7-H1 in monocytes, dendritic cells and keratinocytes after the treatment of interferon-γ [12, 17]. Therefore B7-H1 could be regulated by the local cytokine milieu during the inflammatory process. In fact, the promoter region of human B7-H1 contains several interferon-γ responsive elements [18]. In addition, B7-H1 protein expression is also found in a broad range of human cancer tissues. This is further discussed below.
The mouse B7-H1 gene encodes a type I transmembrane protein of 290 amino acids and has 69% overall amino acid homology with human B7-H1 [19]. RNA analysis revealed that mB7-H1 mRNA was also abundant in heart, spleen, lung, skeletal muscle, and liver, an expression pattern similar to human B7-H1 mRNA. The negligible expression of the B7-H1 mRNA was observed in pancreas and testes. Resting mouse CD3+ T cells did not express B7-H1 by flow cytometry analysis using several independently generated mAb [19, 20] while only a small fraction of B220+ B cells and Mac-1+ macrophages expressed a low level of B7-H1 [18]. A recent study using MIH5 mAb, however, indicates that resting T cells, B cells and macrophages constitutively express B7-H1 [21], a finding different from that of two previously published studies [19, 20]. It is unclear whether MIH5 binds to different epitopes that have a different degree of exposure in resting cells. Stimulation of T cells by anti-CD3 and anti-CD28 increased B7-H1 expression. Activation of B cells and macrophages also greatly increased the expression of B7-H1 on the cell surface [19]. Interestingly, B7-H1 mRNA was also detected in the thymic rudiment of embryo on day 12.5, and the expression of B7-H1 seems to be under the control of whn gene [22]. Therefore, although the expression of B7-H1 is limited to a small fraction of hematopoietic cells, B7-H1 is largely inducible in various tissues and cells.
Immunological functions
Immortalized B7-H1Ig in the presence of anti-CD3 mAb as a mimic antigenic signal, costimulates the proliferation of resting human and mouse T cells, usually two- to three-fold, whereas ligation of CD28 in a similar experimental setting induced proliferation more than ten-fold [12, 19]. Inclusion of soluble B7-H1Ig in allogeneic mixed lymphocyte reactions also moderately increased the proliferation of human T cells [12]. In addition, expression of transfected B7-H1 on P815 cells also costimulates T cell proliferation against allogeneic antigens [19]. B7-H1 Ig induced the proliferation of both CD28−/− and CD28+/+ T cells in a similar degree in the presence of anti-CD3, indicating that B7-H1-mediated costimulation is independent on CD28 signaling. With the same concentration of B7-H1 Ig and anti-CD3, the proliferation of CD4+ T cells appears to be more vigorous than CD8+ T cells [19]. Thus B7-H1 appears to preferentially costimulate the growth of CD4+ T cells.
One of most interesting functions of B7-H1 is selective costimulation of IL-10 secretion in both human and mouse T cells in the presence of anti-CD3 as a surrogate T cell receptor (TCR) signal [12]. Different from B7-1 Ig and anti-CD28, B7-H1Ig induced negligible levels of IL-2 and IL-4 and moderate levels of interferon-γ and granulocyte-macrophage colony-stimulating factor [19]. Similar results were also observed using mouse T cells [17]. A recent observation indicates that immature dendritic cells stimulate the production of IL-10 in T cells that are related with the anergy status of T cells, and the production of IL-10 regulatory T cells could be blocked by anti-B7-H1 mAb [23]. It is thus possible that B7-H1 costimulates a subset of T cells that has the intrinsic capacity to secrete IL-10. Therefore costimulation by B7-H1 may lead to inhibition of T cell-mediated immunity through IL-10. It is thus important to point out that costimulation of T cell proliferation may not always lead to enhanced immunity, and that the outcome of costimulation is also dependent on other factors such as selectivity of functional subsets to be costimulated.
In addition to its costimulatory function that may ultimately lead to inhibition of T cell priming, B7-H1 could also inhibit functions of T cells in the effector phase. It was reported that immobilized B7-H1Ig inhibits cytokine production from T cells that has been fully activated by anti-CD3/CD28 mAb [17]. The mechanisms of this inhibition, however, may be attributed to engagement of an inhibitory receptor, PD-1, that may induce a signal to inhibit cell cycle progression [17, 24]. However, a recent study from our laboratory demonstrated that, upon initial proliferation of human T cells due to costimulation, immobilized B7-H1Ig with optimal anti-CD3 signal induces a rapid increase in T cell apoptosis in 48–72 h of culture [15]. A significant up-regulation of Fas and Fas ligand expression could be detected on activated T cells, in addition to the secretion of IL-10 upon B7-H1Ig costimulation. Inclusion of anti-Fas ligand or anti-IL-10 mAbs partially inhibited the apoptosis. Therefore B7-H1 ligation, probably through different receptors on T cells, inhibits T cell activation in both priming and effector stages through different mechanisms.
