
Abstract
Purpose
The relation between functional imaging and intrapatient genetic heterogeneity remains poorly understood. The aim of our study was to investigate spatial sampling and functional imaging by FDG-PET/MRI to describe intrapatient tumour heterogeneity.
Methods
Six patients with oropharyngeal cancer were included in this pilot study. Two tumour samples per patient were taken and sequenced by next-generation sequencing covering 327 genes relevant in head and neck cancer. Corresponding regions were delineated on pretherapeutic FDG-PET/MRI images to extract apparent diffusion coefficients and standardized uptake values.
Results
Samples were collected within the primary tumour (n = 3), within the primary tumour and the involved lymph node (n = 2) as well as within two independent primary tumours (n = 1). Genetic heterogeneity of the primary tumours was limited and most driver gene mutations were found ubiquitously. Slightly increasing heterogeneity was found between primary tumours and lymph node metastases. One private predicted driver mutation within a primary tumour and one in a lymph node were found. However, the two independent primary tumours did not show any shared mutations in spite of a clinically suspected field cancerosis. No conclusive correlation between genetic heterogeneity and heterogeneity of PET/MRI-derived parameters was observed.
Conclusion
Our limited data suggest that single sampling might be sufficient in some patients with oropharyngeal cancer. However, few driver mutations might be missed and, if feasible, spatial sampling should be considered. In two independent primary tumours, both lesions should be sequenced. Our data with a limited number of patients do not support the concept that multiparametric PET/MRI features are useful to guide biopsies for genetic tumour characterization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aiming for precision medicine, the understanding of inter- and intrapatient diversity of tumours might support successfully personalized therapeutic approaches. In radiation oncology, spatial dose distribution (“dose painting”) to subvolumes of the tumours can be applied [1, 2]. For this purpose, intrapatient tumour heterogeneity can be investigated by imaging features or by next-generation sequencing (NGS).
The relevant interpatient heterogeneity of somatic mutations in head and neck squamous cell carcinomas (HNSCCs) has been shown before [3,4,5]. Furthermore, the extent of intrapatient diversity of solid tumours has been intensely investigated recently, as intratumour genetic heterogeneity (ITH) might substantially contribute to therapy failure and dismal outcome in cancer patients [6,7,8]. Single biopsies are discussed in order to underestimate genetic diversity [9]. McGranahan and Swanton (2017) reviewed several studies of different cancer types and described distinct patterns of evolutionary trees with regard to ITH according to diverse tumour types [7]. Tumours related to exogenous mutagens such as nicotine abuse or ultraviolet light (lung adenocarcinomas of smokers and melanomas) showed increased clonal mutational burden compared to other entities, but comparably reduced ITH proportion. The majority of alterations of these tumours might have been acquired prior to invasiveness and therefore contribute to the large clonal “trunk” while the tree’s “branches” are formed by relatively fewer subclonal mutations [7]. In contrast, gliomas, for example, showed a short “trunk” but a proportionally high subclonal variability (likely acquired during tumour progression) [7]. To investigate tumour diversity, spatial sampling of different biopsies can be applied [10, 11].
Imaging features can be used as an additional evaluation of intratumour diversity [12]. Besides genetic diversity, image-based heterogeneity was also found to correlate with worse outcome [13] and correlations of genetic data and imaging features are gaining increased interest [14]. Imaging features might potentially guide biopsy sampling for NGS. However, studies investigating the spatial relationship of these two approaches remain rare [12].
Functional images can be obtained by magnetic resonance imaging (MRI), positron-emission tomography (PET) or by hybrid imaging combining both technologies (PET/MRI). Diffusion-weighted MRI (DWI) allows for quantification of water diffusion which depends on different tissue characteristics such as cell size, cell density and membrane architecture [15]. Lower apparent diffusion coefficients (ADCs) usually indicate restricted diffusion capability. Besides DWI [16], PET imaging with [18F]-fluorodeoxyglucose (FDG) is an emerging technique for tumour characterization [17] and treatment guidance [18] in HNSCC. Standardized uptake values (SUV) can be used to measure glucose consumption and energy metabolism of the tumour. Therefore, functional imaging techniques are promising to elucidate regional diversity of cancerous lesions.
