
Abstract
Cancer is a major worldwide public health problem and is still the leading cause of death in the United States. There are many types of cancer treatment but completely successful results are oftentimes not attained. It remains a challenge to develop efficacious clinically useful cancer therapies. Therapies targeting dysregulated signal transduction pathways in cancer can be efficacious anti-cancer therapies with minimal adverse effects. In this study, we focus on novel small molecule p53 Activator Wnt Inhibitor-2 (PAWI-2) that was developed by optimizing potency and pharmaceutical properties. PAWI-2 is a nontoxic DNA-damage pathway inhibitor that shows a broad spectrum of potency and significant efficacy in vitro and in vivo. This study focuses on the application of PAWI-2 to four major types of cancers including colorectal cancer (CRC), breast cancer (BC), prostate cancer (PCa), and pancreatic cancer (PC). PAWI-2 shows a novel mechanism of action (MOA) by modulating two mechanisms of cancer invasion. In cancer with unimpaired p53, PAWI-2 activates DNA-damage checkpoint and mitochondrial p53-dependent apoptotic signaling. Consistently observed in most cancer types, PAWI-2 induces phosphorylation of optineurin (OPTN) to cause G2/M cell cycle arrest. These two mechanisms operate regardless of p53 variants and/or KRAS mutation status and also manipulate the effect of PAWI-2 to overcome tumor stemness and drug resistance in PC stem cells (PCSCs). This study summarizes the development of PAWI-2 as an attractive targeted therapeutic for mechanism-driven anti-cancer drug discovery.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer is a major worldwide public health problem and is the second leading cause of death in the United States (US). In 2019, there were ~1,762,450 new cancer cases diagnosed and about 606,880 cancer deaths that occurred in the US (Siegel et al. 2020). There are many types of cancer treatment, including surgery, radiation therapy, chemotherapy, immunotherapy, targeted therapy, hormone therapy, stem cell transplantation, and precision medicine (National Cancer Institute Types of Cancer Treatment 2020). These therapies can be applied either alone or in combination with other drugs in the treatment of cancer. However, many important challenges are still present in the field of cancer therapies that need to be met to improve treatment outcomes for patients.
To date, nonsurgical treatment of cancer (mainly conventional chemotherapy, targeted biological therapies, and radiotherapy) has not generated completely satisfactory results. Conventional chemotherapies have many problems including low target selectivity, drug resistance, inability to effectively address metastatic disease and oftentimes, severe side effects. Targeted biological therapies (i.e., monoclonal antibodies) are promising but are relatively expensive and are not broadly applicable. The pathogenesis of cancer is characterized by clinically relevant genetic alterations leading to either activation of oncogenes or inactivation of tumor suppressor genes (e.g., inactivation of p53 function) (Anastas and Moon 2013; Kahn 2014; Lane et al. 2010; Levine and Oren 2009). Therapies targeting protein components of dysregulated signal transduction pathways can be efficacious anti-cancer therapies with minimal adverse effects (Anastas and Moon 2013; Kahn 2014; Lane et al. 2010; Levine and Oren 2009). For example, tumor suppressor protein p53 plays a critical role in cellular response to DNA damage and other genomic aberrations, and is an attractive target for mechanism-driven anti-cancer drug discovery (Lane et al. 2010). In some cases, targeting p53 has achieved good clinical responses that have affected survival in some cancers.
The present study focuses on a novel small molecule called p53 Activator Wnt Inhibitor-2 (i.e., PAWI-2, Fig. 1) that was developed after numerous rounds of medicinal chemistry refinement to optimize potency and pharmaceutical properties. As previously reported, PAWI-2 is a nontoxic DNA-damage pathway inhibitor that suppresses cancer cell growth and survival (Cashman et al. 2013; Cheng et al. 2018, 2019a, 2019b; Okolotowicz et al. 2018; Cheng and Cashman 2020) in several cancer types including colorectal cancer (CRC) (Cheng et al. 2018), breast cancer (BC) (Okolotowicz et al. 2018), prostate cancer (PCa) (Cheng et al. 2019a), and pancreatic cancer (PC) (Cheng et al. 2019b; Cheng and Cashman 2020). PAWI-2 shows a broad spectrum of potency and significant efficacy in vitro and in vivo. This study also provides overviews regarding the mechanisms of action of PAWI-2 in various types of cancer (Fig. 2).

Chemical structure of PAWI-1 and PAWI-2

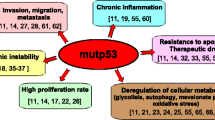
Proposed model depicts a mechanism of PAWI-2 in various types of cancer. Green arrows, stimulations; red lines, inhibition. CRC colorectal cancer, BC breast cancer, PCa prostate cancer, PC pancreatic cancer, PCSCs pancreatic cancer stem cells, KRAS Kirsten rat sarcoma viral oncogene homolog, TBK1 TANK-binding kinase 1, ATM ataxia-telangiectasia mutated serine/threonine kinase, ATR ATM- and Rad3-related serine/threonine kinase, HIPK2 homeodomain interacting protein kinase 2, TCF4 transcription factor 4/immunoglobulin transcription factor 2 (ITF-2), Wnt wingless integration site, p53 tumor protein p53, Bcl-2 B-cell lymphoma 2 protein, Bcl-xL B-cell lymphoma-extra-large protein, Mcl-1 induced myeloid leukemia cell differentiation protein, Bak Bcl-2 homologous antagonist/killer protein, Bax Bcl-2-associated X protein, Bad Bcl-2 associated agonist of cell death protein, Bid BH3 interacting-domain death agonist protein, CASP3 caspase-3, PARP poly (ADP-ribose) polymerase, p62 (p62/SQSTM1) ubiquitin-binding protein/sequestosome-1, OPTN optineurin, p21 (p21/Cip1) cyclin-dependent kinase inhibitor 1/CDK-interacting protein 1, EGFR-TKIs epidermal growth factor receptor-tyrosine kinase inhibitors, G2/M Gap 2 phase/mitotic phase (subphase of interphase in the cell cycle)
SAR of PAWI compounds and ADMET of PAWI-2
“Hit” compound PAWI-1 (Fig. 1) was identified as an inhibitor of canonical Wnt/β-catenin-dependent transcription in a high-throughput screen. After numerous rounds of medicinal chemistry refinement (i.e., chemical synthesis of analogs of PAWI-1), PAWI-2 was identified and developed as a lead compound with greater potency and more promising pharmaceutical properties than hit PAWI-1 (Cashman et al. 2013; Okolotowicz et al. 2018). Compared with PAWI-1, PAWI-2 incorporates a tertiary amine HCl salt (Fig. 1). This increased aqueous solubility of PAWI-2. PAWI-2 also has improved potency for Wnt inhibition, inhibition of cancer cell proliferation (two-fold greater potency), improved modulation of physicochemical properties, and also improved pharmacological properties including ADMET (Cashman et al. 2013; Okolotowicz et al. 2018).
PAWI-2 possesses drug-like ADMET properties. PAWI-2 is chemically stable (i.e., >30 days, no change, pH 7.4). PAWI-2 is not extensively metabolized (i.e., stable for >200 min in mouse, rat, and human liver microsomes + NADPH) although minor (i.e., 5%) N-demethylation was observed in small animal and human liver microsomes supplemented with NADPH. In human liver microsomes, PAWI-2 does not inhibit the metabolism of testosterone suggesting it does not inhibit prominent human P-450s. PAWI-2 has acceptable pharmacokinetic properties including a favorable half-life (i.e., 16 h), Tmax, and bioavailability (i.e., ~20%). PAWI-2 dose-dependently accumulated in tumors based on LCMS analysis. Compared with PAWI-1, the lack of metabolism and greater aqueous solubility of PAWI-2 affords more favorable pharmacokinetic properties. As described below, PAWI-2 is not detectably toxic to small animals. In our view, the efficacy of PAWI-2 to ameliorate multiple forms of cancer (i.e., prostate, breast, colon and pancreatic, and other cancers) points to the importance of inhibition of key cancer-causing signaling pathways and not to some arbitrary toxicity.