B7-H1 receptor
Our previous studies demonstrated that B7-H1 is not the ligand for CD28, cytotoxic T-lymphocyte antigen-4 (CTLA-4) and ICOS [12]. It was reported that B7-H1 binds to PD-1, a molecule originally cloned from T cell hybridoma undergoing apoptosis [17, 25]. PD-1 mRNA expression was found in the murine thymus, as well as in human bone marrow pro-B cells [26]. Human PD-1 gene is localized in chromosome 2q37 [26], and this gene recently was found to be inside the susceptible locus of human systemic lupus erythematosus [27]. In normal mice, PD-1 is expressed in 3–5% of thymocytes and mainly on the surface of CD4–CD8– double-negative cells (34%) [28] but not on resting T or B cells [29]. Activation of T cells by anti-CD3 mAb, however, induces the expression of PD-1 in 27–83% of murine lymph node T cells in 1–5 days [30]. Anti-IgM antibody stimulated the majority of B220+ B cells in spleen to express PD-1 [29]. These results suggest that PD-1 is an inducible molecule on a subset of T cells and on all the B cells upon activation.
The experiments performed in PD-1 deficient mice suggested that PD-1 is a negative regulator for B cell responses [31]. PD-1−/− mice have normal thymus, but moderate splenomegaly accompanied with the increased number of B cells and myeloid cells. Stimulation of B cells from PD-1−/− mice by anti-IgM antibodies leads to more vigorous proliferation than those from control mice. PD-1−/− mice also showed increased serum levels of IgG3. The levels of serum IgG2b and IgA were also increased, albeit in low levels. Furthermore, PD-1−/− mice exhibited significantly augmented IgG3 anti-DNP antibody response to a type 2 T-independent antigen [31]. More importantly, up to 30–40% of PD-1−/− mice in B6 background developed mild proliferative glomerulonephritis at 6 months of age [32]. Significant deposition of IgG3 was detected in glomeruli of PD-1−/− mice along with C3 deposition. At 14 months of age 50% of PD-1−/− mice exhibited typical lupus-like glomerulonephritis. The mild lupus-like phenotype could be accelerated by the introduction of a Fas mutation (lpr). Autoimmune dilated cardiomyopathy also developed rapidly in nearly 30% of PD-1−/− mice in BALB/c background [33]. Intriguingly, all of the affected BALB/c PD-1−/− mice exhibited high-titer circulating IgG autoantibodies reactive to a 33-kDa antigen expressed specifically on the surface of cardiomyocytes. In addition, it has been shown that PD-1 inhibited the B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine [34].
While PD-1 appears to be a negative regulator for B cell responses, the functions of PD-1 in T cells remain elusive. Purified T cells from normal PD-1−/− mice or 2C PD-1−/− mice did not show any increased proliferation response in the presence or absence of anti-CD3 mAb, suggesting that the effect of PD-1 is not T cell autonomous [31, 32]. There was no change in the size of thymus and the numbers of CD3+ T cell in PD-1−/− mice [31]. In some TCR transgenic mice such as Vβ 8 (TCR against a male antigen in the context of H-2Db), H-Y (H-2b/b or d/d), or 2C (H-2b/b) TCR transgenic mice, PD-1 expression was found dramatically increased in double-negative thymocytes [35]. Upon backcrossing to these transgenic mice, PD-1 defect leads to an increased CD4+CD8+ double-positive population in Vβ 8 transgenic β-selection model, while reducing the CD8+ single-positive cells in H-Y and 2C transgenic positive-selection model [35]. These results suggest a potential role of PD-1 in negative regulation of the differentiation from double-negative cells to double-positive cells in the process of β-selection of thymus. However, the precise role of PD-1 on positive and negative selection is still not clear, as the reduced single-positive cells in H-Y or 2C transgenic mouse with PD-1 defect may be a compensation of the increased DP cells in thymus [35]. PD-1 expression is also found in human tonsil T cells and centrocytes in the light zone of germinal centers, suggesting that PD-1 plays a role in germinal center reaction [36].