Studies regarding spatial sampling for NGS in HNSCC are rare [19,20,21,22,23]. Furthermore, the translation between genetic heterogeneity and functional implications remains poorly understood. Therefore, our study addressed intrapatient heterogeneity of somatic mutations in HNSCC and correlated this data with spatially corresponding functional PET/MRI information.
Materials and methods
Ten patients with locally advanced oro- or hyphopharyngeal HNSCC were recruited prospectively for this pilot study (NCT-02666885). However, due to small tumour volumes, spatial sampling of two lesions per patient could be achieved only in six patients who were included in this planned interim investigation. The study was approved by the local ethics committee (reference number 025/2015) and all patients declared their written informed consent.
All patients received tumour resection with intended adjuvant radio(chemo)therapy at primary diagnosis and did not undergo any previous treatment. Ahead of surgery, simultaneous FDG-PET/MRI (Biograph mMR, Siemens Healthineers, Erlangen, Germany, 3 T) was performed for anatomical and functional characterization of the tumours. The imaging protocol included a morphological T2-weighted transverse short tau inversion recovery sequence (STIR, in-plane voxel size 0.7 × 0.7 mm2, slice thickness 4 mm) for tumour delineation as well as an echo-planar imaging-based DWI sequence (b-values of 150 and 800 s/mm2, in-plane voxel size 1.8 × 1.8 mm2, slice thickness 5 mm) and FDG-PET data (84–90 min post injection, voxel size 2.8 × 2.8 × 2 mm3) were acquired. The patients were scanned in a dedicated radiotherapy-planning setup including a thermoplastic mask to ensure maximal reduction of motion artefacts. The exact hardware solution, PET attenuation and DWI distortion correction methods and detailed imaging information were reported previously [24,25,26]. After distortion correction, ADC maps were calculated. FDG-PET activity concentrations were converted to SUVs by normalizing with respect to injected dose and patient weight. For each region of interest (ROI, corresponding to the biopsy sites), mean ADC as well as mean SUV were calculated to investigate correlations with the genetic information.
During surgery biopsies for NGS were collected from the surgical specimen in cooperation with the institute of pathology, to ensure sufficient tumour content. The biopsy sites were delineated manually as ROIs in the corresponding STIR images as shown in Fig. 1. Specimen sampling as well as the delineation of corresponding tumour subvolumes in MR imaging was done by the same observer to ensure maximal conformity. Two samples of each patient were collected. If clinically feasible, both specimens were sampled from the same primary tumour. Otherwise, the second sample was collected from an involved lymph node. In one patient, the two samples were taken from two spatially separated primary tumours (tonsil and contralateral piriform sinus). The tumour samples were fresh frozen and 4 μm thick sections were stained with hematoxylin and eosin (H&E). The tumour cell content was estimated by experienced pathologists.

The T2-weighted short tau inversion recovery sequence (STIR) image and exemplary regions of interest (ROIs) corresponding to the sampling areas in patient 3
For genetic analyses, we applied a comprehensive panel approach (327 genes) that was designed for HNSCC tumours by the German Cancer Consortium (DKTK) partner site in Berlin [27]. Besides, the dedicated gene list can be found on the Charité homepage (experimental radiation oncology). Ethylenediaminetetraacetic acid blood samples of the patients were used for normal tissue controls.
For library preparation and exonic region in-solution capture, the Agilent HaloplexHS technology (Agilent, Santa Clara, CA, USA) was used. Paired-end sequencing was performed on the HiSeq2500 instrument (Illumina, San Diego, CA, USA). “megSAP” (version 0.1-755-g54185f9, https://github.com/imgag/megSAP), an in-house pipeline, was applied to analyse the data. Using the Burrows–Wheeler Alignment tool (BWA, version 0.7.17) [28], the reads were aligned to the human genome reference (GRCh37). Variant-calling and annotation were conducted using Strelka2 (version 2.8.4) [29] and SnpEff/SnpSift (version 4.37) [30]. Nonsynonymous, coding mutations or splice region and splice donor variants with an allele frequency (AF) >5% were reported and compared to identify ubiquitous versus heterogeneous nonsilent mutations.
All reported ubiquitous mutations were found in both samples at the same chromosome position with the identical nucleotide changes. Variants that were only found in one specimen were considered heterogeneous. All reported variants were visually validated in the raw data and cross-checked with the normal tissue as well as the corresponding other tumour sample.