By applying the same anti-cellular viability study used in cancer cell studies, in normal cells, PAWI-2 did not affect cell growth (i.e., immortalized normal mouse epithelial cells) (Cheng et al. 2018). At a supermaximal concentration (i.e., up to 5 µM), PAWI-2 did not show any detectable inhibition of cell viability of normal intestine cells (IEC-6) (Cheng et al. 2018) or normal prostate epithelial cells (11220-hTERT) (Cheng et al. 2019a). The conclusion is that PAWI-2 only operates to selectively kill rapidly dividing cancer cells and not quiescent normal cells. In Ames-like tests, no direct or indirect genotoxicity or cytotoxicity was observed for PAWI-2 (i.e., up to 10 µM) with or without metabolic bioactivation (Cheng et al. 2018).
In small animal studies, it was shown that PAWI-2 was safe in vivo (no toxicity after 7 days, 30 mg/kg/day, i.p. or oral, mice) and nontoxic (at 1000 mg/kg, i.p., rats; 20 mg/kg/day, 30 days, mice). Histopathological and morphometric evaluation of tissues from animals dosed with large amounts of PAWI-2 (i.e., 1000 mg/kg for 24 h (rats) or 20 mg/kg/day for 30 days (mice)) did not show any abnormalities. So far as we can see, PAWI-2 does not appear to show any toxicity at the doses examined in small animals.
Colorectal cancer
CRC is the third leading cause of cancer-related death in the US. It is anticipated to result in an estimated 53,200 deaths during 2020 (Siegel et al. 2020). Therapeutic options for CRC are limited to surgery, radiation, or chemotherapy and are often effective for early-stage disease but not for metastatic CRC. CRC drugs frequently cause untoward gastrointestinal and hematologic side effects with limited clinical benefit (Curtin 2013; Sridharan et al. 2014). Initiation and progression of CRC has been linked to dysregulation of several signaling pathways including Wnt/β-catenin (Morin et al. 1997; Su et al. 1993) and p53 pathways (Fearon 2010; Iacopetta 2003) and mutations of key molecular regulators that promotes a greater metastatic phenotype. Inhibition of Wnt/β-catenin signaling and regulators of p53 (i.e., MDM2, leading to p53 stabilization and activation) are considered attractive anti-CRC approaches (Lane et al. 2010; Novellasdemunt et al. 2015). However, various adverse effects (i.e., side effects, toxicities) limit long-term treatment with first generation anti-CRC drugs and have prompted ongoing development of less toxic drug-like molecules (Anastas and Moon 2013; Kahn 2014; Tisato et al. 2017).
PAWI-2 works against CRC cells targeting both Wnt signaling and ATM/p53
Shown by in vitro characterization, PAWI-2 potently inhibited cell growth of multiple types of CRCs (IC50s, 10–20 nM) through inhibition of Wnt transcription, activation of p53, and binding of tubulin to afford its anti-mitotic and anti-proliferative activity. Although other anti-mitotic agents have been reported (Janssen and Medema 2011; Rao et al. 2012), the effect of PAWI-2 on Wnt and p53 pathways constitutes a novel and unprecedented mechanism. In contrast to other compounds that modulate the β-catenin complex, the action of PAWI-2 is due to its target downstream of Wnt inhibition at the β-catenin complex.
PAWI-2 activates mitotic stress signaling via ATM/ATR-kinase (Fig. 2) in CRC. Phosphorylated ATM (Ser1981) and p53 (Ser15 and Ser46) were induced by the treatment of PAWI-2, and this showed activation of DNA-damage-sensitive cell cycle checkpoints (Cheng et al. 2018). PAWI-2 induced p53-dependent cell apoptosis regardless of p53 mutation types (i.e., HCT-116, WT; DLD-1, Ser241Phe; SW480, Arg273His/Pro309Ser) (Cheng et al. 2018), showing PAWI-2 could restore the tumor suppressor role of p53 in CRC cells with unimpaired p53 status (Fig. 2). Here, we define “unimpaired p53 status” as that standing for both wild-type (WT) p53 and missense mutations of p53 that retain some or all of p53 functional activity. The opposite of this definition is null p53 status. This distinguishes PAWI-2 from other p53-targeted cancer inhibitors (e.g., AMG-232), that work as MDM2 inhibitors but only promote restoration of critical p53 function in WT p53 tumors (Canon et al. 2015).
As has been found in >80% of sporadic CRCs (Novellasdemunt et al. 2015; Masuda et al. 2015; Yang et al. 2006), inactive adenomatous polyposis coli (APC) mutations (i.e., in DLD-1, SW480 cells) prevent β-catenin degradation. Generally, this limits use of Wnt inhibitors that target upstream of the core pathway (β-catenin complex) in clinical treatment of CRC. PAWI-2 modulates mitotic stress signaling and that leads to inhibition of Wnt responsiveness but works downstream of β-catenin by regulating disengagement of TCF proteins from chromatin (Fig. 2) (Cheng et al. 2018). CRC cell proliferation inhibition by PAWI-2 was independent of APC mutations. This suggests PAWI-2 may be useful as a tumor-specific Wnt inhibitor (i.e., Wnt-activated tumors with APC mutants) that works far downstream from the β-catenin complex.
For the mechanism of action (MOA) of PAWI-2, the linkage between inhibition of Wnt signaling and ATM/p53 activation was associated with activation of homeodomain interacting protein kinase 2 (HIPK2) by PAWI-2 (Fig. 2) (Cheng et al. 2018). HIPK2 activation caused inhibition of Wnt transcription via phosphorylation of TCF proteins (D’Orazi et al. 2002; Hikasa and Sokol 2011) and p53 activation via phosphorylation of p53 at Ser46 (D’Orazi et al. 2002). However, this linkage was observed in HCT-116 cells but not p53 null cells (Cheng et al. 2018). The lack of stimulation of cell apoptosis by PAWI-2 in p53-deficient cells (i.e., 10.1 cell line) (Cheng et al. 2018) showed that p53 played a significant role in the MOA of PAWI-2 to induce cell apoptosis (Fig. 2).
In CRC cells, PAWI-2 also induced immediate morphological cell rounding similar to paclitaxel and colchicine and arrested the cell cycle in the G2/M phase (Fig. 2) (Cheng et al. 2018). CRC cells treated with PAWI-2 showed a dose-dependent decrease in acetylated tubulin with a concomitant increase in tyrosinated tubulin (Cheng et al. 2018). These effects show one of the targets of PAWI-2 could be tubulin (De Brabander et al. 1986) via tubulin destabilization (Janke 2014; Janke and Bulinski 2011). We also showed that PAWI-2 bound to tubulin at a colchicine-binding site that is mechanistically distinct from paclitaxel or vinblastine (Cheng et al. 2018). These properties distinguish PAWI-2 from other reported tubulin inhibitors (Yoshimatsu et al. 1997). In addition, the inhibitory effect of PAWI-2 on CRC cell migration, invasion, and EMT processes further showed the utility of PAWI-2 as a candidate for the treatment of metastatic CRC (Kinzler and Vogelstein 1996).