There is ample evidence indicating that costimulatory function of B7-H1 is not mediated through PD-1. First, the costimulatory effects of immobilized B7-H1Ig could not be blocked by the addition of soluble PD-1 Ig protein [15]. Second, B7-H1 Ig is capable of costimulating proliferation and cytokine secretion of T cells from PD-1−/− mice. Third, our recent studies using functional mapping and molecular modeling show that mutants of B7-H1 with complete loss of binding capacity to PD-1 can still costimulate T cell proliferation (Wang et al., submitted). Therefore, although this putative receptor has not yet been identified, costimulatory function of B7-H1 could be uncoupled from the binding of PD-1. Finally, B7-H1 mediated apoptosis could be induced in PD-1 negative human T cells [15]. Taken together, these results support that a second receptor other than PD-1 is involved in the costimulatory function of B7-H1.
B7-H1 expression by cancer cells
B7-H1 is originally identified in an expressed sequence tag clone of human ovary tumor [12], suggesting the expression of B7-H1 by tumor cells. We have initially demonstrated the expression of B7-H1 on the cell surface of several human tumor lines by specific mAb [37]. This finding is subsequently confirmed by other laboratories [24]. B7-H1 expression was found only in a small fraction of human tumor lines derived from different tissue origins using an anti-B7-H1 mAb. However, the majority of B7-H1 negative tumor lines can be up-regulated to express B7-H1 upon treatment with interferon-γ [15]. Immunohistochemistry analysis demonstrated B7-H1 immunoreactivity in a majority of freshly isolated carcinomas of human lung, ovarian, colon, melanoma, head and neck cancers, and breast cancers. We noticed that some tumor-infiltrating macrophages or dendritic-like cells also expressed B7-H1 on their membrane (Fig. 1). The expression of B7-H1 could be found in both plasma membrane and cytoplasm of cancer cells [15].

Expression of B7-H1 by human tumor cells and infiltrating macrophage/dendritic-like cells. In colon cancer tissues B7-H1 could be detected on the membranes of human colon cancer cells but not the adjuvant normal tissues. In the human ovarian cancer tissues B7-H1 expressed on the membrane and inside the cytoplasm of ovarian cancer cells. B7-H1 positive cancer cells formed a boundary between cancer cells and infiltrating lymphocytes and macrophages/dendritic-like cells. The infiltrating macrophages or dendritic-like cells expressed B7-H1 on their membrane, while the infiltrating lymphocytes did not express B7-H1. Arrow One of the B7-H1 positive macrophages
B7-H1 in the evasion of tumor immunity
We showed recently that transfection to express B7-H1 into a B7-H1 negative human melanoma line, 624mel, induced increased apoptosis of human CD8+ CTL clone, which recognizes an HLA-A2 restricted gp100 epitope presented by 624mel cells. The apoptosis could be partially prevented by inclusion of neutralizing mAb to B7-H1 [15]. These results thus support a role for tumor-associated B7-H1 in the programmed cell death of effector T cells. Furthermore, B7-H1 positive melanoma cells were also more resistant to specific CTL, while nearly all B7-H1 negative tumor cells were eliminated in the cultures. The expression of B7-H1 on 624mel cells, however, does not affect the sensitivity of these cells to CTL in a standard 4-h 51Cr release assay. The inclusion of anti-B7-H1 mAb also partially inhibited the apoptosis of a CTL clone specific for HLA-B7 restricted CEA epitope, after coculture with the CEA positive breast cancer cells, which constitutively expressed B7-H1 on their surface. Taken together, our results support apoptosis of effector T cells as a major mechanism for the resistance of tumor cells.
To examine the role of tumor-associated B7-H1 in vivo we first examined the effect of B7-H1 expression on tumorigenicity. Transfection to express mouse B7-H1 into a B7-H1 negative P815 tumor cells did not change the tumorigenicity of P815 cells in both syngeneic DBA/2 mice and immunodeficient RAG-2−/− mice in comparison with wild type or mock-transfected P815. The growth of P815 tumor cells only elicited a weak T cell response, which presumably does not induce the expression of B7-H1 counterreceptor on effector T cells. We demonstrated previously that B7-1 transfected P815 cells could induce vigorous T cell activation and lead to complete regression of tumor after injection into syngeneic mice [38]. Interestingly, the expression of B7-H1 in B7-1 positive P815 cells induced a dramatic tumor progression by abrogating tumor immunity [15]. Our results thus demonstrated that expression of B7-H1 evades tumor immunity, even if strong CD28 costimulation is provided.