To identify predicted driver mutations, all somatic variants were uploaded to the Cancer Genome Interpreter (CGI) [31]. The genetic variability within the same patient was reported by the percentage of divergent genetic variants: (number of heterogenous variants / (shared variants + heterogenous variants)) × 100 (%).
Results
Six male patients (52–66 years) were included in this prospective biomarker study. All patients were active smokers at the time of primary diagnosis and had primary tumours located in the oropharynx. One patient had a second primary tumour of the uvula and another patient presented with three invasive lesions (tonsil, piriform sinus and vallecula). The patient and tumour characteristics are summarized in Table 1.
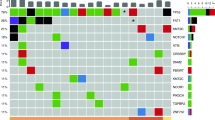
The panel sequencing results including heterogeneous and ubiquitous somatic mutations as well as annotated predicted or known driver mutations according to the CGI are shown in Fig. 2. TP53 mutations were found in all patients. Furthermore, NOTCH1 variants (drivers and passengers) could be detected in several patients. Apart from these variants, a distinct interpatient variability was found.

Nonsilent, coding mutations of the tumours according to spatial sampling. Shared and private mutations are shown in the Venn diagrams and known or predicted driver mutations according to the Cancer Genome Interpreter (CGI) database are printed in bold. Copy number variations are not included
Considering intrapatient heterogeneity, in three patients (Patients 1, 2 and 3), both sequenced samples were collected within the same primary tumour. In these patients, the majority of variants were ubiquitous. Little diversity could be observed, but one predicted driver mutation in ATRX was found to be heterogeneous. In a further patient (Patient 4), the primary tumour and the corresponding lymph node metastasis were investigated and also showed predominant concordance.
In two patients, more than one primary tumour was found. Patient 5 had two primary tumours, namely a squamous cell carcinoma of the uvula (partially keratinizing, 4.5 mm) and of the base of tongue (basaloid, 10 mm). A lymph node metastasis showed extracapsular extension and was morphologically associated with the base of the tongue carcinoma. Due to the limited tissue of the uvula specimen, sequencing was only conducted for the base of the tongue carcinoma and the involved lymph node. Three shared mutations in TP53, LRP1B and SYNE1 were found. However, several additional private mutations as well as one predicted driver mutation in PDGFRA in the lymph node biopsy were detected.
Patient 6 had three separate primary tumours. Samples were collected from the tonsil cancer and the contralateral tumour of the piriform sinus. A field cancerosis was clinically suspected in this patient. However, no common mutations were found in the two samples. A potentially druggable, private mutation was found in the sample of the piriform sinus (BRCA2).
No distinct associations between the genetic heterogeneity and corresponding variation in mean ADC or SUV values were found (Figs. 3 and 4; Table 2). Table 2 shows the ADC and SUV values according to the spatial sampling sides and the relative differences between the two biopsy areas per patient as well as the respective genetic variability of the samples. FDG uptake and DWI measurements showed some remarkable heterogeneity within the same primary tumour in spite of predominant concordance of the genetic profiles. In contrast, comparably similar imaging features were found in the patient with two separate primary tumours (Patient 6) and missing ubiquitous somatic variants. Of note, the lymph node metastasis of Patient 4 showed extensive necrosis, wherefore ADC as well as SUV values should be interpreted with caution for this patient. The other lesions investigated did not show major necrotic areas. Fig. 5 shows exemplary imaging of patient 5 including the oropharyngeal tumour (base of the tongue) and the respective involved lymph node.

Correlation of mean apparent diffusion coefficients (ADCs) and the genetic heterogeneity between the two samples per patient (given by the fraction of divergent genetic variants to the total number of genetic variants). The numbers correspond to the individual patients. In Patient 4, the high ADC value corresponds to a region of interest in a mainly necrotic lymph node and should be interpreted with caution

Correlation of mean [18F]-fluorodeoxyglucose (FDG) standardized uptake values (SUV) and the genetic heterogeneity between the two samples per patient in percent

Imaging of patient 5 showing the oropharyngeal tumour at the base of the tongue and the involved lymph node (arrows): a,b positron-emission tomography, c turbo inversion recovery magnitude (TIRM) imaging and d,e diffusion-weighted imaging with the apparent diffusion coefficient (ADC) map
Discussion
The model of clonal evolution of tumours and associated variant sublines was previously described by P.C. Nowell in 1976 [32]. However, recent sequencing technologies allow further insight into intrapatient heterogeneity as ubiquitous and private mutations can be determined [10, 33].