PAWI-2 inhibited CRC tumor growth in xenograft models
PAWI-2 is chemically and metabolically stable with acceptable PK for efficient drug delivery/clearance and good bioavailability (Cheng et al. 2018). PAWI-2 did not cause any adverse behavioral or toxic effects. In a xenograft model, compared with vehicle control, PAWI-2 (20 mg/kg/day, 28 consecutive days, i.p.) decreased growth of implanted HCT-116 tumors greater than four-fold in nude mice (Fig. 3a, b) (Cheng et al. 2018). Serum clinical data suggested that treatment of animals with PAWI-2 was not toxic to liver, kidney, or blood. Post-xenograft tumor tissue analysis (i.e., immunoblotting and histology) showed a significantly greater apoptotic effect in the PAWI-2 treated group (Cheng et al. 2018). PAWI-2 was also more potent than vinblastine, another microtubule (MT) destabilizer, at inhibition of tumor growth (Koyanagi et al. 1994) but therapeutic use of vinblastine is limited because of its toxicity (Zhou and Rahmani 1992).

Effect of PAWI-2 on HCT-116 CRC tumor growth in a subcutaneous xenograft model in nude mice. a The effect of 20 mg/kg/day PAWI-2 (solid squares, n = 6) or vehicle (open circles, n = 5) on tumor volume of HCT-116 CRC xenografts. Treatment was administered every day for 28 days starting on day 7 by intraperitoneal injection. b The weight of excised tumors at day 34 from the same animals of a and representative photographs of excised tumors. Data are mean ± SD. P values were estimated by Student’s t test. This figure was revised based on Fig. 6 in Cheng et al. 2018
Concluding remarks for effects of PAWI-2 on CRC
PAWI-2 binds tubulin and potently activates mitotic stress signaling to stabilize p53 and inhibit Wnt/β-catenin transactivation of downstream genes in CRC cells. To our knowledge, PAWI-2 is the first reported potent small molecule that inhibits Wnt/β-catenin signaling and activates p53 signaling regardless of p53 mutation status without acute or chronic toxicity (Cheng et al. 2018). Therefore, the design of PAWI-2 for targeting multiple pathways to modulate cross-talk between molecular signaling pathways supports a novel CRC treatment strategy.
Breast cancer
BC is the most common cancer type and the second leading cause of cancer-related deaths in women in the US (Siegel et al. 2020). It is estimated that around 41,760 women died from this disease in 2019 (DeSantis et al. 2019). Although early-stage BC is curable, the 5-year survival rate of patients with metastatic BC is only 20% (DeSantis et al. 2019). Common anti-BC therapeutic modalities typically use a combination of surgery, radiation, and chemotherapeutics. Currently used chemotherapies include anthracyclines and taxanes (e.g., doxorubicin and paclitaxel) and are usually used in combination with other chemotherapeutics including fluorouracil and cyclophosphamide (Chemotherapy Medicines 2018).
Among all the BC subtypes, triple-negative BC (TNBC) is the most lethal subtype and is characteristically aggressive with significant recurrence, is metastatic, and has high mortality rates (Bauer et al. 2007; Foulkes et al. 2010). However, most of these drugs for TNBC treatment have problems with toxicity and may not be able to efficiently nullify activation of multiple growth-promoting pathways associated with TNBC (Carey et al. 2007). Due to the lack of druggable targets (i.e., estrogen receptor (ER), human epidermal growth factor receptor 2 (HER2)), surgery, radiation, and chemotherapy are still the “gold standards” in the treatment of TNBC. However, TNBC patients that do not respond to these therapies will eventually develop metastatic BC, which is virtually incurable (Carey et al. 2007; Dent et al. 2007; Liedtke et al. 2008). PAWI-2 is a nontoxic small molecule that uses a completely new approach to treat advanced BC (i.e., TNBC).
PAWI-2 works against BC cells regardless of BC types
PAWI-2 inhibits cell viability and stimulated apoptosis of non-TNBC (MCF-7) and TNBC (including MDA-MB-231, HS578T, BT-549, T47D, and MDA-MB-468) cells in vitro (IC50s, 20–200 nM). In cisplatin (cP)-resistant MCF-7 cells, PAWI-2 also potently inhibited cell viability with an IC50 similar to that observed for parental MCF-7 cells (unpublished results). Specifically targeting drug-resistant BC cells is clinically important because drug resistance is a hallmark of metastatic BC. An increase in the expression level of phospho-ATM (an upstream kinase for p53 phosphorylation, Fig. 2) in both MCF-7 and MDA-MB-231 cells after treatment of cells with PAWI-2 (Okolotowicz et al. 2018) was also observed.
One of the most common chemotherapeutics for treatment of BC is doxorubicin and is usually given in combination with other chemotherapeutics, including paclitaxel or 5-fluorouracil (Chemotherapy Medicines 2018). PAWI-2 showed a synergistic effect on doxorubicin to inhibit proliferation of BC cells. Most patients with TNBC (78%) overexpress the transmembrane epidermal growth factor receptor (EGFR) (Arteaga and Truica 2004). This suggests EGFR is a potential therapeutic target. However, early phase clinical trials of anti-EGFR therapies failed to show significant efficacy in TNBC (Arteaga and Truica 2004). We tested synergism between PAWI-2 and 12 types of FDA-approved EGFR-tyrosine kinase inhibitors (TKIs) or vascular endothelial growth factor receptor (VEGFR)-TKI (i.e., erlotinib, afatinib, etc.) (National Cancer Institute Developmental Therapeutics Program 2020) in MDA-MB-231 cells and analyzed the results with Chou–Talalay synergism analysis (Chou 2010). PAWI-2 synergized 10 of the 12 drugs effectively to inhibit cell viability in MDA-MB-231 cells (lower value “combination index” < 1; unpublished result). This provides a powerful basis to develop PAWI-2 for the treatment of TNBC. To our knowledge, no EGFR therapies are efficient for treatment of TNBC (Nakai et al. 2016).
PAWI-2 inhibited BC tumor growth in xenograft models
PAWI-2 potently inhibited TNBC tumor growth in vivo when tested in a subcutaneous xenograft TNBC (MDA-MB-231 cells) tumor model (Fig. 4). Moreover, co-administration of PAWI-2 (20 mg/kg/day, 16 consecutive days, i.p.) plus doxorubicin (5 mg/kg/week, 16 days, i.p.) decreased tumor growth rate 8.8-fold compared with vehicle-treated animals (Fig. 4a) (Okolotowicz et al. 2018). At the end of the 16-day study treatment with PAWI-2 and doxorubicin dramatic, decrease in tumor volume and weight was observed compared with vehicle-treated mice (Fig. 4b, c) (Okolotowicz et al. 2018). Immunoblot analysis of tumor tissues showed considerably greater levels of p53 and phospho(Ser15)-p53 proteins and PARP cleavage (P < 0.05) compared with vehicle-treated samples (Okolotowicz et al. 2018).

Effect of PAWI-2 on MDA-MB-231 BC xenograft tumor growth in a subcutaneous model in nude mice. a Average tumor growth, b tumor volume, and c weight for excised tumors of animals treated with vehicle, doxorubicin, PAWI-2, or doxorubicin plus PAWI-2. Treatment was administered for 16 days starting on day 35 by intraperitoneal injection. Dose treatment: vehicle control (aqueous-PEG), n = 7; doxorubicin (5 mg/kg/week), n = 8; PAWI-2 (20 mg/kg/day), n = 6; or doxorubicin (5 mg/kg/week) plus PAWI-2 (20 mg/kg/day), n = 5; Dox doxorubicin. Data are mean ± SEM. P values were estimated by one-way ANOVA test in a (***P < 0.001) and by Student’s t test in b and c (*P < 0.05, **P < 0.01, ****P < 0.0001). This figure was revised based on Fig. 3 in Okolotowicz et al. 2018
Concluding remarks for effects of PAWI-2 on BC
PAWI-2 is a potent pathway modulator that suppressed BC cell viability and in combination with other chemotherapeutics or EGFR-TKIs also showed the ability to synergistically inhibit cell growth of MDA-MB-231 in vitro and in vivo. These studies warrant further preclinical investigation of PAWI-2 for the treatment of BC especially aggressive BC types (i.e., TNBC).