We also employed a T cell adoptive transfer system to determine the function and mechanisms of tumor-associated B7-H1 in vivo. 2C TCR transgenic T cells [39], which recognize a p2Ca peptide in the context of Ld MHC class I molecule on the P815 tumor line, were used in this evaluation. In this model mock-transfected P815 cells (mock/P815) or B7-H1-transfected P815 cells (B7-H1/P815) were injected into immunodeficient RAG-1−/− mice to establish progressively growing tumors. Activated 2C T cells were transferred into P815-bearing mice. Following initial contact with antigen, the ratio of 2C T cell population greatly increased in mice harboring mock/P815 cells, but not in mice with B7-H1/P815 cells [19]. Furthermore, 2C T cells in mice harboring B7-H1/P815 tumor cells underwent significant apoptosis. The rapid increase in T cell apoptosis after being exposed to B7-H1-transfected tumor cells is plausible evidence for in vivo deletion of activated T cells by tumor-associated B7-H1. Following tumor growth in peritoneal cavities after adoptive transfer of 2C T cells, we found increased numbers of B7-H1/P815 cells. In contrast, the proliferation of mock/P815 cells was largely inhibited. Administration of mouse B7-H1 monoclonal antibody (clone 10B5) inhibited the growth of B7-H1/P815 in vivo [15]. These results support a potential role for tumor-associated B7-H1 in the evasion of active immunity in vivo.
Our studies may help to address the dilemma in cancer immunotherapy that cancer vaccines are quite effective in preventing the initial growth of a tumor, but usually fail to induce regression of established tumors [40]. The current strategy for cancer vaccination is focusing on enhancing the efficacy of antigen processing and presentation. Our results indicate that, even using highly effective T cells that are elicited by cancer vaccine, confrontation of such T cells with a negative signal such as B7-H1 in tumor sites could hamper the effect of activated T cells. Therefore specific mAbs or B7-H1 inhibitors should offer a novel therapeutic approach to improve the effectiveness of cancer vaccines.
Strategies to augment tumor immunity by manipulation of B7-H1 pathway
Direct implication from the study of B7-H1 is that blockade of B7-H1 pathway should be part of the strategy to enhance efficacy of active immune responses. As discussed above, B7-H1 may be involved in the downregulation of cell-mediated immune responses in priming state by inducing inhibitory cytokines such as IL-10. In addition, engagement of fully activated T cells by B7-H1 may lead to increased programmed cell death. Theoretically, blockade of B7-H1 pathway could be used in conjunction with all active and passive immunotherapy aiming to stimulate activation of T cells. For example, tumor antigen-based vaccinations are undergoing extensive testing in human clinical trials [41, 42]. In addition, adoptive transfer of in vitro activated T cells, which were isolated from cancer patients, has also been used in clinical treatment of advanced human cancers [43]. Furthermore, infusion of mAb that stimulates T cell responses, such as CD137 and CTLA-4 mAb, are under development for clinical trials [44, 45, 46]. Blockade of B7-H1 by neutralizing mAb thus constitutes an attractive approach to use in conjunction with other immunotherapy approaches as mentioned above to enhance and sustain T cell immunity against cancers. In fact, this B7-H1 blockade regimen was confirmed in a mouse tumor model conducted by Iwai et al. recently [47].
Conclusion
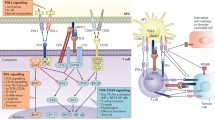
Recent studies identify a new B7-H1 pathway, which negatively regulates T cell immune responses by selectively costimulating a putative T cell subset that secretes IL-10, and increases programmed cell death of activated T cells (Fig. 2). Therefore B7-H1 could affect T cell responses in both priming and effector phase. Understanding the mechanisms of B7-H1 mediated suppression, identification and characterization of its receptors and signaling events should shed light on the selective manipulation of this pathway. In conjunction with cancer vaccine or adoptive immunotherapy, blockade of the B7-H1 pathway could lead to a new strategy for therapeutic purposes in the control of human cancer and autoimmune diseases.