For the estimation of genetic intrapatient heterogeneity and correlations between somatic mutations and imaging information, we focussed on coding (and splice site), nonsynonymous single-nucleotide variants or small insertions/deletions with an AF >5%. An AF of 5% is a common threshold and reflects the limitation of the method used to safely detect variants. Regarding these variants, as previously stated by Reiter et al. [34], predicted functional changes can be estimated and are of targetable interest, while functional implications of intronic regions, copy number variations (CNVs) or noncoding variants are currently not well understood. We reported heterogeneous versus ubiquitous mutations based on a panel approach and did not calculate phylogenetic trees as we focussed on functional impact rather than on tumour evolution.
Our investigation of inter- and intrapatient variability of somatic mutations in patients with oropharyngeal cancer confirms relevant heterogeneity between different patients. Furthermore, no shared mutations were found in a patient with two distant primary tumours (i.e. tonsil and contralateral piriform sinus), in spite of a clinically suspected field cancerosis.
However, disregarding this particular setting of two separate primary tumours, limited diversity between different tumour samples within a single patient was found. This is clinically important focussing on precision medicine, target therapies and potential dose painting as our results suggest minor diversity if samples were taken from the same primary tumour and some increasing heterogeneity if the primary tumour and the lymph node metastasis were aligned. Especially predicted or known driver gene mutations were predominantly found ubiquitously which indicates common functional changes. Heterogeneous mutations were mainly classified as predicted passenger mutations according to the CGI database. However, in one patient, a predicted driver mutation in ATRX was found only in one of the two samples originating from the same primary tumour. In another patient, a private predicted driver mutation in PDGFRA was found in the lymph node. Thus, in some patients, relevant variants might be missed by single sampling but the majority of driver mutations could be detected by a single biopsy in our cohort.
Our findings of rather limited intrapatient genetic diversity are in line with a recent publication about metastases in solid tumours that showed only minor “functional driver gene heterogeneity” between the different metastatic sites [34]. The vast majority of functionally relevant mutations within individual patients was commonly found in all metastatic lesions.
The majority of ubiquitous mutations compared to relatively less heterogeneous variants within the primary tumours may be explained by the long-time exposition to exogenous mutagens as all patients were active smokers for years at time of diagnosis. These findings are well in line with the review of McGranahan and Swanton (2017), indicating comparably small ITH proportion in exogenous mutagen-driven cancers [7].
Due to the complex approach, studies about spatial sampling in patients with primary carcinomas of the oropharynx are rare and of very limited patient numbers. Ledgerwood et al. reported seven patients with carcinomas of the tongue, the floor of the mouth and the larynx and suggested the heterogeneity may vary with respect to the tumour localization [19]. However, our results are in line with a report concerning five patients with oral squamous cell carcinomas showing high interpatient but low intrapatient tumour heterogeneity [21]. Similar results were recently shown in spatial sampling of 13 tumours of the oral cavity, whereas patterns of oropharyngeal (n = 6), pharyngeal (n = 3) and laryngeal cancers (n = 1) that were matched to paired relapse tumour samples (recurrence after chemoradiation) showed greater heterogeneity [23]. The study of Hedberg et al. (2016) reported six oral, three pharyngeal and four laryngeal carcinomas with relevant mutational similarity between the primary tumours and their corresponding synchronous lymph node metastases [22]. Thus, our results are widely consistent with the published literature about HNSCC tumour heterogeneity. Furthermore, to the best of our knowledge, this is one of the largest cohorts of spatial sampling in patients with oropharyngeal carcinomas reported to date despite of our limited patient number.