Prostate cancer
In 2019, PCa was the second leading cause of cancer-related death for men and resulted in an estimated 31,620 deaths (Siegel et al. 2020). PCa is often initially responsive to anti-androgen hormone therapies and thus characterized as castration-sensitive PCa. However, in 35% of patients, PCa recurs and is often transformed to castrate-resistant PCa (CRPCa), thus rendering hormone therapies ineffective (Gandhi et al. 2018; Howlader et al. 2019). However, effective drugs to treat CRPCa are lacking. Standard-of-care treatment options for CRPCa are limited to radiation or hormone therapy (e.g., enzalutamide) (Schalken and Fitzpatrick 2016; Tran et al. 2009) or in combination with chemotherapy (e.g., docetaxel) (Mukherji et al. 2014).
Androgen receptor (AR) signaling is a critical survival pathway for PCa cells. Expression of AR in PCa is heterogeneous. PCa cells are often classified as AR expressing (i.e., AR+, LNCaP) and AR low- or nonexpressing (i.e., AR−/lo, PC-3) (van Bokhoven et al. 2003). Blockade of AR was shown to be an effective PCa therapeutic strategy (Schalken and Fitzpatrick 2016; Tran et al. 2009). But AR−/lo PCa cells are resistant to most commonly used therapy for androgen ablation (Katzenwadel and Wolf 2015). Most AR+ PCa reportedly respond to androgen ablation therapies but often leads to androgen-depletion independent status (Katzenwadel and Wolf 2015; Karantanos et al. 2013). This incurable stage of PCa (CRPCa) remains a challenge to treat (Jernberg et al. 2017; Ni et al. 2013; Ritch and Cookson 2016; Yang et al. 2009) and >90% of patients develop metastases that cause PCa-related deaths (Gandhi et al. 2018). For example, enzalutamide is an FDA-approved AR antagonist to treat PCa (Azvolinsky 2012). Combination of enzalutamide with abiraterone is the most widespread first-line treatment for CRPCa (de Bono et al. 2011). However, clinical studies showed this multi-component therapy only modestly extended overall survival with many untoward side effects (Gandhi et al. 2018) and high rate of relapse for patients (de Bono et al. 2011; Dhingra et al. 2013; Scher et al. 2012). Therefore, developing novel therapeutic approaches to overcome castrate-resistance are urgently needed in the treatment of CRPCa.
PAWI-2 works against both androgen-sensitive and androgen-independent PCa cells
We observed that PAWI-2 was effective against androgen-sensitive PCa (LNCaP) and androgen-insensitive CRPCa (PC-3). P53-dependence of PAWI-2 was also observed in PCa cells because apoptosis induced by PAWI-2 was significantly greater in LNCaP (WT p53) than that in PC-3 (similar to null p53 status) (Cheng et al. 2019a). However, there was no apparent relationship observed between p53 mutation status and potency of PAWI-2 for inhibition of in vitro PCa cell growth (similar IC50s, ~15 nM) (Cheng et al. 2019a).
In the presence of PAWI-2, inhibition of PCa cell viability was markedly increased compared with enzalutamide alone or a combination of enzalutamide with abiraterone (Cheng et al. 2019a). This was consistently observed in both enzalutamide-sensitive LNCaP cells and enzalutamide-resistant PC-3 cells. PAWI-2 sensitized these cells to enzalutamide (but not abiraterone) (Cheng et al. 2019a). PAWI-2 inhibited PCa cell migration and invasion regardless of AR response status and also synergized/resensitized the effect of enzalutamide. PAWI-2 is capable of interrupting highly invasive and metastatic properties of CRPCa. Considering cancer metastasis is a hallmark of malignancy in CRPCa, this is clinically relevant. However, the synergistic effect of PAWI-2 on enzalutamide in PCa cells was not completely dependent on p53 activation. Other effectors likely contribute to potency.
In PCa cells, PAWI-2 caused loss of mitochondrial membrane potential and affected mitochondrial membrane dynamics and import/translocation of proteins (degradation of several mitochondrial localized Bcl-2 family proteins) (Cheng et al. 2019a). This effect was independent of AR status and also highly associated with synergistic effects of PAWI-2 on enzalutamide. PAWI-2 selectively affected Bcl-2 family proteins (Fig. 2) that can be linked to non-genomic signaling of AR in PCa (i.e., control mitochondrial function and retrograde signaling (Massie et al. 2011; Zarif and Miranti 2016)). Accordingly, upregulation of pro-survival factors (i.e., Bcl-2, Bcl-xL, and Mcl-1) and acquired enzalutamide resistance is a hallmark of CRPCa (Li et al. 2018). The imbalance of pro-survival and anti-survival factors caused by PAWI-2 (Fig. 1) through affecting mitochondrial membrane potential/function may be a controlling mechanism in synergizing/resensitizing the effect of enzalutamide to overcome enzalutamide resistance.
PAWI-2 inhibited PCa tumor growth in xenograft models
Efficacy of PAWI-2 was examined either as a single agent or in combination with enzalutamide in a PC-3 xenograft animal model. PAWI-2 (20 mg/kg/day, 21 consecutive days, i.p.) or enzalutamide (5 mg/kg/day, 21 consecutive days, i.p.) with PAWI-2 decreased PC-3 tumor growth rate in mice but at the dose examined, enzalutamide alone did not inhibit PC-3 tumor growth (Fig. 5a–c) (Cheng et al. 2019a). Thus, PAWI-2 significantly decreased PC-3 tumor growth in vivo and also resensitized the effect of enzalutamide inhibition without any apparent abnormal effect on liver or kidney function (Cheng et al. 2019a). Amongst the few medications or treatments registered for CRPCa (including docetaxel, cabazitaxel, abiraterone, enzalutamide, and radium-223) (Karantanos et al. 2013), enzalutamide is well tolerated and has a favorable toxicity profile (Tran et al. 2009). However, enzalutamide treatment outcome remains modest. It is notable that PAWI-2 was more efficacious than the most commonly used clinical treatment in an animal model of PCa.

Effect of PAWI-2 on PC-3 PCa tumor growth in a subcutaneous xenograft model in nude mice. a Average tumor growth, b tumor volume, and c weight for excised tumors of animals treated with vehicle, enzalutamide, PAWI-2, or enzalutamide plus PAWI-2. Treatment was administered every day for 21 days starting on day 6 by intraperitoneal injection. Dose treatment: vehicle control (aqueous-PEG), n = 9; enzalutamide (5 mg/kg/day), n = 9; PAWI-2 (20 mg/kg/day), n = 9; or enzalutamide (5 mg/kg/day) plus PAWI-2 (20 mg/kg/day), n = 6; Enza enzalutamide. Data are mean ± SEM. P values were estimated by one-way ANOVA test in a (***P < 0.001) and by Student’s t test in b and c (*P < 0.05, **P < 0.01). This figure was revised based on Fig. 4 in Cheng et al. 2019a
Concluding remarks for effects of PAWI-2 on PCa
PAWI-2 is a highly efficacious compound for PCa with decreased side effects. PAWI-2 also provides a molecule for both androgen-dependent and androgen-resistant PCa treatment. PAWI-2 showed considerable synergism with enzalutamide. PAWI-2 effectively inhibited tumor growth in a xenograft model of CRPCa cells (PC-3) as a single agent and also in combination with enzalutamide. Because of its novel MOA, PAWI-2 has broad utility to treat more aggressive CRPCa.