Mechanisms of B7-H1 mediated inhibition of cell-mediated responses. In the peripheral lymphoid organs, B7-H1 on the surface of antigen-presenting cells (APC) costimulates a subset of naive or early primed T cells in the presence of antigen, by binding through a B7-H1 counterreceptor (B7-H1R) but not PD-1. This subset preferentially releases IL-10 that induces tolerance/anergy of T cells. In the peripheral tissues, when the effecter (fully activated) T cells contact the targeting tissues/cells (tumor cells) or activated APCs, the B7-H1 on these targeting cells deliver death signals to terminate a T cell response through B7-H1R and/or PD-1. As a result B7-H1 could prevent the generation of self-reactive T cells by inducing anergy at their priming stage and protects the peripheral tissues from bystander or antigen specific destruction mediated by activated effecter T cells
Abbreviations
- B7-H1 :
-
B7-homolog 1
- CTL :
-
Cytolytic T cells
- IL :
-
Interleukin
- mAb :
-
Monoclonal antibody
- PD :
-
Programmed death
- TCR :
-
T cell receptor
References
Jantzer P, Schendel DJ (1998) Human renal cell carcinoma antigen-specific CTLs: antigen-driven selection and long-term persistence in vivo. Cancer Res 58:3078–3086
D'Souza S, Rimoldi D, Lienard D, Lejeune F, Cerottini JC, Romero P (1998) Circulating Melan-A/Mart-1 specific cytolytic T lymphocyte precursors in HLA-A2+ melanoma patients have a memory phenotype. Int J Cancer 78:699–706
Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M, Davis MM (1999) Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med 5:677–685
Cohen PA, Peng L, Kjaergaard J, Plautz GE, Finke JH, Koski GK, Czerniecki BJ, Shu S (2001) T-cell adoptive therapy of tumors: mechanisms of improved therapeutic performance. Crit Rev Immunol 21:215–248
Abken H, Hombach A, Heuser C, Kronfeld K, Seliger B (2002) Tuning tumor-specific T-cell activation: a matter of costimulation? Trends Immunol 23:240–245
Molldrem JJ, Lee PP, Wang C, Felio K, Kantarjian HM, Champlin RE, Davis MM (2000) Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med 6:1018–1023
Romero P, Dunbar PR, Valmori D, Pittet M, Ogg GS, Rimoldi D, Chen JL, Lienard D, Cerottini JC, Cerundolo V (1998) Ex vivo staining of metastatic lymph nodes by class I major histocompatibility complex tetramers reveals high numbers of antigen-experienced tumor-specific cytolytic T lymphocytes. J Exp Med 188:1641–1650
Hanson HL, Donermeyer DL, Ikeda H, White JM, Shankaran V, Old LJ, Shiku H, Schreiber RD, Allen PM (2000) Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity 13:265–276
Smyth MJ, Godfrey DI, Trapani JA (2001) A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol 2:293–299
Khong HT, Restifo NP (2002) Natural selection of tumor variants in the generation of "tumor escape" phenotypes. Nat Immunol 3:999–1005
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3:991–998
Dong H, Zhu G, Tamada K, Chen L (1999) B7–H1, a third member of the B7 family, costimulates T-cell proliferation and secretion of interleukin-10. Nat Med 5:1365–1369
Jeannin P, Delneste Y, Lecoanet-Henchoz S, Gauchat JF, Ellis J, Bonnefoy JY (1997) CD86 (B7–2) on human B cells. A functional role in proliferation and selective differentiation into IgE- and IgG4-producing cells. J Biol Chem 272:15613–15619
Dong H, Strome SE, Matteson EL, Moder KG, Flies DB, Zhu G, Tamura H, Driscoll CL, Chen L (2003) Costimulating aberrant T cell responses by B7–H1 autoantibodies in rheumatoid arthritis. J Clin Invest 111:363–370
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L (2002) Tumor-associated B7–H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8:793–800
Petroff MG, Chen L, Phillips TA, Hunt JS (2002) B7 family molecules: novel immunomodulators at the maternal-fetal interface. Placenta [Suppl A]:S95–101
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192:1027–1034
Mazanet MM, Hughes CC (2002) B7–H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol 169:3581–3588
Tamura H, Dong H, Zhu G, Sica GL, Flies DB, Tamada K, Chen L (2001) B7–H1 costimulation preferentially enhances CD28-independent T-helper cell function. Blood 97:1809–1816
Ishida M, Iwai Y, Tanaka Y, Okazaki T, Freeman GJ, Minato N, Honjo T (2002) Differential expression of PD-L1 and PD-L2, ligands for an inhibitory receptorPD-1, in the cells of lymphohematopoietic tissues. Immunol Lett 84:57–62
Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, Shin T, Tsuchiya H, Pardoll DM, Okumura K, Azuma M, Yagita H (2002) Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 169:5538–5545
Bleul CC, Boehm T (2001) Laser capture microdissection-based expression profiling identifies PD1-ligand as a target of the nude locus gene product. Eur J Immunol 31:2497–2503