Therefore, with full acknowledgement of the caveat due to our limited sample size, we suggest that intrapatient heterogeneity in oropharyngeal cancer might increase from rather similar mutation patterns within single primary tumours to slightly ascending diversity between the primary tumour and lymph node metastases. In both constellations, little functional driver gene heterogeneity was observed, but two driver variants (ATRX, PDGFRA) could have been missed by single sampling. For daily practice this is a relevant contribution to the unresolved question of how many tumour samples are required in which patient, e.g. for target therapy approaches. In some patients, single sampling might be sufficient to identify the functionally relevant, potentially druggable driver mutations, but if clinically and financially feasible, spatial sampling should be considered. In contrast, two anatomically different primary tumours within one single patient (oro- and hypopharynx) showed entire heterogeneity and several private driver gene mutations were found. Therefore, in clinical practice, in multilocular, potentially independent tumours, spatial sequencing should be intended. Otherwise, relevant information for precision medicine or targeted therapies might be missed.
There are few data about correlations of functional imaging and genetic tumour heterogeneity. Moon et al. investigated a genetic heterogeneity index and according PET features in lung cancer in single biopsies [35]. Only in one subgroup of patients with small cell lung cancer (SCLC) was a correlation between the surface SUV entropy and genetic heterogeneity found in involved lymph nodes. In lung tissue of SCLC, in adenocarcinomas and squamous cell carcinomas as well as for all other investigated PET features, no distinct correlation between genetic heterogeneity and PET parameters was found. Another group investigated glioblastoma heterogeneity by MRI and correlated CNVs of spatial tissue sampling [36]. Intratumour CNV heterogeneity was found in 7 of 13 investigated patients. Several correlations between selected driver genes and diverse MRI parameters were reported.
In our cohort of HNSCC patients, we did not find conclusive accordance between genetic heterogeneity and heterogeneity in the image-derived parameters ADC and SUV. Thus, tumour microstructure and glucose consumption might not be completely determined by somatic mutations. This could be explained by multiple epigenetic and microenvironmental influences affecting cell density and energy metabolism. Even though biological validation of imaging biomarkers is desirable, not all imaging features might directly correlate with an equivalent pathologic tissue [37] or genetic profile. In this way, image-guided spatial sampling based on ADC/SUV variations to predefine and anticipate genetic heterogeneity does not seem to be promising. If our preliminary findings of independent biomarkers can be confirmed in larger biomarker trials, mutation patterns, DW and PET imaging could be used complementary for tumour characterization.
Besides the small cohort and limited sample size, the limitations of this study include the use of a dedicated HNSCC cancer panel instead of whole-exome or whole-genome sequencing. Therefore, some variants might be missed. On the contrary, this panel approach focused on previously described, functionally relevant HNSCC genes in which a deeper sequencing and detection of low-level mosaicism was feasible improving the reliability of results. Furthermore, for feasibility reasons, no image-guided, stereotactic biopsies were applied. Thus, some minor inconsistencies with regard to the exact anatomic correlation between genomics and image-derived features cannot be ruled out.
Conclusion
Inter- and intrapatient heterogeneity are major challenges for successful tumour treatment. In our HNSCC patients, spatial genetic heterogeneity within primary tumours was limited. Slightly increased heterogeneity could be observed between primary tumours and involved lymph nodes. Most functional driver mutations were ubiquitously found and a single biopsy might be sufficient to identify druggable targets in some HNSCC patients. However, if feasible, spatial sampling should be considered, as few driver mutations might be missed. In multilocular, potentially independent primary tumours, multiple sampling should be intended for a comprehensive genetic characterization. Mean ADC and FDG-SUV did not translate directly into genetic variability in our cohort and might therefore provide complementary information about tumour biology. ADC/SUV-based image-guided sampling to anticipate genetic heterogeneity does not seem to be promising.