Pancreatic cancer
PC has the poorest prognosis of any major malignancy with a 5-year survival rate about 5% (Siegel et al. 2020). PC is the third leading cause of cancer death in the US and soon will be the second most common cause of mortality due to cancer (Siegel et al. 2020; Rahib et al. 2014). Pancreatic ductal adenocarcinoma (PDAC) is the most common form of PC (>95%) (Siegel et al. 2020). Patients with PDAC are often diagnosed at late stages with extensive local tumor invasion and early metastasis, presenting a major obstacle to all forms of therapy. First-line chemotherapy (i.e., gemcitabine, 5-fluorouracil or FOLFIRINOX, etc.) has made minimal impact on PDAC treatment (Burris and Storniolo 1997; Burris et al. 1997; Conroy et al. 2011; Moore et al. 2007) and a majority of PC patients are often resistant to clinical therapies (Burris et al. 1997). Thus, it remains a challenge to develop an efficacious clinically useful PC therapy.
Mutations of tumor suppressor p53 are among the most common genetic changes in cancer (Deer et al. 2010). Approximately 75% of PDAC patients harbor intragenic p53 mutations (Moore et al. 2007; Fiorini et al. 2015) that makes PDAC cells resistant to chemotherapeutic regimens (Fiorini et al. 2015) due to loss of functional effects activated by p53 (i.e., growth arrest, apoptosis, and senescence). Mutations of p53 in tumor cells can cause loss of WT p53 and gain of novel oncogenic functions (i.e., regulation of DNA-damage-induced apoptotic response), leading to metastasis of cancer cells (Freed-Pastor and Prives 2012) and poor clinical response to cancer chemotherapies (Xu et al. 2014). An overarching challenge is to develop a drug that potently inhibits PDAC growth by restoring tumor suppressor function. Extra-nuclear p53 apoptotic cell death mechanisms are dependent on transactivation-deficient p53 localization to cytosol or mitochondria-associated membrane and/or ER (Chipuk and Green 2004; Moll et al. 2005; Vaseva and Moll 2009). In mitochondrial-controlled cell apoptosis (intrinsic pathway), trafficking of mitochondria-bound Bcl-2 family members (i.e., Bax, Bad, Bak, etc.) cause the release of mitochondrial cytochrome c into cytosol to induce further cell apoptosis (Chipuk et al. 2006; Suen et al. 2008).
Recently, cancer stem cells (CSCs) have come into focus as potential therapeutic targets in multiple cancers (Nassar and Blanpain 2016). Accumulation of mutations in CSCs enhances chemo/radiation resistance that often ablates the effect of therapy, leading to cancer recurrence, characteristic of PDAC (Hermann et al. 2007). Conversely, because CSCs are a unique subset of a tumor cell population, targeting these cells may lead to the identification of effective drugs for poorly treatable cancers such as PDAC (Nassar and Blanpain 2016). Human PC stem cells (hPCSCs) reported previously (i.e., FGβ3 cells) are a validated human CSC model (Desgrosellier et al. 2009; Seguin et al. 2014, 2017) that overexpress integrin αvβ3. In FGβ3 cells, integrin αvβ3 recruits Kirsten rat sarcoma viral oncogene homolog GTPase (KRAS) and RAS like proto-oncogene B (RalB) to activate serine/threonine kinase TANK-binding kinase 1 (TBK1, IκB kinase (IKK)-related kinase) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) to trigger dysregulated KRAS-RalB-NF-κB. This pathway was reported to be a pharmacological target to reverse CSC-like properties or resensitize drug resistance for established FGβ3 tumors (Desgrosellier et al. 2009; Seguin et al. 2014, 2017).
PAWI-2 potently decreased PDAC cell proliferation by activating DNA-damage checkpoint and apoptotic pathways (e.g., the mitochondrial (intrinsic) control of apoptosis) (Cheng et al. 2019b). PAWI-2 inhibited tumor growth of a syngeneic, orthotopic model of PDAC (Cheng et al. 2019b). PC stem cells (PCSCs) may provide an important target of significant clinical utility to treat PC. In this context, it is notable that PAWI-2 was observed to afford a novel treatment strategy that targeted hPCSCs or their extrinsic and intrinsic regulators. PAWI-2 ameliorates drug-resistant hPCSCs (i.e., FGβ3 cells) and synergizes erlotinib by targeting optineurin (OPTN)-dependent cell cycle arrest (Cheng and Cashman 2020). Development of PAWI-2 as an anti-PC drug candidate addresses an unmet clinical need. PAWI-2 could improve standard of care for patients because it synergizes eradication of hPCSCs.
PAWI-2 inhibits invasive PC by restoring cell apoptosis through activation of mitochondrial p53
PAWI-2 showed significant potency in inhibition of cell proliferation and activation of cell apoptosis in PDAC cell models (i.e., MIA PaCa-2, HPAC-1, BxPC-3, etc.) including two patient-derived PDAC cell lines (i.e., 779E and 1334E) (Cheng et al. 2019b). Resistance to other chemotherapy (i.e., gemcitabine, 5-fluorouracil, etc.) normally observed in cancer cells (Burris and Storniolo 1997; Burris et al. 1997; Conroy et al. 2011; Moore et al. 2007) with mutant p53 was not observed in PDAC cells (with different p53 mutant status) treated with PAWI-2. The dominant MOA through upstream DNA damage via ATR/ATM-kinase activation and p53-dependent apoptosis was still observed for PAWI-2 in PC (Fig. 2) (Cheng et al. 2019b) as we observed in CRC cells (Cheng et al. 2018). Shown by synergism analysis, p53 activation of apoptosis contributes to the MOA of PAWI-2 in the presence of chemotoxins. Specifically, phosphorylation of p53 at Ser15 likely is a factor that controls different synergistic effects of PAWI-2 observed in different PDAC cells (Cheng et al. 2019b).
Compared with WT p53 cells (i.e., HPAC), cell apoptosis of mutant p53 cancer cells (i.e., LM-P, MIA PaCa-2, etc.) was induced by PAWI-2 but without significant upregulation of p53 or phosphorylation of p53 (Cheng et al. 2019b). PAWI-2 activated p53 mediated transcription-independent mitochondrial-apoptotic pathways in both WT p53 and mutant p53 PDAC cells (Fig. 2) (Cheng et al. 2019b). PAWI-2 disrupts the interactions of pro-survival factors (i.e., Bcl-xL) and pro-apoptotic Bcl-2 family members (i.e., Bax) and p53. This action releases p53/Bax to activate translocation of pro-apoptotic Bcl-2 family proteins from the cytosol to the mitochondria (Cheng et al. 2019b). Activation of apoptotic systems by treatment of PAWI-2 in PDAC cells also involves loss of mitochondrial membrane integrity and opening of permeability transition pores related to mitochondrial outer-membrane permeabilization. As a result, this further induces cytochrome c, Smac, and HSP60 release into cytosol that neutralizes inhibition of apoptosis (caspases) and causes incipient apoptotic signaling (i.e., PARP cleavage) (Vaseva and Moll 2009; Chandra et al. 2002; Galluzzi et al. 2008). Such a working model differentiates PAWI-2 from other p53-targeted inhibitors in tumor suppression. P53-dependent cell death checkpoint inhibitors (e.g., nutlin-3a and pfithrins) do not induce apoptosis in mutant p53-bearing PCs (Sriraman et al. 2016).