Gray CP, Arosio P, Hersey P (2002) Heavy chain ferritin activates regulatory T cells by induction of changes in dendritic cells. Blood 99:3326–3334
Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ (2001) PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2:261–268
Ishida Y, Agata Y, Shibahara K, Honjo T (1992) Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 11:3887–3895
Finger LR, Pu J, Wasserman R, Vibhakar R, Louie E, Hardy RR, Burrows PD, Billips LG (1997) The human PD-1 gene: complete cDNA, genomic organization, and developmentally regulated expression in B cell progenitors. Gene 197:177–187
Prokunina L, Castillejo-Lopez C, Oberg F, Gunnarsson I, Berg L, Magnusson V, Brookes AJ, Tentler D, Kristjansdottir H, Grondal G, Bolstad AI, Svenungsson E, Lundberg I, Sturfelt G, Jonssen A, Truedsson L, Lima G, Alcocer-Varela J, Jonsson R, Gyllensten UB, Harley JB, Alarcon-Segovia D, Steinsson K (2002) Alarcon-Riquelme ME. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat Genet 32:666–669
Nishimura H, Agata Y, Kawasaki A, Sato M, Imamura S, Minato N, Yagita H, Nakano T, Honjo T (1996) Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4-CD8-) thymocytes. Int Immunol 8:773–780
Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T (1996) Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol 8:765–772
Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, Collins M, Honjo T, Freeman GJ, Carreno BM (2002) PD-1: PD-L inhibitory pathway affects both CD4 (+) and CD8 (+) T cells and is overcome by IL-2. Eur J Immunol 32:634–643
Nishimura H, Minato N, Nakano T, Honjo T (1998) Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol 10:1563–1572
Nishimura H, Nose M, Hiai. H, Minato N, Honjo T (1999) Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11:141–151
Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T (2001) Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291:319–322
Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T (2001) PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci USA 98:13866–13871
Nishimura H, Honjo T, Minato N (2000) Facilitation of beta selection and modification of positive selection in the thymus of PD-1-deficient mice. J Exp Med 191:891–898
Iwai Y, Okazaki T, Nishimura H, Kawasaki A, Yagita H, Honjo T (2002) Microanatomical localization of PD-1 in human tonsils. Immunol Lett 83:215–220
Ferrone S, Finerty JF, Jaffee EM, Nabel GJ (2000) How much longer will tumour cells fool the immune system? Immunol Today 21:70–72
Chen L, McGowan P, Ashe S, Johnston JV, Hellstrom I, Hellstrom KE (1994) B7–1/CD80-transduced tumor cells elicit better systemic immunity than wild-type tumor cells admixed with Corynebacterium parvum. Cancer Res 54:5420–5423
Sykulev Y, Brunmark A, Tsomides TJ, Kageyama S, Jackson M, Peterson PA, Eisen HN (1994) High-affinity reactions between antigen-specific T-cell receptors and peptides associated with allogeneic and syngeneic major histocompatibility complex class I proteins. Proc Natl Acad Sci USA 91:11487–11491
Lollini PL, Forni G (1999) Specific and nonspecific immunity in the prevention of spontaneous tumors. Immunol Today 20:347–350
Sinkovics JG, Horvath JC (2000) Vaccination against human cancers. Int J Oncol 16:81–96
Pecher G, Haring A, Kaiser L, Thiel E (2002) Mucin gene (MUC1) transfected dendritic cells as vaccine: results of a phase I/II clinical trial. Cancer Immunol Immunother 51:669–673
Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA (2002) Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298:850–854
Ye Z, Hellstrom I, Hayden-Ledbetter M, Dahlin A, Ledbetter JA, Hellstrom KE (2002) Gene therapy for cancer using single-chain Fv fragments specific for 4-1BB. Nat Med 8:343–348
Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, Mittler RS, Chen L (1997) Monoclonal antibodies against the 4–1BB T-cell activation molecule eradicate established tumors. Nat Med 3:682–685
Chambers CA, Kuhns MS, Egen JG, Allison JP (2001) CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 19:565–594
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N (2002) Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA 99:12293–12297
Acknowledgements
We thank all members in this laboratory for their contribution to the work described in the article. This work was supported by the National Institutes of Health grant CA97085 and by Mayo Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dong, H., Chen, L. B7-H1 pathway and its role in the evasion of tumor immunity. J Mol Med 81, 281–287 (2003). https://doi.org/10.1007/s00109-003-0430-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-003-0430-2