References
Bentzen SM, Gregoire V (2011) Molecular imaging-based dose painting: a novel paradigm for radiation therapy prescription. Semin Radiat Oncol 21(2):101–110. https://doi.org/10.1016/j.semradonc.2010.10.001
Grönlund E, Johansson S, Montelius A, Ahnesjö A (2017) Dose painting by numbers based on retrospectively determined recurrence probabilities. Radiother Oncol 122(2):236–241. https://doi.org/10.1016/j.radonc.2016.09.007
Cancer Genome Atlas Network (2015) Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517(7536):576–582. https://doi.org/10.1038/nature14129
Leemans CR, Snijders PJF, Brakenhoff RH (2018) The molecular landscape of head and neck cancer. Nat Rev Cancer 18(5):269–282. https://doi.org/10.1038/nrc.2018.11
Zwirner K, Hilke FJ, Demidov G, Socarras Fernandez J, Ossowski S, Gani C, Thorwarth D, Riess O, Zips D, Schroeder C, Welz S (2019) Radiogenomics in head and neck cancer: correlation of radiomic heterogeneity and somatic mutations in TP53, FAT1 and KMT2D. Strahlenther Onkol 195(9):771–779. https://doi.org/10.1007/s00066-019-01478-x
Greaves M (2015) Evolutionary determinants of cancer. Cancer Discov 5(8):806–820. https://doi.org/10.1158/2159-8290.CD-15-0439
McGranahan N, Swanton C (2017) Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168(4):613–628. https://doi.org/10.1016/j.cell.2017.01.018
Morris LG, Riaz N, Desrichard A, Şenbabaoğlu Y, Hakimi AA, Makarov V, Reis-Filho JS, Chan TA (2016) Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget 7(9):10051–10063. https://doi.org/10.18632/oncotarget.7067
Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, Martinez P, Phillimore B, Begum S, Rabinowitz A, Spencer-Dene B, Gulati S, Bates PA, Stamp G, Pickering L, Gore M, Nicol DL, Hazell S, Futreal PA, Stewart A, Swanton C (2014) Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 46(3):225–233. https://doi.org/10.1038/ng.2891
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366(10):883–892. https://doi.org/10.1056/NEJMoa1113205
Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, Seth S, Chow CW, Cao Y, Gumbs C, Gold KA, Kalhor N, Little L, Mahadeshwar H, Moran C, Protopopov A, Sun H, Tang J, Wu X, Ye Y, William WN, Lee JJ, Heymach JV, Hong WK, Swisher S, Wistuba II, Futreal PA (2014) Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346(6206):256–259. https://doi.org/10.1126/science.1256930
O’Connor JP, Rose CJ, Waterton JC, Carano RA, Parker GJ, Jackson A (2015) Imaging intratumor heterogeneity: role in therapy response, resistance, and clinical outcome. Clin Cancer Res 21(2):249–257. https://doi.org/10.1158/1078-0432.CCR-14-0990
Aerts HJ, Velazquez ER, Leijenaar RT, Parmar C, Grossmann P, Carvalho S, Bussink J, Monshouwer R, Haibe-Kains B, Rietveld D, Hoebers F, Rietbergen MM, Leemans CR, Dekker A, Quackenbush J, Gillies RJ, Lambin P (2014) Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat Commun 5:4006. https://doi.org/10.1038/ncomms5006
Jansen RW, van Amstel P, Martens RM, Kooi IE, Wesseling P, de Langen AJ, Menke-Van der Houven van Oordt CW, Jansen BHE, Moll AC, Dorsman JC, Castelijns JA, de Graaf P, de Jong MC (2018) Non-invasive tumor genotyping using radiogenomic biomarkers, a systematic review and oncology-wide pathway analysis. Oncotarget 9(28):20134–20155. https://doi.org/10.18632/oncotarget.24893
Lin G, Keshari KR, Park JM (2017) Cancer metabolism and tumor heterogeneity: imaging perspectives using MR imaging and spectroscopy. Contrast Media Mol Imaging 2017:6053879. https://doi.org/10.1155/2017/6053879
Leibfarth S, Winter RM, Lyng H, Zips D, Thorwarth D (2018) Potentials and challenges of diffusion-weighted magnetic resonance imaging in radiotherapy. Clin Transl Radiat Oncol 13:29–37. https://doi.org/10.1016/j.ctro.2018.09.002
Becker M, Zaidi H (2014) Imaging in head and neck squamous cell carcinoma: the potential role of PET/MRI. Br J Radiol 87(1036):20130677. https://doi.org/10.1259/bjr.20130677