As observed in CRC cells, PAWI-2 exerts its effect on Wnt signaling inhibition via HIPK2 and TCF proteins linked to p53 activation via phospho(Ser46)-p53 (Cheng et al. 2018). However, in PDAC cells with mutant p53 status, no detectable effect of PAWI-2 on Wnt target gene expression was observed. Likewise, no activation of phospho(Ser46)-p53 or phosphorylated-HIPK2 and TCF3 by PAWI-2 was observed in PDAC cells examined (Cheng et al. 2019b). Inhibition of Wnt-dependent transcription by PAWI-2 is unlikely to be a major pathway in the MOA of PAWI-2 in PDAC cells (Fig. 2).
PAWI-2 overcomes tumor stemness and drug resistance via cell cycle arrest in PCSCs
PAWI-2 is selective for killing hPCSC tumor spheroids responsible for hPCSC drug resistance (Cheng and Cashman 2020). In a well-established hPCSC model (FGβ3 cells) (Desgrosellier et al. 2009; Seguin et al. 2014, 2017), association of TBK1 with RalB of the major oncogene (RAS) in a dysregulated integrin αvβ3-KRAS-NF-κB signaling pathway promotes tumorigenesis and CSC-like properties (Desgrosellier et al. 2009; Seguin et al. 2014, 2017). We observed that PAWI-2 inhibited KRAS-NF-κB-RalB signaling regardless of KRAS or Ral status (Cheng and Cashman 2020). Given the fact that >90% of KRAS is activated by mutations in PC (Deer et al. 2010) and RAS or Ral inhibitors have not proven effective clinically (Seguin et al. 2014), this suggests that PAWI-2 may possess advantages in the clinic.
TBK1 is a serine/threonine kinase that phosphorylates p62 or OPTN (Pilli et al. 2012; Wild et al. 2011). TBK1 is involved in tumor suppression via a mitophagy pathway (Richter et al. 2016). However, we excluded the role of mitophagy initiated via TBK1/OPTN for the MOA of PAWI-2. Instead, phosphorylation of OPTN at Ser177 plays a pivotal role in mitotic progression and induces OPTN translocation into the nucleus (Ying et al. 2010). OPTN-dependent G2/M cell cycle arrest induced by PAWI-2 in FGβ3 cells parallels this process (Cheng and Cashman 2020). PAWI-2-induced OPTN phosphorylation negatively regulates TBK1 functional activity (dephosphorylation) that further regulated KRAS-NF-κB signaling. Moreover, phosphorylation of OPTN also controlled synergism between PAWI-2 and other validated drugs (i.e., erlotinib) (Cheng and Cashman 2020). OPTN may work as an overarching branch point for PAWI-2 inhibition of cell viability to overcome self-renewal capacity in FGβ3 cells and also to synergize other pathway inhibitors (Fig. 2).
Anti-mitotic agents may perturb the mitotic spindle through either disruption (e.g., vinblastine) or stabilization (e.g., paclitaxel) of MTs (Janssen and Medema 2011). PAWI-2 was previously found to disrupt MT structure in PDAC cells in a similar manner to that observed in other cancer types (Cheng et al. 2018, 2019b). However, we observed PAWI-2 was a dose-dependent MT stabilizer (<50 nM) and destabilizer (>100 nM) (Cheng and Cashman 2020). This further differentiates PAWI-2 from other MT disturbing agents that may also contribute to the considerable efficacy and lack of toxicity observed for PAWI-2 (Cheng et al. 2018). Phosphorylation of OPTN was closely associated with MT stabilization because this effect was also observed in cells treated with other MT stabilizers (e.g., paclitaxel or docetaxel). Accumulation of pS177-OPTN in the presence of MT stabilizers may be due to the essential role of MTs in coordinating and organizing many crucial cellular steps (Brouhard and Rice 2018). Thus, OPTN phosphorylation induced by PAWI-2 or other MT stabilizers could modulate synergism effects to overcome drug resistance and combat more aggressive CSCs.
PAWI-2 inhibited tumor growth in an orthotopic model of invasive PDAC
The efficacy of PAWI-2 was examined in an orthotopic PDAC (LM-P, invasive PDAC murine cell line) animal model (Tseng et al. 2010). PAWI-2 significantly decreased PDAC growth in vivo. Compared with vehicle-treated mice, excised tumor volumes and tumor weights from PAWI-2-treated mice (20 mg/kg/day; 28 days; i.p.) were significantly lower (i.e., P < 0.05; 65% and 41%, respectively; Fig. 6) (Cheng et al. 2019b). Compared with vehicle-treated animals, no difference was observed for tumor tissue morphology but a significant apoptotic effect (TUNEL staining) was observed in PAWI-2 treated animals (Cheng et al. 2019b). In clinical treatment of PDAC tumors, gemcitabine plus nab-paclitaxel is a standard systemic chemotherapy (Frese et al. 2012). PAWI-2 was equal or more efficacious compared with this most commonly used clinical combination treatment (Von Hoff et al. 2011, 2013) in an animal model of PDAC.

Effect of PAWI-2 on LM-P PDAC tumor growth in a syngeneic orthotopic model in mice. Tumor volume and weight for tumor samples excised from orthotopic mice treated with vehicle or two different doses of PAWI-2 (10 or 20 mg/kg/day) and representative photographs of excised tumors. Treatment was administered every day for 28 days starting on day 6 by intraperitoneal injection. Dose treatment: vehicle control (aqueous-DMSO-captisol), n = 7; PAWI-2 (10 mg/kg/day), n = 6; or PAWI-2 (20 mg/kg/day), n = 7. Data are mean ± SEM. P values were estimated by Student t tests (*P < 0.05). This figure was revised based on Fig. 4 in Cheng et al. 2019b
Concluding remarks for effects of PAWI-2 on PC
In summary, PAWI-2 is a nontoxic, highly efficacious treatment of PDAC that activates damage checkpoint and mitochondrial p53-dependent apoptosis. PAWI-2 showed considerable synergism with commonly used combination therapy in the treatment of PDAC and also showed synergism with specific pathway inhibitors (e.g., TBK1 inhibitors, EGFR inhibitors) against PCSCs. PAWI-2 inhibited tumor growth in an orthotopic model of invasive PDAC cells (LM-P). Moreover, selective pharmacological potency of PAWI-2 against PCSCs showed the utility of PAWI-2 to inhibit CSCs versus bulk cancer cells. This observation provides a basis for PAWI-2 as an efficient treatment of PC, especially in highly aggressive/metastatic cancer with stem-like properties and intrinsic or acquired drug resistance.
Overall conclusion
Cancer is still the leading cause of death in the US. Development of new ways to treat cancer is a significant challenge. Late-stage diagnosis of cancer renders current therapies ineffective often due to their drug-resistant nature. The effectiveness of relatively new “targeted treatments” remains to be shown. PAWI-2 affords a completely different focus on inhibition of key molecular pathways by a safe compound. PAWI-2 showed great efficacy in four relevant but different preclinical cancer models. PAWI-2 does not depend on any particular cancer cell mutation profile to work effectively. PAWI-2 may synergize existing standard of care thus increasing efficacy of standard of care anti-cancer agents with improved safety. Our work on PAWI-2 provides fundamental information about a novel therapeutic strategy to treat cancer and will further enhance standard of care with minimal side effects.
References
Anastas JN, Moon RT (2013) WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 13(1):11–26
Arteaga CL, Truica CI (2004) Challenges in the development of anti-epidermal growth factor receptor therapies in breast cancer. Semin Oncol 31(1 Suppl 3):3–8
Azvolinsky A (2012) FDA approves enzalutamide (Xtandi) for late-stage prostate cancer. CancerNetwork. https://www.cancernetwork.com/articles/fda-approves-enzalutamide-xtandi-late-stage-prostate-cancer
Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V (2007) Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer 109(9):1721–1728
van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, Nordeen SK, Miller GJ, Lucia MS (2003) Molecular characterization of human prostate carcinoma cell lines. Prostate 57(3):205–225
de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman Jr OB, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Flechon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI, Investigators, C.-A.- (2011) Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 364(21):1995–2005
De Brabander M, Geuens G, Nuydens R, Willebrords R, Aerts F, De Mey J (1986) Microtubule dynamics during the cell cycle: the effects of taxol and nocodazole on the microtubule system of Pt K2 cells at different stages of the mitotic cycle. Int Rev Cytol 101:215–274
Brouhard GJ, Rice LM (2018) Microtubule dynamics: an interplay of biochemistry and mechanics. Nat Rev Mol Cell Biol 19(7):451–463
Burris H, Storniolo AM (1997) Assessing clinical benefit in the treatment of pancreas cancer: gemcitabine compared to 5-fluorouracil. Eur J Cancer 33(Suppl 1):S18–S22
Burris III HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD (1997) Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 15(6):2403–2413
Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, Eksterowicz J, Ye Q, Jin L, Chen A, Zhou J, Cordover D, Kaufman S, Kendall R, Oliner JD, Coxon A, Radinsky R (2015) The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol Cancer Ther 14(3):649–658
Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML, Perou CM (2007) The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res 13(8):2329–2334
Cashman JR, Mercola M, Schade D, Tsuda M (2013) Compounds for inhibition of cancer cell proliferation. US Patent 13/748,770
Chandra D, Liu JW, Tang DG (2002) Early mitochondrial activation and cytochrome c up-regulation during apoptosis. J Biol Chem 277(52):50842–50854
Chemotherapy medicine form Breastcancer.org. http://www.breastcancer.org/treatment/chemotherapy/medicines. Accessed 19 Sep 2018
Cheng J, Cashman JR (2020) PAWI-2 overcomes tumor stemness and drug resistance via cell cycle arrest in integrin β3-KRAS-dependent pancreatic cancer stem cells. Sci Rep. https://doi.org/10.1038/s41598-020-65804-5
Cheng J, Dwyer M, Okolotowicz KJ, Mercola M, Cashman JR, Novel A (2018) Inhibitor targets both Wnt signaling and ATM/p53 in colorectal cancer. Cancer Res 78(17):5072–5083
Cheng J, Moore S, Gomez-Galeno J, Lee DH, Okolotowicz KJ, Cashman JR (2019a) A novel small molecule inhibits tumor growth and synergizes effects of enzalutamide on prostate cancer. J Pharm Exp Ther 371(3):703–712
Cheng J, Okolotowicz KJ, Ryan D, Mose E, Lowy AM, Cashman JR (2019b) Inhibition of invasive pancreatic cancer: restoring cell apoptosis by activating mitochondrial p53. Am J Cancer Res 9(2):390–405
Chipuk JE, Bouchier-Hayes L, Green DR (2006) Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ 13(8):1396–1402
Chipuk JE, Green DR (2004) Cytoplasmic p53: bax and forward. Cell Cycle 3(4):429–431
Chou TC (2010) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70(2):440–446
Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M, Groupe Tumeurs Digestives of, U., Intergroup, P. (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364(19):1817–1825
Curtin JC (2013) Novel drug discovery opportunities for colorectal cancer. Expert Opin drug Discov 8(9):1153–1164
Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ (2010) Phenotype and genotype of pancreatic cancer cell lines. Pancreas 39(4):425–435
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA (2007) Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 13(15 Pt 1):4429–4434
DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding Sauer A, Jemal A, Siegel RL (2019) Breast cancer statistics, 2019. CA Cancer J Clin 69(6):438–451
Desgrosellier JS, Barnes LA, Shields DJ, Huang M, Lau SK, Prevost N, Tarin D, Shattil SJ, Cheresh DA (2009) An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat Med 15(10):1163–1169
Dhingra R, Sharma T, Singh S, Sharma S, Tomar P, Malhotra M, Bhardwaj TR (2013) Enzalutamide: a novel anti-androgen with prolonged survival rate in CRPC patients. Mini Rev Med Chem 13(10):1475–1486
D’Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, Piaggio G, Fanciulli M, Appella E, Soddu S (2002) Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol 4(1):11–19
Fearon ER (2010) Molecular genetics of colorectal cancer. Annu Rev Pathol 6:479–507
Fiorini C, Cordani M, Padroni C, Blandino G, Di Agostino S, Donadelli M (2015) Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim Biophys Acta 1853(1):89–100
Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363(20):1938–1948
Freed-Pastor WA, Prives C (2012) Mutant p53: one name, many proteins. Genes Dev 26(12):1268–1286
Frese KK, Neesse A, Cook N, Bapiro TE, Lolkema MP, Jodrell DI, Tuveson DA (2012) nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov 2(3):260–269
Galluzzi L, Morselli E, Kepp O, Tajeddine N, Kroemer G (2008) Targeting p53 to mitochondria for cancer therapy. Cell Cycle 7(13):1949–1955
Gandhi J, Afridi A, Vatsia S, Joshi G, Joshi G, Kaplan SA, Smith NL, Khan SA (2018) The molecular biology of prostate cancer: current understanding and clinical implications. Prostate Cancer Prostatic Dis 21(1):22–36
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1(3):313–323
Hikasa H, Sokol SY (2011) Phosphorylation of TCF proteins by homeodomain-interacting protein kinase 2. J Biol Chem 286(14):12093–12100
Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369(18):1691–1703
Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias JL, Zhang H, Soon-Shiong P, Shi T, Rajeshkumar NV, Maitra A, Hidalgo M (2011) Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 29(34):4548–4554
Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (2019) SEER cancer statistics review, 1975-2016. National Cancer Institute, Bethesda, MD, Basedon November 2018 SEER data submission https://seer.cancer.gov/csr/1975_2016/
Iacopetta B (2003) TP53 mutation in colorectal cancer. Hum Mutat 21(3):271–276
Janke C (2014) The tubulin code: molecular components, readout mechanisms, and functions. J cell Biol 206(4):461–472
Janke C, Bulinski JC (2011) Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 12(12):773–786
Janssen A, Medema RH (2011) Mitosis as an anti-cancer target. Oncogene 30(25):2799–2809
Jernberg E, Bergh A, Wikstrom P (2017) Clinical relevance of androgen receptor alterations in prostate cancer. Endocr Connect 6(8):R146–R161
Kahn M (2014) Can we safely target the WNT pathway? Nat Rev Drug Discov 13(7):513–532
Karantanos T, Corn PG, Thompson TC (2013) Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 32(49):5501–5511
Katzenwadel A, Wolf P (2015) Androgen deprivation of prostate cancer: leading to a therapeutic dead end. Cancer Lett 367(1):12–17
Kinzler KW, Vogelstein B (1996) Lessons from hereditary colorectal cancer. Cell 87(2):159–170
Koyanagi N, Nagasu T, Fujita F, Watanabe T, Tsukahara K, Funahashi Y, Fujita M, Taguchi T, Yoshino H, Kitoh K (1994) In vivo tumor growth inhibition produced by a novel sulfonamide, E7010, against rodent and human tumors. Cancer Res 54(7):1702–1706
Lane DP, Cheok CF, Lain S (2010) p53-based cancer therapy. Cold Spring Harb Perspect Biol 2(9):a001222
Levine AJ, Oren M (2009) The first 30 years of p53: growing ever more complex. Nat Rev Cancer 9(10):749–758