Mehanna H, Wong WL, McConkey CC, Rahman JK, Robinson M, Hartley AG, Nutting C, Powell N, Al-Booz H, Robinson M, Junor E, Rizwanullah M, von Zeidler SV, Wieshmann H, Hulme C, Smith AF, Hall P, Dunn J (2016) PET-CT surveillance versus neck dissection in advanced head and neck cancer. N Engl J Med 374(15):1444–1454. https://doi.org/10.1056/NEJMoa1514493
Ledgerwood LG, Kumar D, Eterovic AK, Wick J, Chen K, Zhao H, Tazi L, Manna P, Kerley S, Joshi R, Wang L, Chiosea SI, Garnett JD, Tsue TT, Chien J, Mills GB, Grandis JR, Thomas SM (2016) The degree of intratumor mutational heterogeneity varies by primary tumor sub-site. Oncotarget 7(19):27185–27198. https://doi.org/10.18632/oncotarget.8448
Zhang XC, Xu C, Mitchell RM, Zhang B, Zhao D, Li Y, Huang X, Fan W, Wang H, Lerma LA, Upton MP, Hay A, Méndez E, Zhao LP (2013) Tumor evolution and intratumor heterogeneity of an oropharyngeal squamous cell carcinoma revealed by whole-genome sequencing. Neoplasia 15(12):1371–1378
Tabatabaeifar S, Thomassen M, Larsen MJ, Larsen SR, Kruse TA, Sørensen JA (2017) The subclonal structure and genomic evolution of oral squamous cell carcinoma revealed by ultra-deep sequencing. Oncotarget 8(10):16571–16580. https://doi.org/10.18632/oncotarget.15014
Hedberg ML, Goh G, Chiosea SI, Bauman JE, Freilino ML, Zeng Y, Wang L, Diergaarde BB, Gooding WE, Lui VW, Herbst RS, Lifton RP, Grandis JR (2016) Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J Clin Invest 126(4):1606. https://doi.org/10.1172/JCI86862
de Roest RH, Mes SW, Poell JB, Brink A, van de Wiel MA, Bloemena E, Thai E, Poli T, Leemans CR, Brakenhoff RH (2019) Molecular characterization of locally relapsed head and neck cancer after concomitant chemoradiotherapy. Clin Cancer Res 25(23):7256–7265. https://doi.org/10.1158/1078-0432.CCR-19-0628
Winter RM, Leibfarth S, Schmidt H, Zwirner K, Monnich D, Welz S, Schwenzer NF, la Fougere C, Nikolaou K, Gatidis S, Zips D, Thorwarth D (2018) Assessment of image quality of a radiotherapy-specific hardware solution for PET/MRI in head and neck cancer patients. Radiother Oncol. https://doi.org/10.1016/j.radonc.2018.04.018
Winter RM, Schmidt H, Leibfarth S, Zwirner K, Welz S, Schwenzer NF, la Fougere C, Nikolaou K, Gatidis S, Zips D, Thorwarth D (2017) Distortion correction of diffusion-weighted magnetic resonance imaging of the head and neck in radiotherapy position. Acta Oncol 56(11):1659–1663. https://doi.org/10.1080/0284186X.2017.1377347
Zwirner K, Thorwarth D, Winter RM, Welz S, Weiss J, Schwenzer NF, Schmidt H, la Fougere C, Nikolaou K, Zips D, Gatidis S (2018) Voxel-wise correlation of functional imaging parameters in HNSCC patients receiving PET/MRI in an irradiation setup. Strahlenther Onkol 194(8):719–726. https://doi.org/10.1007/s00066-018-1292-4
Eder T, Hess AK, Konschak R, Stromberger C, Johrens K, Fleischer V, Hummel M, Balermpas P, von der Grun J, Linge A, Lohaus F, Krause M, Baumann M, Stuschke M, Zips D, Grosu AL, Abdollahi A, Debus J, Belka C, Pigorsch S, Combs SE, Budach V, Tinhofer I (2019) Interference of tumour mutational burden with outcome of patients with head and neck cancer treated with definitive chemoradiation: a multicentre retrospective study of the German Cancer Consortium Radiation Oncology Group. Eur J Cancer 116:67–76. https://doi.org/10.1016/j.ejca.2019.04.015
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P, Saunders CT (2018) Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15(8):591–594. https://doi.org/10.1038/s41592-018-0051-x
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‑2; iso‑3. Fly (Austin) 6(2):80–92. https://doi.org/10.4161/fly.19695
Tamborero D, Rubio-Perez C, Deu-Pons J, Schroeder MP, Vivancos A, Rovira A, Tusquets I, Albanell J, Rodon J, Tabernero J, de Torres C, Dienstmann R, Gonzalez-Perez A, Lopez-Bigas N (2018) Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med 10(1):25. https://doi.org/10.1186/s13073-018-0531-8
Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194(4260):23–28
de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, Jamal-Hanjani M, Shafi S, Murugaesu N, Rowan AJ, Gronroos E, Muhammad MA, Horswell S, Gerlinger M, Varela I, Jones D, Marshall J, Voet T, Van Loo P, Rassl DM, Rintoul RC, Janes SM, Lee SM, Forster M, Ahmad T, Lawrence D, Falzon M, Capitanio A, Harkins TT, Lee CC, Tom W, Teefe E, Chen SC, Begum S, Rabinowitz A, Phillimore B, Spencer-Dene B, Stamp G, Szallasi Z, Matthews N, Stewart A, Campbell P, Swanton C (2014) Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346(6206):251–256. https://doi.org/10.1126/science.1253462
Reiter JG, Makohon-Moore AP, Gerold JM, Heyde A, Attiyeh MA, Kohutek ZA, Tokheim CJ, Brown A, DeBlasio RM, Niyazov J, Zucker A, Karchin R, Kinzler KW, Iacobuzio-Donahue CA, Vogelstein B, Nowak MA (2018) Minimal functional driver gene heterogeneity among untreated metastases. Science 361(6406):1033–1037. https://doi.org/10.1126/science.aat7171
Moon SH, Kim J, Joung JG, Cha H, Park WY, Ahn JS, Ahn MJ, Park K, Choi JY, Lee KH, Kim BT, Lee SH (2018) Correlations between metabolic texture features, genetic heterogeneity, and mutation burden in patients with lung cancer. Eur J Nucl Med Mol Imaging. https://doi.org/10.1007/s00259-018-4138-5
Hu LS, Ning S, Eschbacher JM, Baxter LC, Gaw N, Ranjbar S, Plasencia J, Dueck AC, Peng S, Smith KA, Nakaji P, Karis JP, Quarles CC, Wu T, Loftus JC, Jenkins RB, Sicotte H, Kollmeyer TM, O’Neill BP, Elmquist W, Hoxworth JM, Frakes D, Sarkaria J, Swanson KR, Tran NL, Li J, Mitchell JR (2017) Radiogenomics to characterize regional genetic heterogeneity in glioblastoma. Neuro-oncology 19(1):128–137. https://doi.org/10.1093/neuonc/now135
O’Connor JPB (2017) Cancer heterogeneity and imaging. Semin Cell Dev Biol 64:48–57. https://doi.org/10.1016/j.semcdb.2016.10.001
Acknowledgements
We acknowledge the support of Prof. B. Sipos, Institute of Pathology and Neuropathology; Department of General and Molecular Pathology and Pathological Anatomy, Tübingen, Germany and Prof. I. Tinhofer, Department of Radiation Oncology and Radiotherapy, Charité Berlin, Germany. Furthermore, we would like to thank C. Goltermann for the language editing.
Funding
This project was funded by the Center for Personalised Medicine (ZPM) Tübingen/Germany (grant number 025/2015BO1). Kerstin Clasen was supported by the Fortüne/PATE Program of the Medical Faculty, Eberhard Karls University Tübingen (grant number: 2447-0-0). O. Riess and S. Ossowski receive funding by the German Research Foundation (DFG) as an NGS Competence Center (INST 37/1049-1). Parts of the research leading to these results have received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013), ERC Grant Agreement no. 335367. None of our funding sources was involved in the study design, data analysis or data interpretation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
C. la Fougère has research collaborations with Siemens Healthineers. D. Zips and D. Thorwarth have research collaborations with Elekta, Philips and Siemens. K. Clasen, S. Leibfarth, F.J. Hilke, J. Admard, R.M. Winter, S. Welz, S. Gatidis, D. Nann, S. Ossowski, T. Breuer, K. Nikolaou, O. Riess and C. Schroeder declare that they have no competing interests.
Ethical standards
The study was approved by the local ethics committee (reference number 025/2015) and all patients declared their written informed consent. The study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments.
Rights and permissions
About this article

Cite this article
Clasen, K., Leibfarth, S., Hilke, F.J. et al. PET/MRI and genetic intrapatient heterogeneity in head and neck cancers. Strahlenther Onkol 196, 542–551 (2020). https://doi.org/10.1007/s00066-020-01606-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00066-020-01606-y