Li Q, Deng Q, Chao HP, Liu X, Lu Y, Lin K, Liu B, Tang GW, Zhang D, Tracz A, Jeter C, Rycaj K, Calhoun-Davis T, Huang J, Rubin MA, Beltran H, Shen J, Chatta G, Puzanov I, Mohler JL, Wang J, Zhao R, Kirk J, Chen X, Tang DG (2018) Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat Commun 9(1):3600
Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, Pusztai L (2008) Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol 26(8):1275–1281
Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N, Bon H, Zecchini V, Smith DM, Denicola GM, Mathews N, Osborne M, Hadfield J, Macarthur S, Adryan B, Lyons SK, Brindle KM, Griffiths J, Gleave ME, Rennie PS, Neal DE, Mills IG (2011) The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J 30(13):2719–2733
Masuda M, Sawa M, Yamada T (2015) Therapeutic targets in the Wnt signaling pathway: feasibility of targeting TNIK in colorectal cancer. Pharm Ther 156:1–9
Moll UM, Wolff S, Speidel D, Deppert W (2005) Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol 17(6):631–636
Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W, National Cancer Institute of Canada Clinical Trials, G. (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25(15):1960–1966
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW (1997) Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275(5307):1787–1790
Mukherji D, Omlin A, Pezaro C, Shamseddine A, de Bono J (2014) Metastatic castration-resistant prostate cancer (CRPC): preclinical and clinical evidence for the sequential use of novel therapeutics. Cancer Metastasis Rev 33(2-3):555–566
Nakai K, Hung MC, Yamaguchi H (2016) A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res 6(8):1609–1623
Nassar D, Blanpain C (2016) Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol 11:47–76
National Cancer Institute Developmental Therapeutics Program. https://dtp.cancer.gov/organization/dscb/obtaining/available_plates.htm. Accessed 10 Apr 2020
National Cancer Institute Types of Cancer Treatment. https://www.cancer.gov/about-cancer/treatment/types. Accessed 19 Apr 2020
Ni L, Llewellyn R, Kesler CT, Kelley JB, Spencer A, Snow CJ, Shank L, Paschal BM (2013) Androgen induces a switch from cytoplasmic retention to nuclear import of the androgen receptor. Mol Cell Biol 33(24):4766–4778
Novellasdemunt L, Antas P, Li VS (2015) Targeting Wnt signaling in colorectal cancer. A review in the theme: cell signaling: proteins, pathways and mechanisms. Am J Physiol Cell Physiol 309(8):C511–C521
Okolotowicz KJ, Dwyer M, Ryan D, Cheng J, Cashman EA, Moore S, Mercola M, Cashman JR (2018) Novel tertiary sulfonamides as potent anti-cancer agents. Bioorg Med Chem 26(15):4441–4451
Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, Bruun JA, Hansen TE, Johansen T, Deretic V (2012) TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37(2):223–234
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM (2014) Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74(11):2913–2921
Rao CV, Kurkjian CD, Yamada HY (2012) Mitosis-targeting natural products for cancer prevention and therapy. Curr drug targets 13(14):1820–1830
Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, Youle RJ, Dikic I (2016) Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA 113(15):4039–4044
Ritch CR, Cookson MS (2016) Advances in the management of castration resistant prostate cancer. BMJ 355:i4405
Schalken J, Fitzpatrick JM (2016) Enzalutamide: targeting the androgen signalling pathway in metastatic castration-resistant prostate cancer. BJU Int 117(2):215–225
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Flechon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, de Bono JS, Investigators, A. (2012) Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 367(13):1187–1197
Seguin L, Camargo MF, Wettersten HI, Kato S, Desgrosellier JS, von Schalscha T, Elliott KC, Cosset E, Lesperance J, Weis SM, Cheresh DA (2017) Galectin-3, a druggable vulnerability for KRAS-addicted cancers. Cancer Discov 7(12):1464–1479
Seguin L, Kato S, Franovic A, Camargo MF, Lesperance J, Elliott KC, Yebra M, Mielgo A, Lowy AM, Husain H, Cascone T, Diao L, Wang J, Wistuba II, Heymach JV, Lippman SM, Desgrosellier JS, Anand S, Weis SM, Cheresh DA (2014) An integrin beta(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat cell Biol 16(5):457–468
Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70(1):7–30
Sridharan M, Hubbard JM, Grothey A (2014) Colorectal cancer: how emerging molecular understanding affects treatment decisions. Oncology 28(2):110–118
Sriraman A, Radovanovic M, Wienken M, Najafova Z, Li Y, Dobbelstein M (2016) Cooperation of nutlin-3a and a Wip1 inhibitor to induce p53 activity. Oncotarget 7(22):31623–31638
Su LK, Vogelstein B, Kinzler KW (1993) Association of the APC tumor suppressor protein with catenins. Science 262(5140):1734–1737
Suen DF, Norris KL, Youle RJ (2008) Mitochondrial dynamics and apoptosis. Genes Dev 22(12):1577–1590
Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G (2017) MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol 10(1):133
Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL (2009) Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324(5928):787–790
Tseng WW, Winer D, Kenkel JA, Choi O, Shain AH, Pollack JR, French R, Lowy AM, Engleman EG (2010) Development of an orthotopic model of invasive pancreatic cancer in an immunocompetent murine host. Clin Cancer Res 16(14):3684–3695
Vaseva AV, Moll UM (2009) The mitochondrial p53 pathway. Biochim Biophys Acta 1787(5):414–420
Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333(6039):228–233
Xu J, Wang J, Hu Y, Qian J, Xu B, Chen H, Zou W, Fang JY (2014) Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis 5:e1108
Yang J, Zhang W, Evans PM, Chen X, He X, Liu C (2006) Adenomatous polyposis coli (APC) differentially regulates beta-catenin phosphorylation and ubiquitination in colon cancer cells. J Biol Chem 281(26):17751–17757
Yang JC, Ok JH, Busby JE, Borowsky AD, Kung HJ, Evans CP (2009) Aberrant activation of androgen receptor in a new neuropeptide-autocrine model of androgen-insensitive prostate cancer. Cancer Res 69(1):151–160
Ying H, Shen X, Park B, Yue BY (2010) Posttranslational modifications, localization, and protein interactions of optineurin, the product of a glaucoma gene. PLoS ONE 5(2):e9168
Yoshimatsu K, Yamaguchi A, Yoshino H, Koyanagi N, Kitoh K (1997) Mechanism of action of E7010, an orally active sulfonamide antitumor agent: inhibition of mitosis by binding to the colchicine site of tubulin. Cancer Res 57(15):3208–3213
Zarif JC, Miranti CK (2016) The importance of non-nuclear AR signaling in prostate cancer progression and therapeutic resistance. Cell Signal 28(5):348–356
Zhou XJ, Rahmani R (1992) Preclinical and clinical pharmacology of vinca alkaloids. Drugs 44(4):1–16. Discussion 66-9
Acknowledgements
We acknowledge all the coworkers and collaborators cited in the references that contributed to this work. We are grateful to the financial support of the National Institutes of Health and the California Institute for Regenerative Medicine (CIRM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Dedication: This study is dedicated to Professor Robert Hanzlik on the occasion of his retirement from the University of Kansas. As a graduate student working under Professor Hanzlik and later as a practicing scientist, I learned a great deal from Bob. He provided very thoughtful perspectives on science, teamwork, and professional networking. Most importantly, he taught me about the joy of science. He was kind, good-hearted, and firm—all virtues of a compassionate teacher and wonderful mentor. Because I worked on a sulfur-containing compound under his direction, it is only fitting that the story told herein is about a new approach to addressing cancer with sulfur-containing compound PAWI-2.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cheng, J., Cashman, J.R. PAWI-2: A novel inhibitor for eradication of cancer. Med Chem Res 29, 1147–1159 (2020). https://doi.org/10.1007/s00044-020-02575-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-020-02575-8